Dupuytren’s Disease Is Mediated by Insufficient TGF-β1 Release and Degradation

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
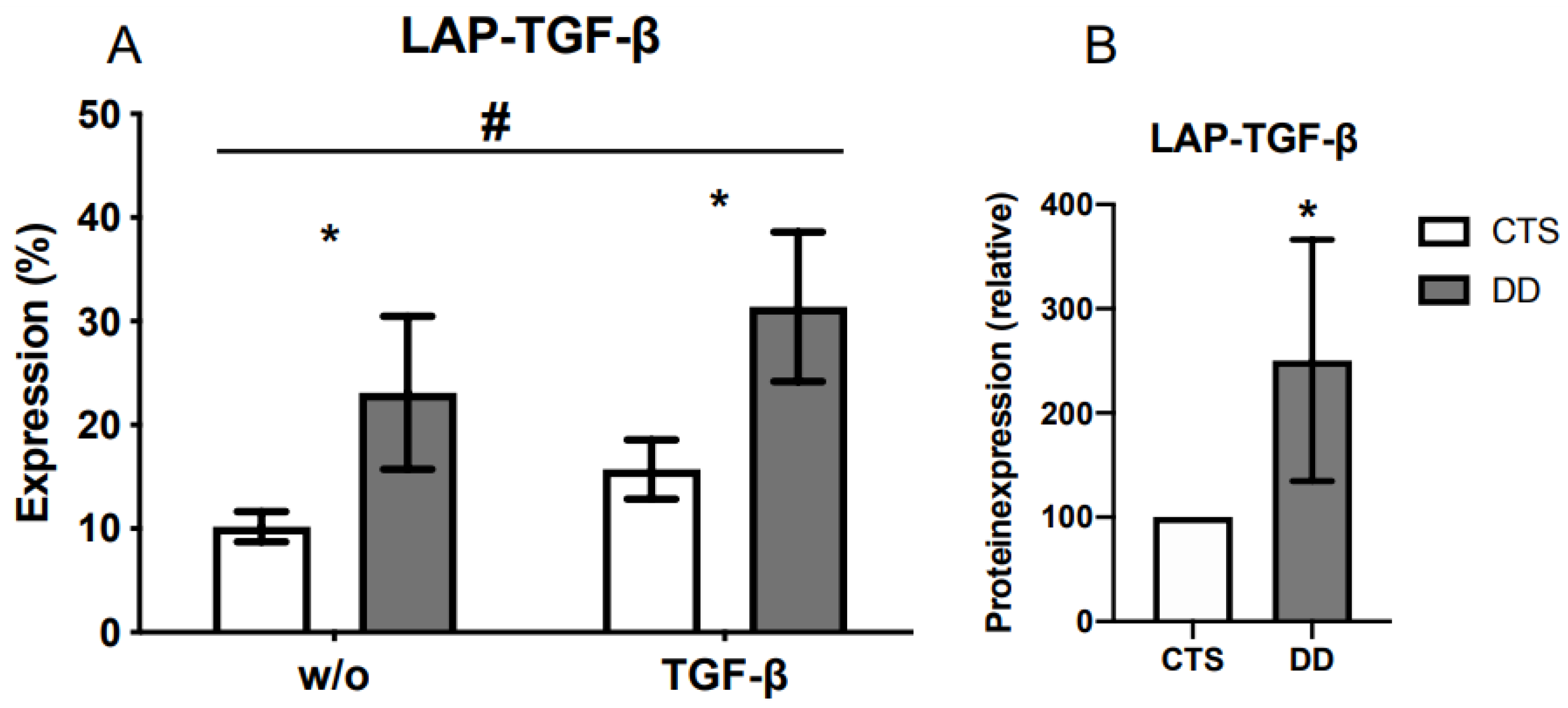
2.1. Comparison of Latency-Associated Peptide (LAP)-Transforming Growth Factor-β (TGF-β) Expression
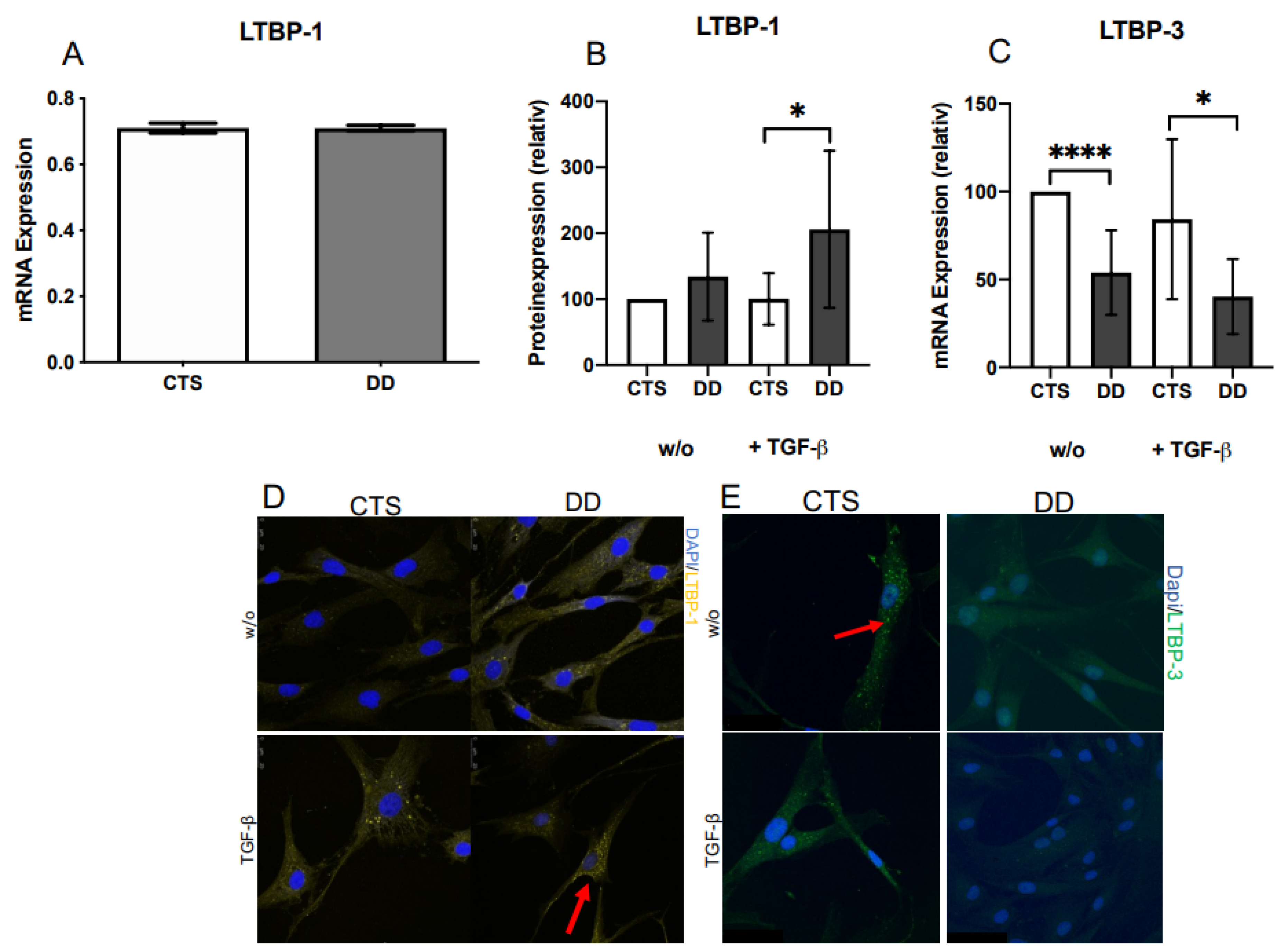
2.2. Evaluation of Latent TGF-β Binding Protein (LTBP)-1 and LTBP-3 Expression
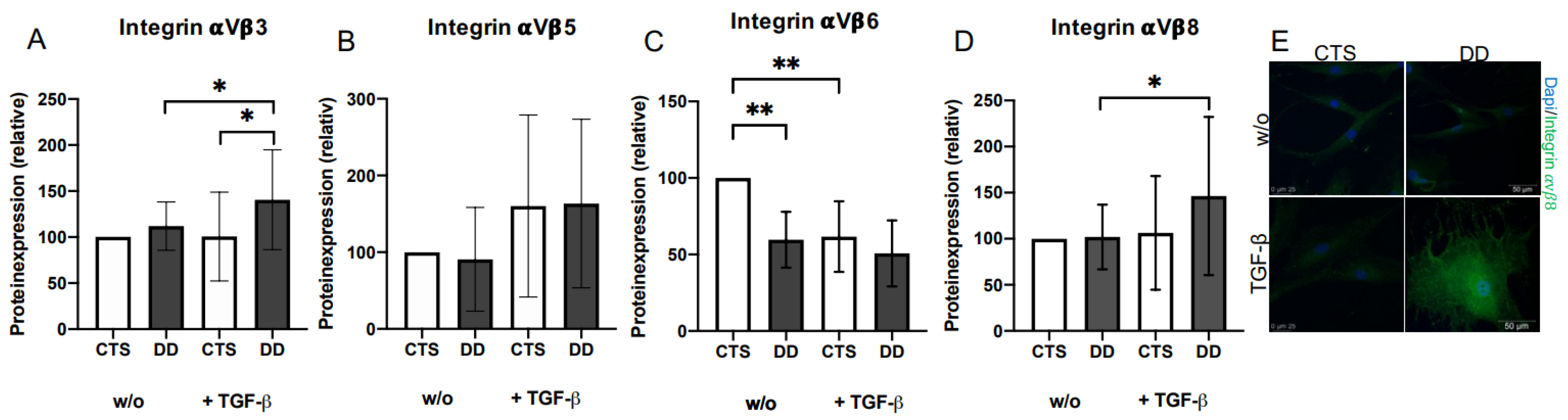
2.3. Evaluation of the Impact of 𝛼Vβ3, 𝛼Vβ5, 𝛼Vβ6, and 𝛼Vβ 8 Integrins on DD’s Myofibroblastogenesis
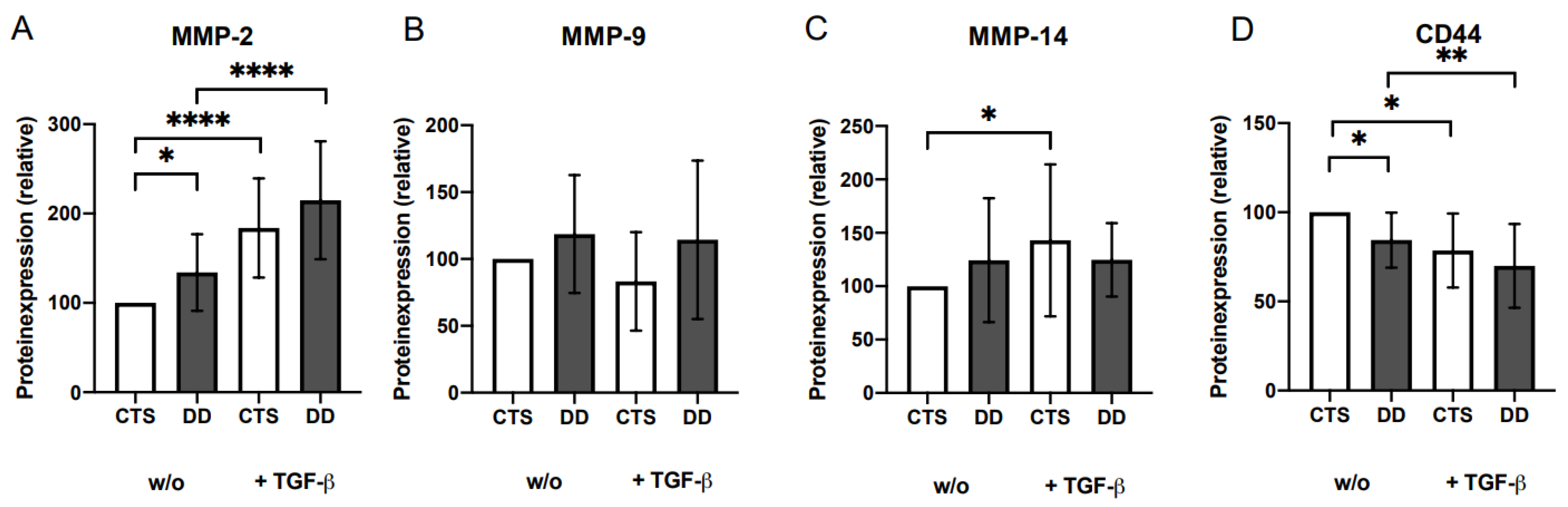
2.4. Analysis of Matrix Metalloproteinases (MMP)-2, MMP-9, and MMP-14 and CD44 Expressions
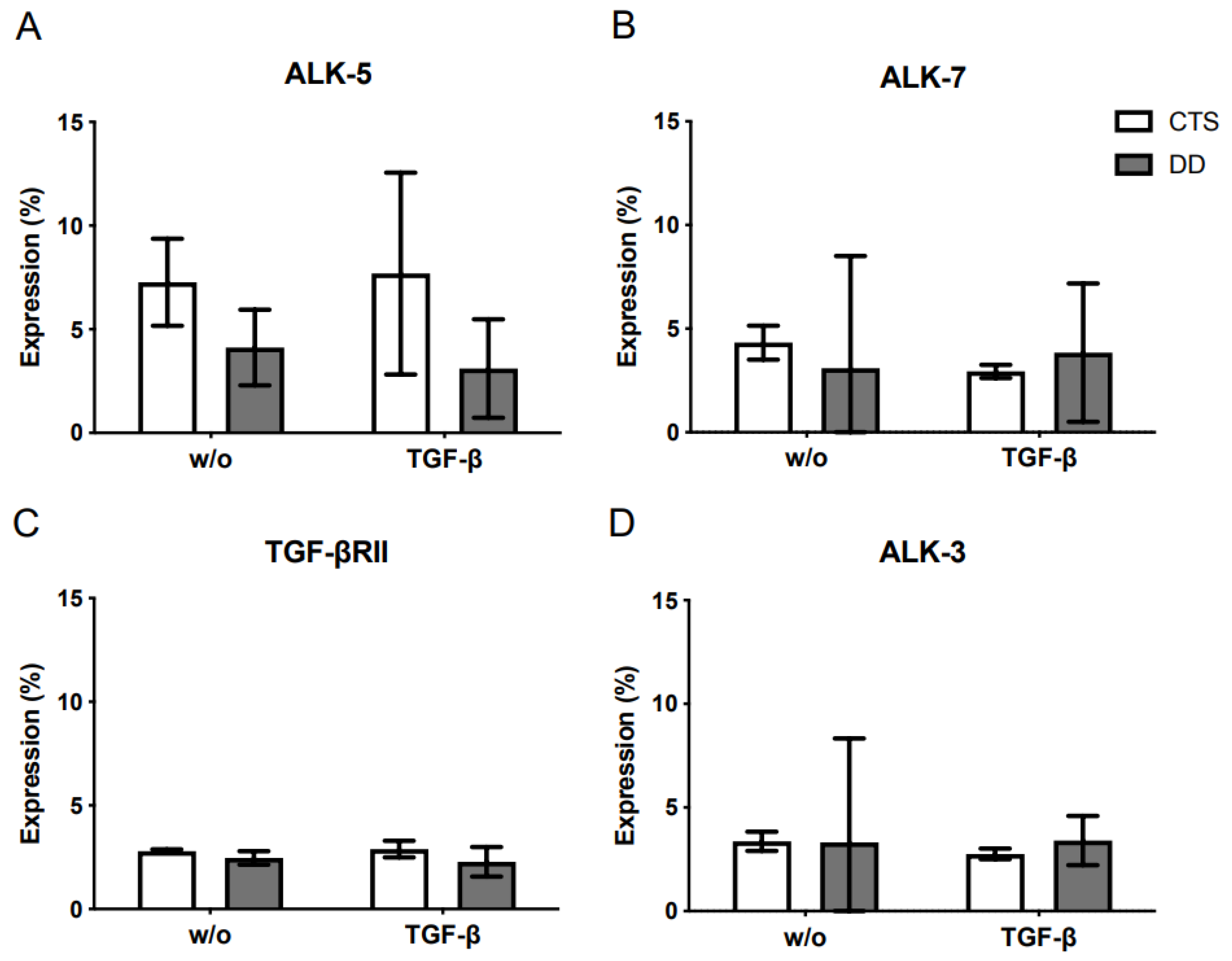
2.5. Analysis of the TGF-β Receptor Expression
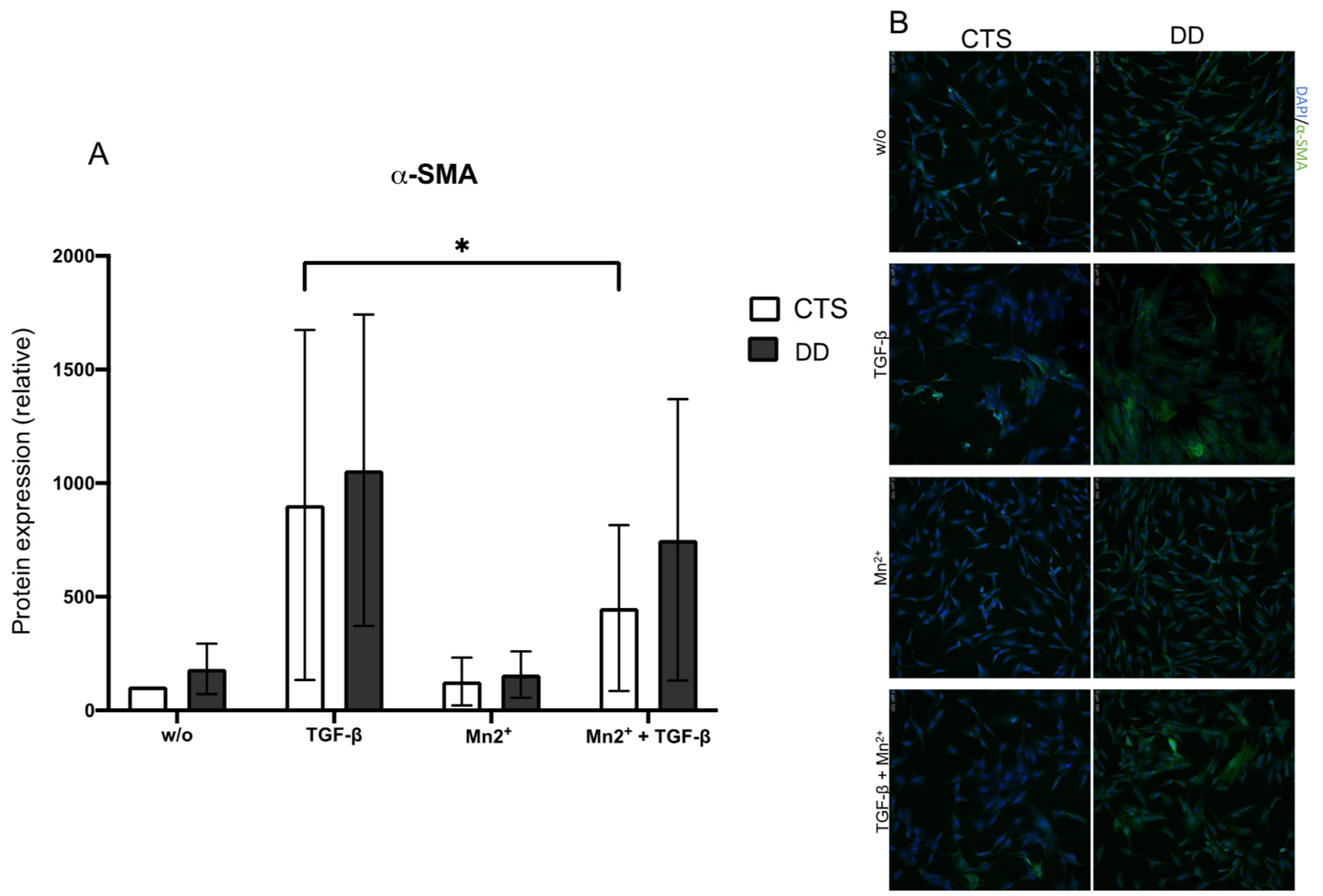
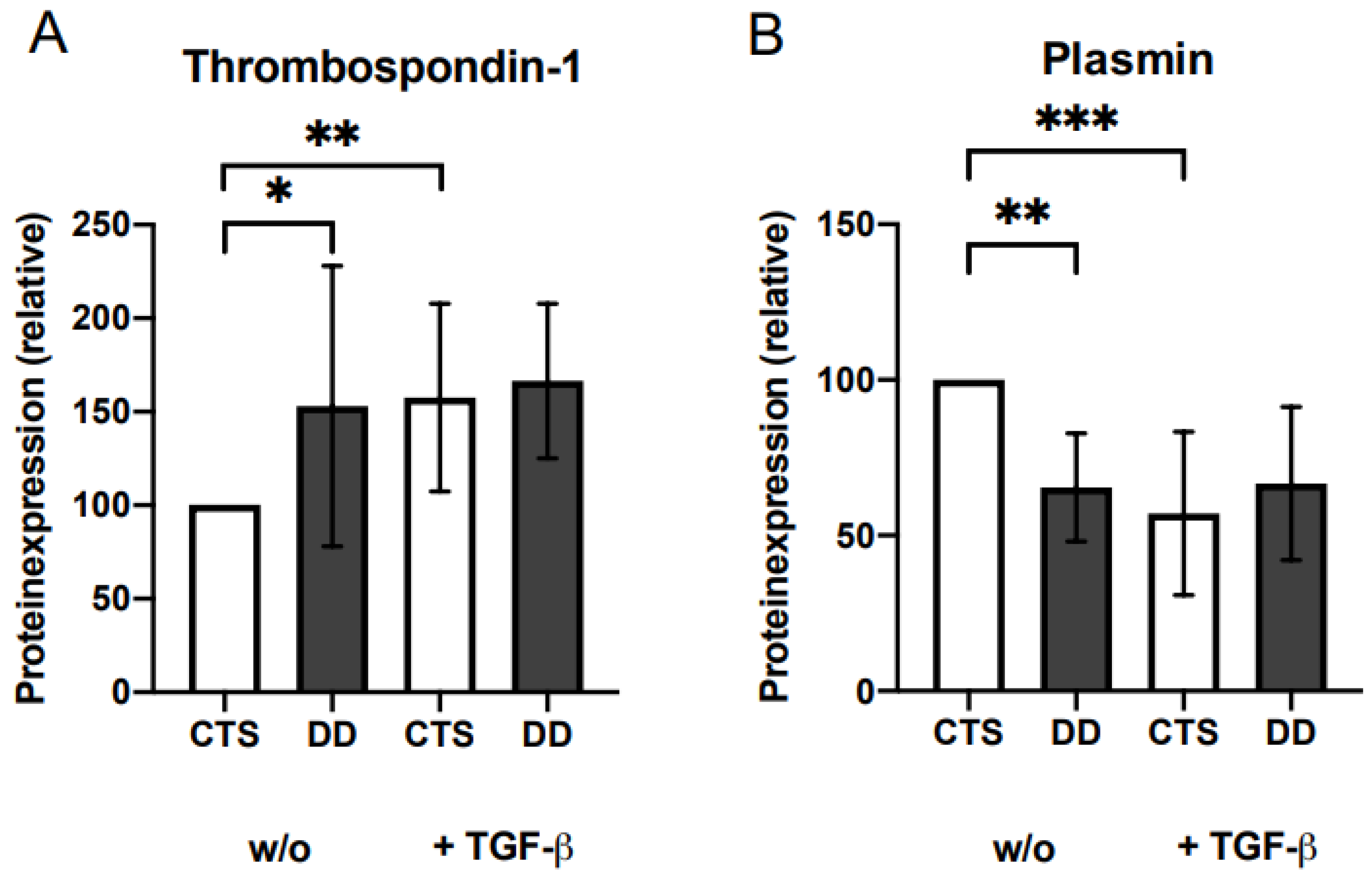
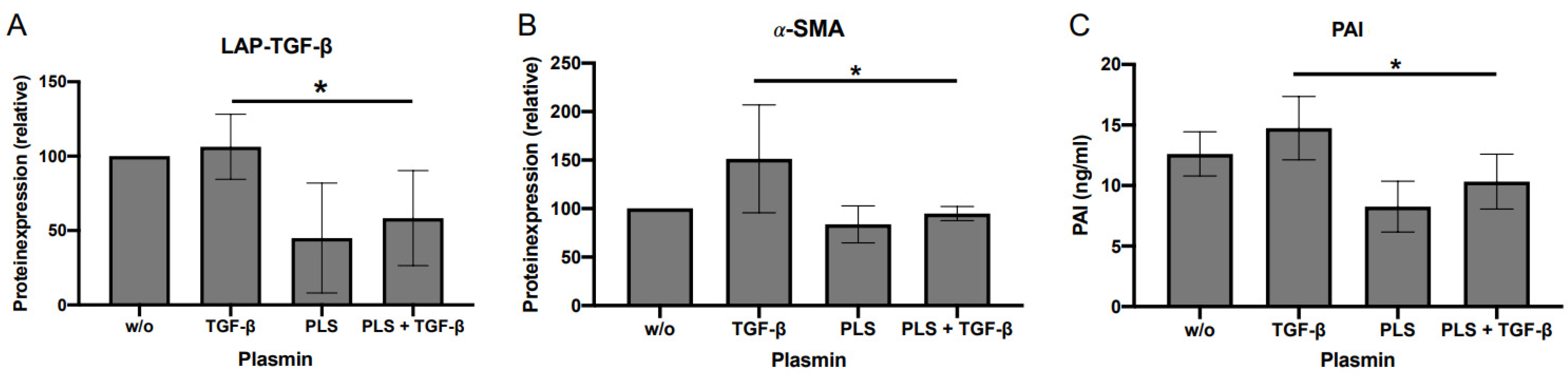
2.6. Evaluation of the Impact of Plasmin and Thrombospondin (TSP)-1 on DD’s Myofibroblastogenesis
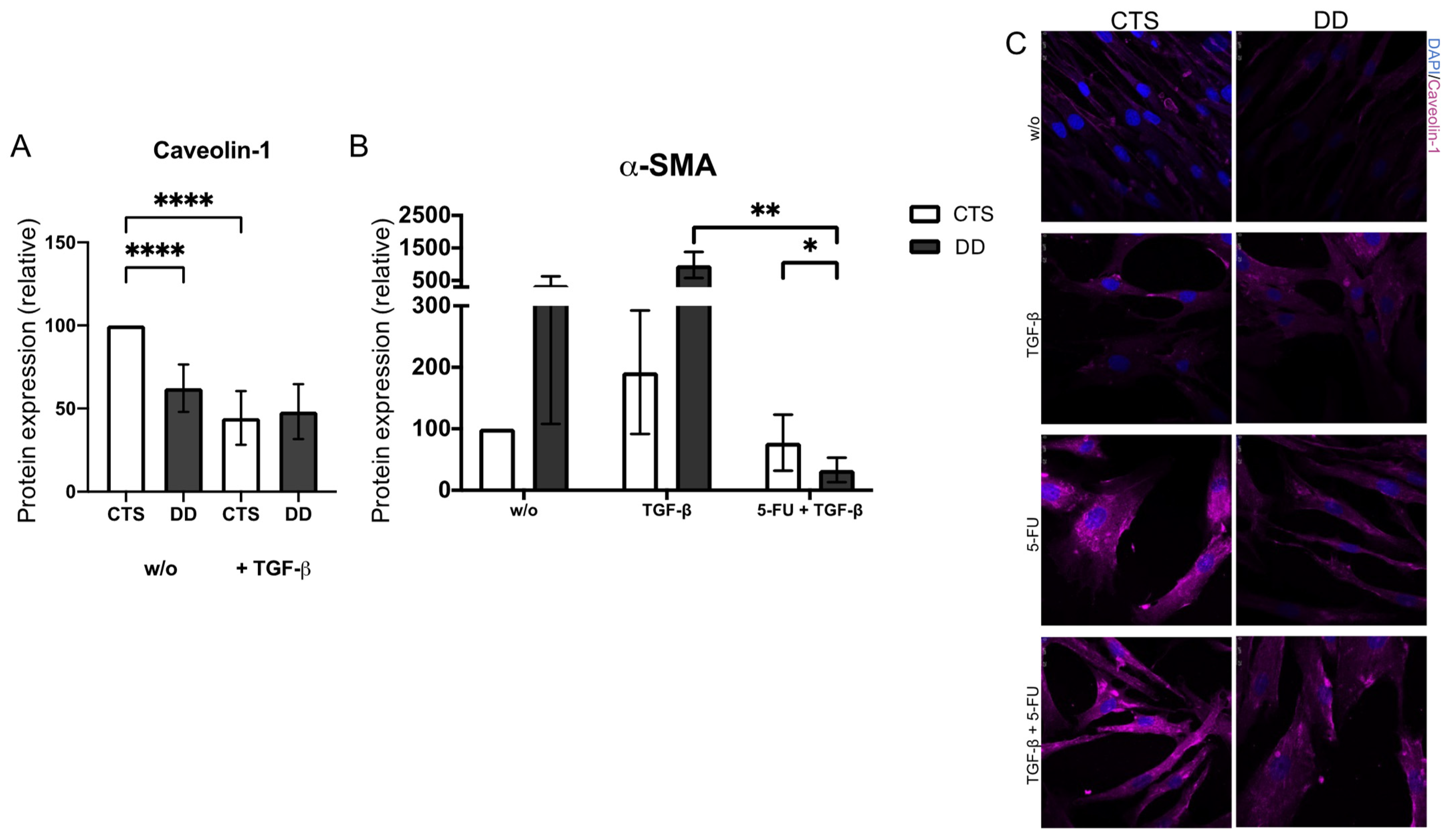
2.7. Evaluation of Caveolin-1 Expression in DD
3. Discussion
Limitations of the Study
4. Materials and Methods
4.1. Ethics Approval and Consent to Participate
4.2. Materials
4.3. Isolation and Cultivation of Human Fibroblasts (FBs)
4.4. Serpin E1/Plasminogen Activator Inhibitor (PAI)–Enzyme Linked Immunosorbent Assay (ELISA)
4.5. Flow Cytometry Analysis of Receptor Expression
4.6. Western Blot Analysis
4.7. mRNA Expression Analysis of LTBP-1 and LTBP-3
4.8. Immunofluorescence
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henry, M. Dupuytren’s disease: Current state of the art. Hand 2014, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hindocha, S.; John, S.; Stanley, J.K.; Watson, S.J.; Bayat, A. The heritability of Dupuytren’s disease: Familial aggregation and its clinical significance. J. Hand Surg. Am. 2006, 31, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Brenner, P.; Krause-Bergmann, A.; Van, V.H. Dupuytren contracture in North Germany. Epidemiological study of 500 cases. Der Unfallchirurg 2001, 104, 303–311. [Google Scholar] [CrossRef]
- Hindocha, S. Risk Factors, Disease Associations, and Dupuytren Diathesis. Hand Clin. 2018, 34, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Gabbiani, G. Modulation of α-smooth muscle actin expression in fibroblasts by transforming growth factor-β isoforms: An in vivo and in vitro study. Wound Repair Regen. 1996, 4, 278–287. [Google Scholar] [CrossRef]
- Vaughan, M.B.; Howard, E.W.; Tomasek, J.J. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 2000, 257, 180–189. [Google Scholar] [CrossRef]
- Mustoe, T.A.; Pierce, G.F.; Thomason, A.; Gramates, P.; Sporn, M.B.; Deuel, T.F. Accelerated healing of incisional wounds in rats induced by transforming growth factor-beta. Science 1987, 237, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massagué, J. Type beta transforming growth factor controls the adipogenic differentiation of 3T3 fibroblasts. Proc. Natl. Acad. Sci. USA 1985, 82, 8530–8534. [Google Scholar] [CrossRef]
- Twardzik, D.R. Differential expression of transforming growth factor-alpha during prenatal development of the mouse. Cancer Res. 1985, 45 Pt 1, 5413–5416. [Google Scholar]
- Knowles, M.A.; Hicks, R.M.; Dave, H.; Harvey, A.E.; Roberts, A.B.; Sporn, M.B. Transforming growth factors induce markers of neoplasia in cultured adult rat bladder. Carcinogenesis 1985, 6, 595–604. [Google Scholar] [CrossRef]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H.; et al. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar] [CrossRef] [PubMed]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.; Rayan, G.M. Correlation of alpha-smooth muscle actin expression and contraction in Dupuytren’s disease fibroblasts. J. Hand Surg. Am. 1995, 20, 450–455. [Google Scholar] [CrossRef]
- Lyons, R.M.; Gentry, L.E.; Purchio, A.F.; Moses, H.L. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J. Cell Biol. 1990, 110, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Olofsson, A.; Colosetti, P.; Heldin, C.H. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO J. 1991, 10, 1091–1101. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; Kojima, S.; Abe, M.; Rifkin, D.B. Activation of latent transforming growth factor beta. Adv. Pharmacol. 1993, 24, 51–76. [Google Scholar]
- Taipale, J.; Miyazono, K.; Heldin, C.H.; Keski-Oja, J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J. Cell Biol. 1994, 124, 171–181. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol 2016, 8, a021907. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar]
- Brown, P.D.; Wakefield, L.M.; Levinson, A.D.; Sporn, M.B. Physicochemical activation of recombinant latent transforming growth factor-beta’s 1, 2, and 3. Growth Factors 1990, 3, 35–43. [Google Scholar] [CrossRef]
- Lyons, R.M.; Keski-Oja, J.; Moses, H.L. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J. Cell Biol. 1988, 106, 1659–1665. [Google Scholar] [CrossRef]
- Ahamed, J.; Burg, N.; Yoshinaga, K.; Janczak, C.A.; Rifkin, D.B.; Coller, B.S. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-beta1. Blood 2008, 112, 3650–3660. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J. Immunol. 2005, 175, 7708–7718. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Tamaki, K. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am. J. Pathol. 2006, 168, 499–510. [Google Scholar] [CrossRef]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G. The role of proteases in transforming growth factor-beta activation. Int. J. Biochem. Cell Biol. 2008, 40, 1068–1078. [Google Scholar] [CrossRef]
- Schultz-Cherry, S.; Murphy-Ullrich, J.E. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J. Cell Biol. 1993, 122, 923–932. [Google Scholar] [CrossRef]
- Echarri, A.; Del Pozo, M.A. Caveolae—Mechanosensitive membrane invaginations linked to actin filaments. J. Cell Sci. 2015, 128, 2747–2758. [Google Scholar] [CrossRef]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef]
- Koli, K.; Ryynänen, M.J.; Keski-Oja, J. Latent TGF-beta binding proteins (LTBPs)-1 and -3 coordinate proliferation and osteogenic differentiation of human mesenchymal stem cells. Bone 2008, 43, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Hyytiäinen, M.; Penttinen, C.; Keski-Oja, J. Latent TGF-beta binding proteins: Extracellular matrix association and roles in TGF-beta activation. Crit. Rev. Clin. Lab. Sci. 2004, 41, 233–264. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Lundin, L.; Oberg, K.; Wilander, E.; Miyazono, K.; Heldin, C.H.; Funa, K. Involvement of transforming growth factor-beta in the formation of fibrotic lesions in carcinoid heart disease. Am. J. Pathol. 1993, 142, 71–78. [Google Scholar] [PubMed]
- Maeda, J.; Ueki, N.; Ohkawa, T.; Iwahashi, N.; Nakano, T.; Hada, T.; Higashino, K. Local production and localization of transforming growth factor-beta in tuberculous pleurisy. Clin. Exp. Immunol. 1993, 92, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Chen, Y.; Prijatelj, P.; Sakai, T.; Fässler, R.; Sakai, L.Y.; Rifkin, D.B. Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J. 2005, 19, 1798–1808. [Google Scholar] [CrossRef]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J.; Hinz, B. The single-molecule mechanics of the latent TGF-β1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef]
- Howard, J.C.; Varallo, V.M.; Ross, D.C.; Faber, K.J.; Roth, J.H.; Seney, S.; Gan, B.S. Wound healing-associated proteins Hsp47 and fibronectin are elevated in Dupuytren’s contracture. J. Surg. Res. 2004, 117, 232–238. [Google Scholar] [CrossRef]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef]
- Vehviläinen, P.; Koli, K.; Myllärniemi, M.; Lindholm, P.; Soini, Y.; Salmenkivi, K.; Kinnula, V.L.; Keski-Oja, J. Latent TGF-β binding proteins (LTBPs) 1 and 3 differentially regulate transforming growth factor-β activity in malignant mesothelioma. Hum. Pathol. 2011, 42, 269–278. [Google Scholar] [CrossRef]
- Saharinen, J.; Keski-Oja, J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell 2000, 11, 2691–2704. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.; Jenkins, G. Role of integrin-mediated TGFβ activation in the pathogenesis of pulmonary fibrosis. Biochem. Soc. Trans. 2009, 37, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Puthawala, K.; Hadjiangelis, N.; Jacoby, S.C.; Bayongan, E.; Zhao, Z.; Yang, Z.; Devitt, M.L.; Horan, G.S.; Weinreb, P.H.; Lukashev, M.E.; et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 82–90. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak-Wielgomas, K.; Gosk, J.; Rabczyński, J.; Augoff, K.; Podhorska-Okołów, M.; Gamian, A.; Rutowski, R. Expression of MMP-2, TIMP-2, TGF-β1, and Decorin in Dupuytren’s Contracture. Connect. Tissue Res. 2012, 53, 469–477. [Google Scholar] [CrossRef]
- Ulrich, D.; Ulrich, F.; Piatkowski, A.; Pallua, N. Expression of matrix metalloproteinases and their inhibitors in cords and nodules of patients with Dupuytren’s disease. Arch. Orthop. Trauma Surg. 2009, 129, 1453–1459. [Google Scholar] [CrossRef]
- Taipale, J.; Lohi, J.; Saarinen, J.; Kovanen, P.T.; Keski-Oja, J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J. Biol. Chem. 1995, 270, 4689–4696. [Google Scholar] [CrossRef]
- Adams, J.C.; Lawler, J. The thrombospondins. Cold Spring Harb. Perspect. Biol. 2011, 3, a009712. [Google Scholar] [CrossRef]
- Grainger, D.J.; Frow, E.K. Thrombospondin 1 does not activate transforming growth factor beta1 in a chemically defined system or in smooth-muscle-cell cultures. Biochem. J. 2000, 350 Pt 1, 291–298. [Google Scholar] [CrossRef]
- Schultz-Cherry, S.; Chen, H.; Mosher, D.F.; Misenheimer, T.M.; Krutzsch, H.C.; Roberts, D.D.; Murphy-Ullrich, J.E. Regulation of transforming growth factor-beta activation by discrete sequences of thrombospondin 1. J. Biol. Chem. 1995, 270, 7304–7310. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Wagner, A.; Hohenstein, B.; Hugo, C. Thrombospondin-2 therapy ameliorates experimental glomerulonephritis via inhibition of cell proliferation, inflammation, and TGF-beta activation. Am. J. Physiol. Renal. Physiol. 2009, 297, F1299–F1309. [Google Scholar] [CrossRef]
- Song, C.Z.; Siok, T.E.; Gelehrter, T.D. Smad4/DPC4 and Smad3 mediate transforming growth factor-beta (TGF-beta) signaling through direct binding to a novel TGF-beta-responsive element in the human plasminogen activator inhibitor-1 promoter. J. Biol. Chem. 1998, 273, 29287–29290. [Google Scholar] [CrossRef] [PubMed]
- Kruglikov, I.L.; Scherer, P.E. Caveolin-1 as a target in prevention and treatment of hypertrophic scarring. NPJ Regen. Med. 2019, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, N.; Li, W.; Liu, P.; Chen, Q.; Situ, H.; Zhong, S.; Guo, L.; Lin, Y.; Shen, J.; et al. Caveolin-1 mediates chemoresistance in breast cancer stem cells via β-catenin/ABCG2 signaling pathway. Carcinogenesis 2014, 35, 2346–2356. [Google Scholar] [CrossRef]
- Del Galdo, F.; Sotgia, F.; de Almeida, C.J.; Jasmin, J.F.; Musick, M.; Lisanti, M.P.; Jiménez, S.A. Decreased expression of caveolin 1 in patients with systemic sclerosis: Crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum 2008, 58, 2854–2865. [Google Scholar] [CrossRef]
- Bisson, M.A.; McGrouther, D.A.; Mudera, V.; Grobbelaar, A.O. The different characteristics of Dupuytren’s disease fibroblasts derived from either nodule or cord: Expression of alpha-smooth muscle actin and the response to stimulation by TGF-beta1. J. Hand Surg. 2003, 28, 351–356. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oezel, L.; Wohltmann, M.; Gondorf, N.; Wille, J.; Güven, I.; Windolf, J.; Thelen, S.; Jaekel, C.; Grotheer, V. Dupuytren’s Disease Is Mediated by Insufficient TGF-β1 Release and Degradation. Int. J. Mol. Sci. 2023, 24, 15097. https://doi.org/10.3390/ijms242015097
Oezel L, Wohltmann M, Gondorf N, Wille J, Güven I, Windolf J, Thelen S, Jaekel C, Grotheer V. Dupuytren’s Disease Is Mediated by Insufficient TGF-β1 Release and Degradation. International Journal of Molecular Sciences. 2023; 24(20):15097. https://doi.org/10.3390/ijms242015097
Chicago/Turabian StyleOezel, Lisa, Marie Wohltmann, Nele Gondorf, Julia Wille, Irmak Güven, Joachim Windolf, Simon Thelen, Carina Jaekel, and Vera Grotheer. 2023. "Dupuytren’s Disease Is Mediated by Insufficient TGF-β1 Release and Degradation" International Journal of Molecular Sciences 24, no. 20: 15097. https://doi.org/10.3390/ijms242015097
APA StyleOezel, L., Wohltmann, M., Gondorf, N., Wille, J., Güven, I., Windolf, J., Thelen, S., Jaekel, C., & Grotheer, V. (2023). Dupuytren’s Disease Is Mediated by Insufficient TGF-β1 Release and Degradation. International Journal of Molecular Sciences, 24(20), 15097. https://doi.org/10.3390/ijms242015097