
Unravelling Novel SCN5A Mutations Linked to Brugada Syndrome: Functional, Structural, and Genetic Insights
 ,
,  , , , , , , , ,
, , , , , , , ,  , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Results
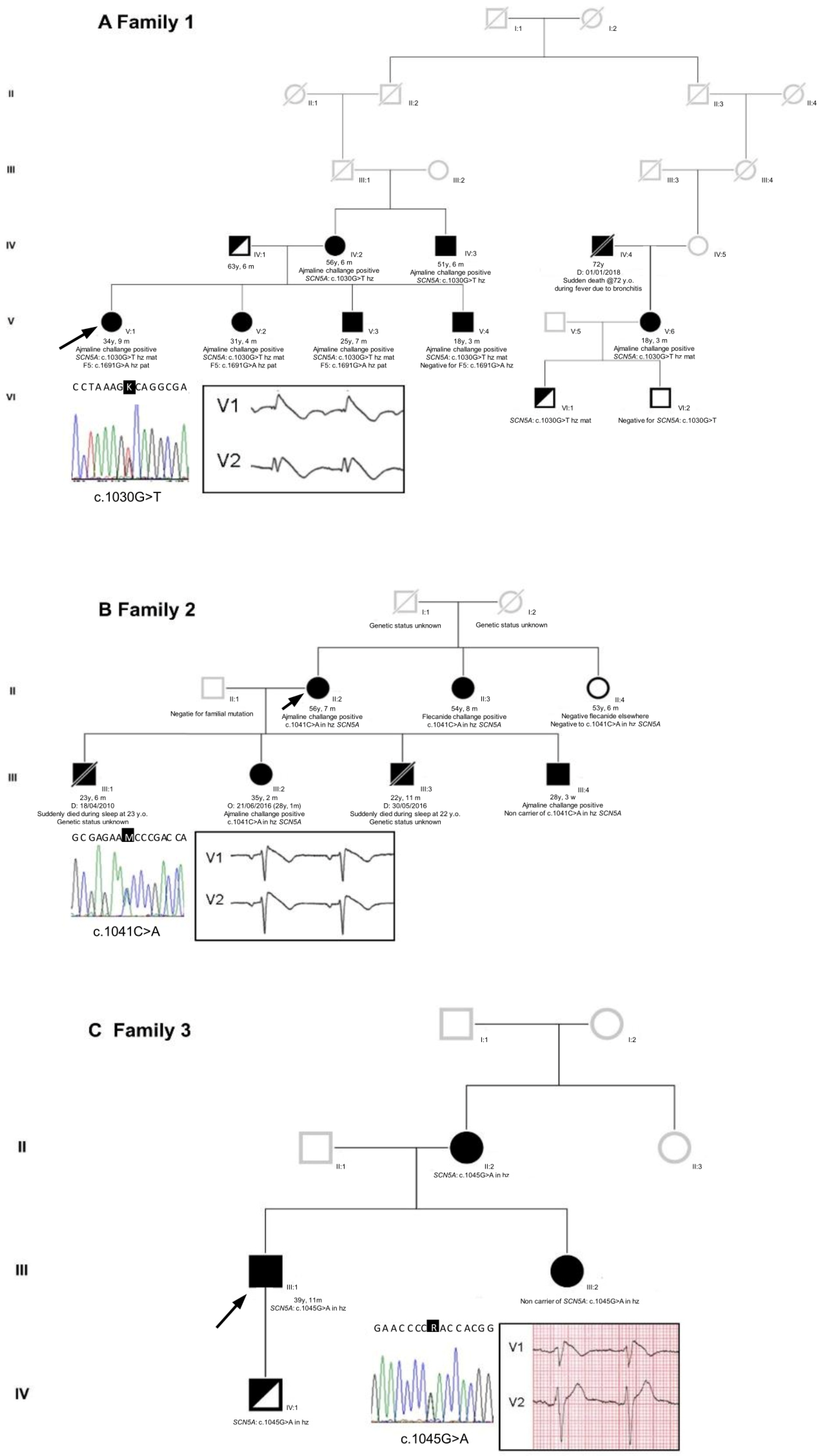
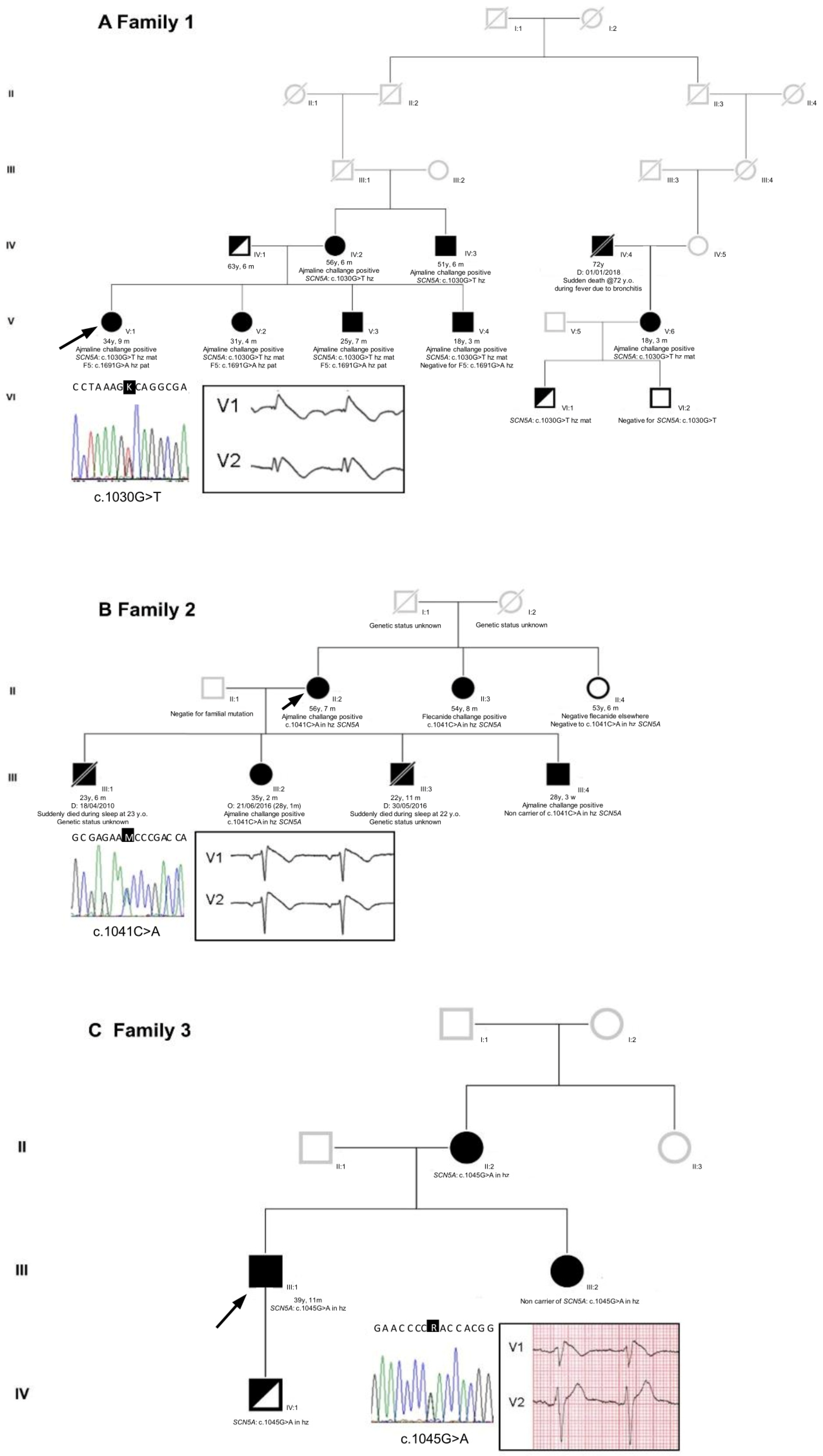
2.1. Proband Patient Characteristics
2.2. Genetic Testing Results and In Silico Prediction
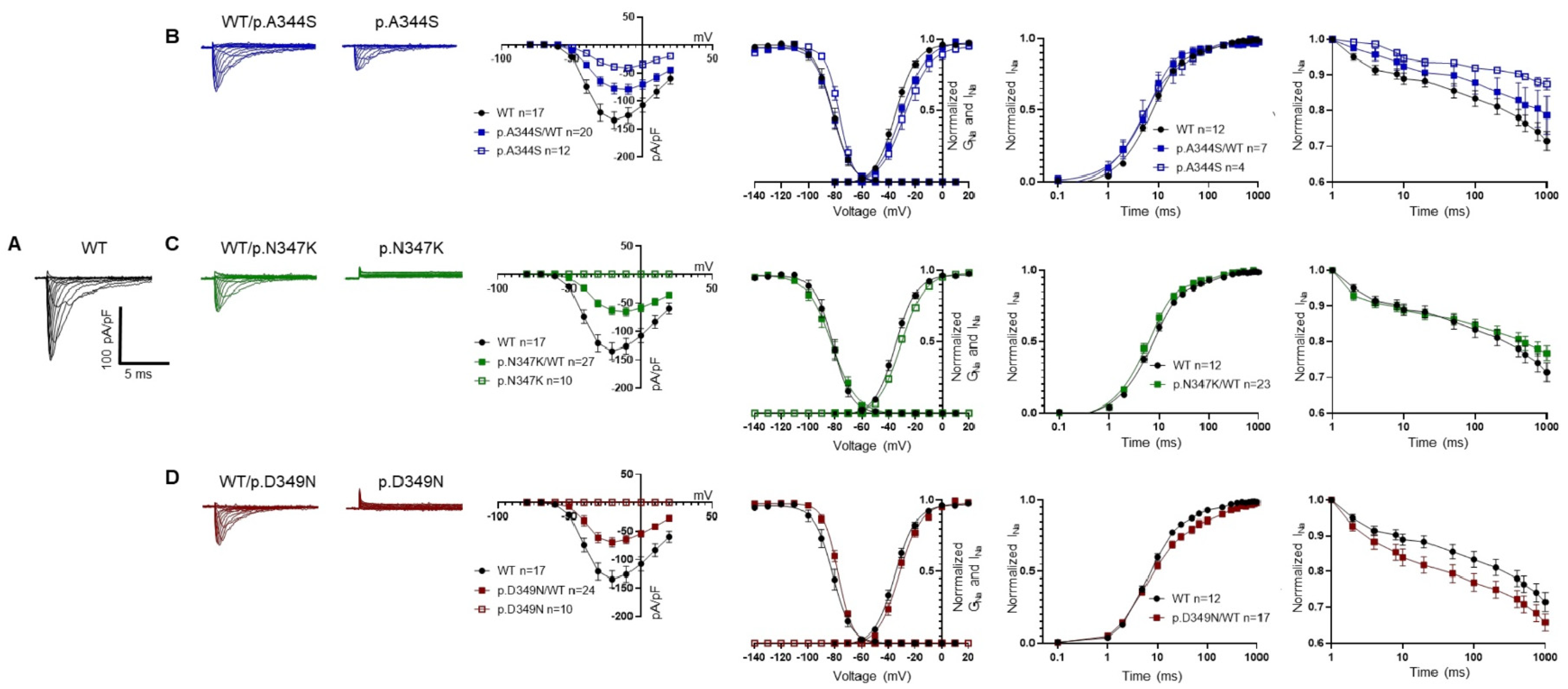
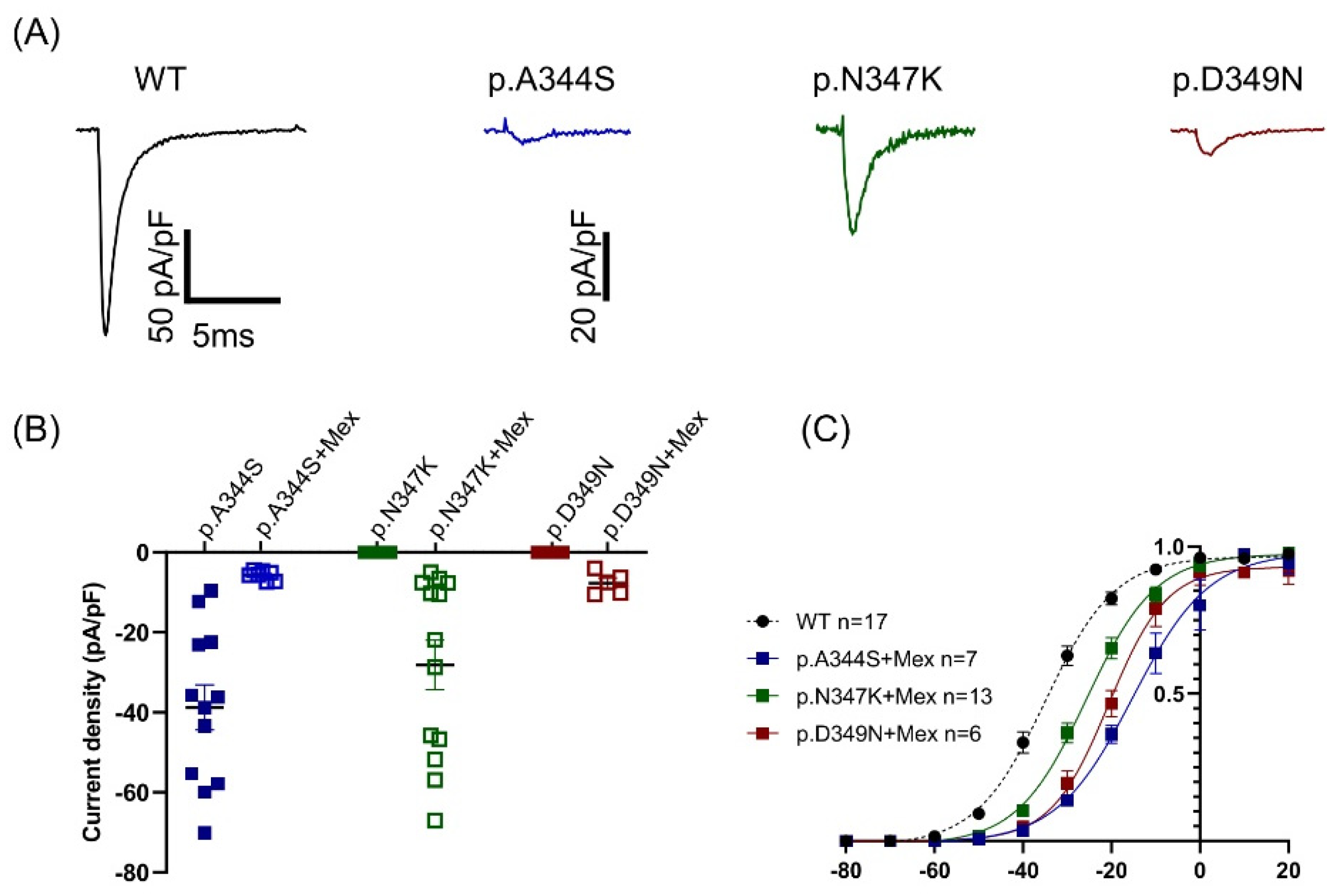
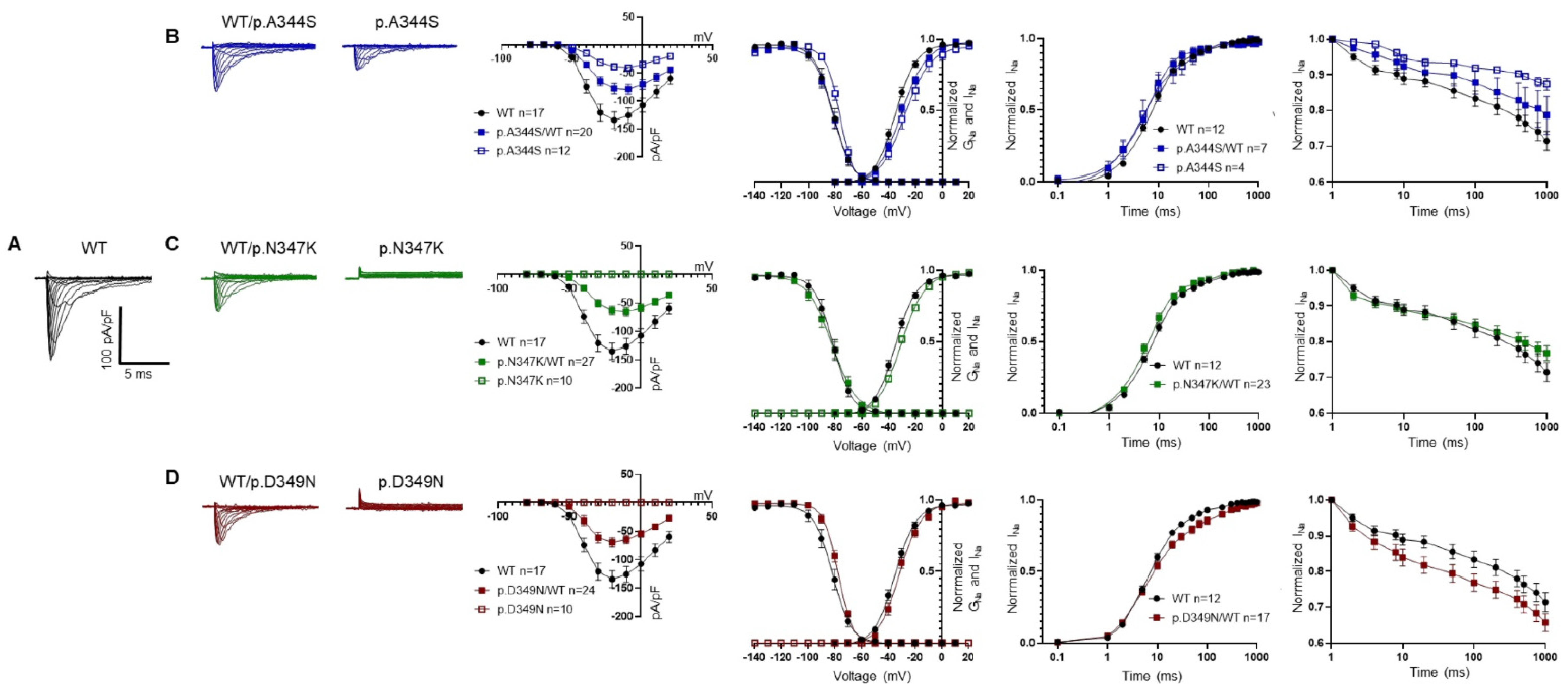
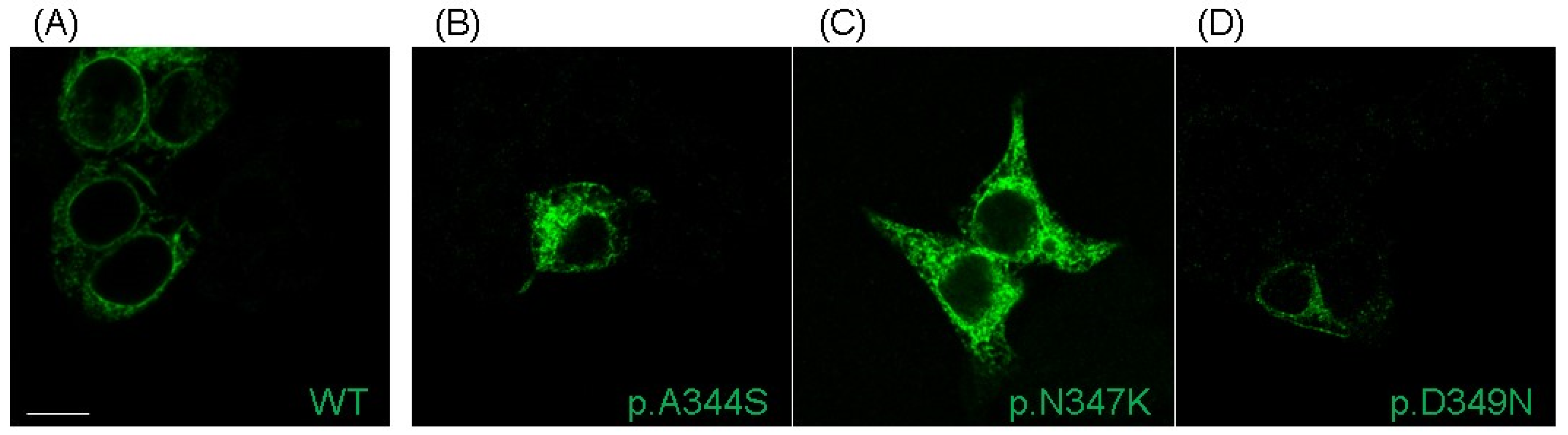
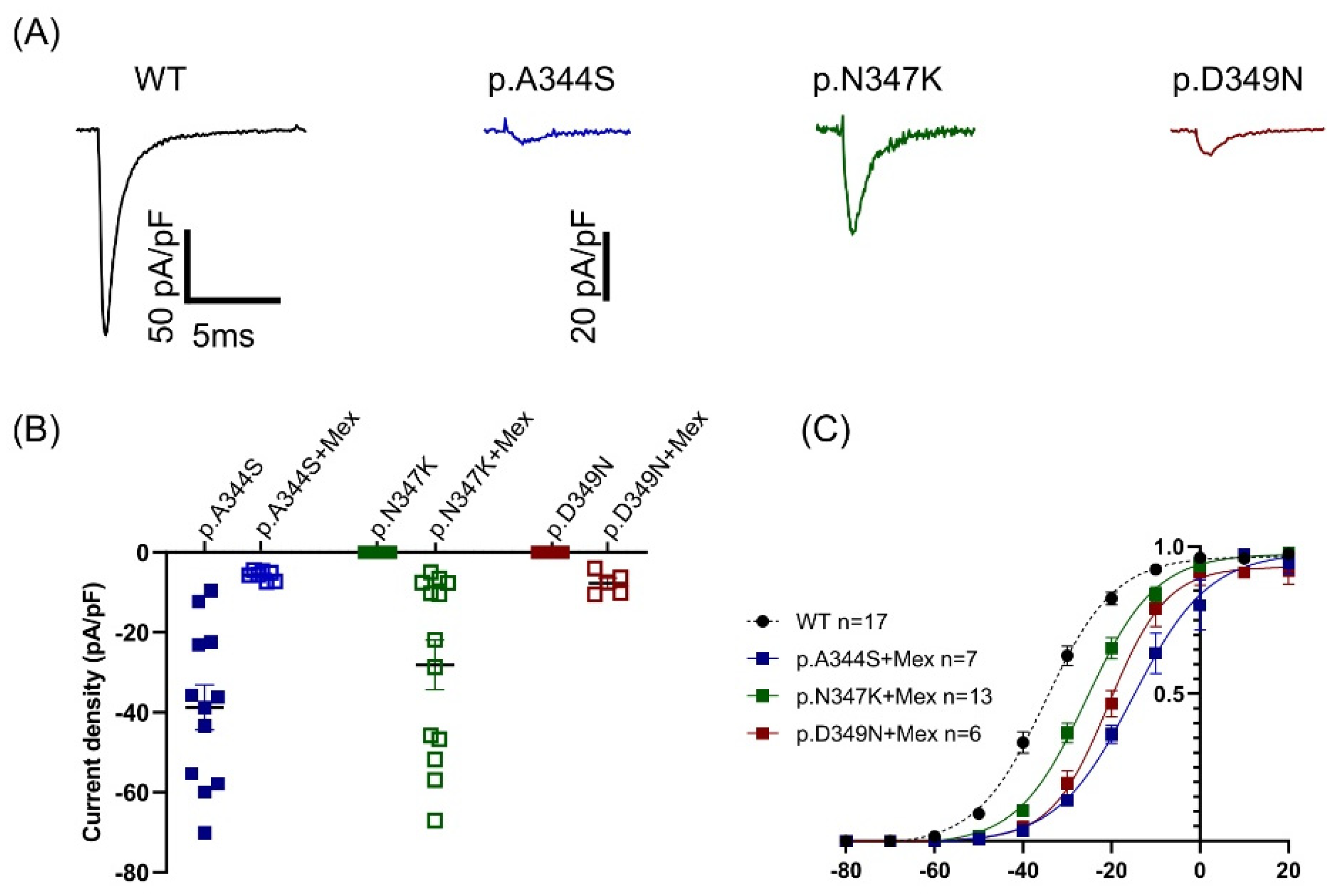
2.3. Functional Analysis and Protein Localization
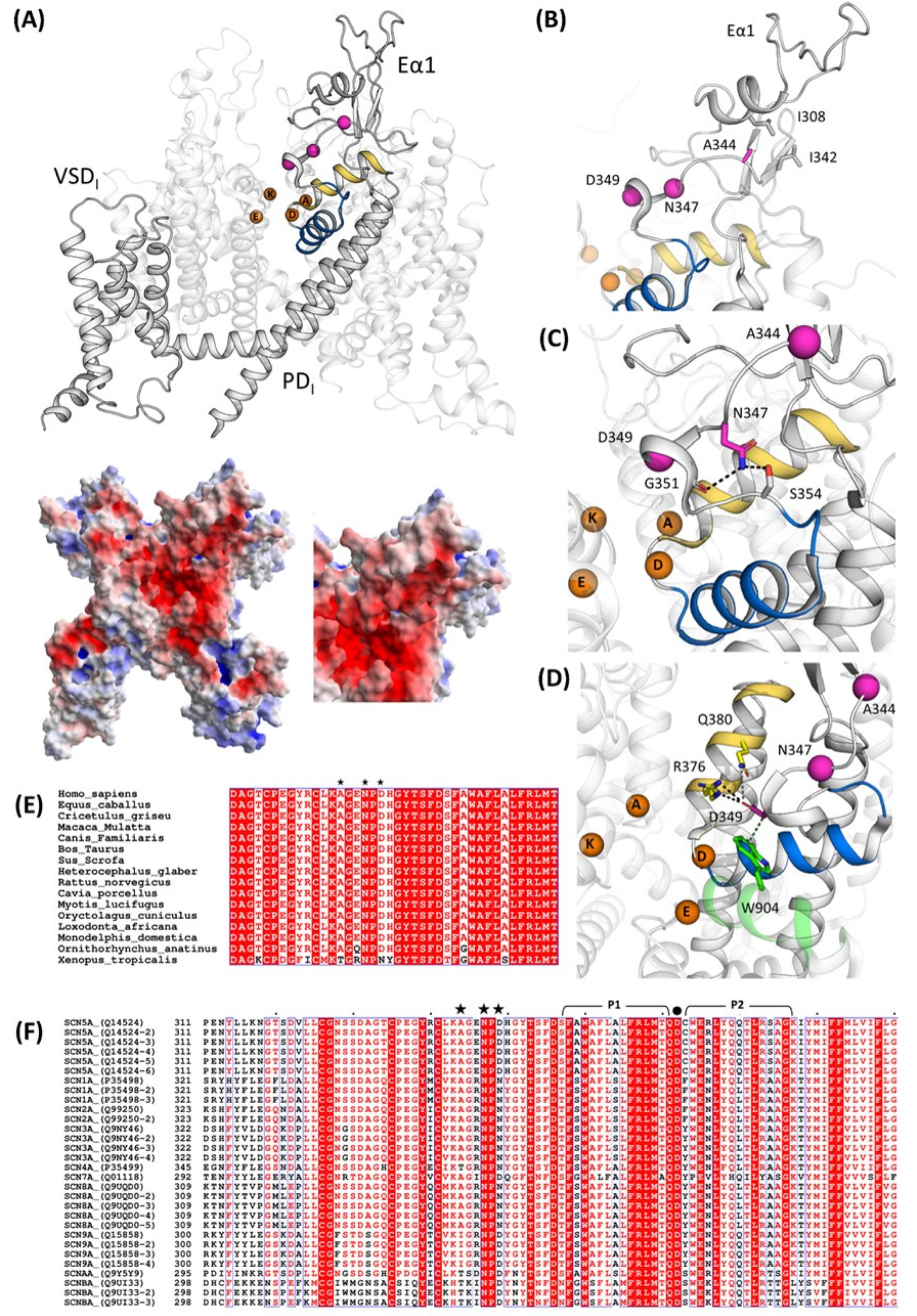
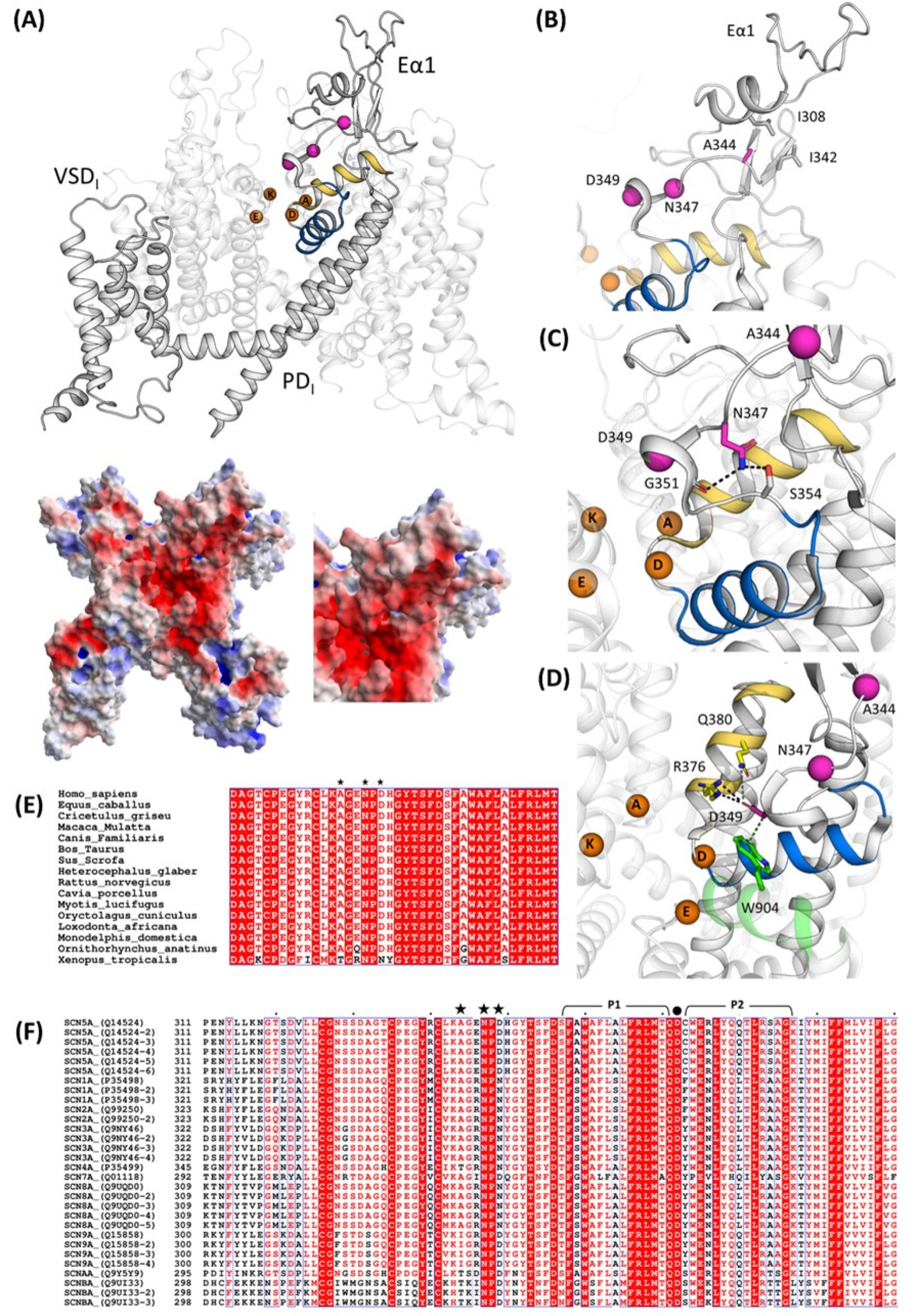
2.4. Structural Analysis
3. Discussion
Limitations of the Study
4. Materials and Methods
4.1. Subjects
4.2. DNA Extraction and Genetic Analysis
4.3. Plasmid Generation
4.4. Cell Culture and Transfection
4.5. Functional Analysis
4.5.1. Automated Patch Clamp
4.5.2. Manual Patch Clamp
4.5.3. Statistical Analysis
4.6. Immunofluorescence Staining
4.7. Structural Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pappone, C.; Santinelli, V. Brugada Syndrome: Progress in Diagnosis and Management. Arrhythmia Electrophysiol. Rev. 2019, 8, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Yan, G.-X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Europace 2017, 19, 665–694. [Google Scholar] [CrossRef] [PubMed]
- Sarquella-Brugada, G.; Campuzano, O.; Arbelo, E.; Brugada, J.; Brugada, R. Brugada syndrome: Clinical and genetic findings. Genet. Med. 2016, 18, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Marsman, E.M.J.; Postema, P.G.; Remme, C.A. Brugada syndrome: Update and future perspectives. Heart 2022, 108, 668–675. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Locati, E.T.; Pappone, C. Evaluating the Use of Genetics in Brugada Syndrome Risk Stratification. Front. Cardiovasc. Med. 2021, 8, 652027. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Brignole, M.; Giordano, U.; Giovannini, T.; Menozzi, C.; Bloise, R.; et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation 2000, 102, 2509–2515. [Google Scholar] [CrossRef]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef]
- Offerhaus, J.A.; Bezzina, C.R.; Wilde, A.A.M. Epidemiology of inherited arrhythmias. Nat. Rev. Cardiol. 2020, 17, 205–215. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef]
- Roden, D.M.; Balser, J.R.; George, A.L., Jr.; Anderson, M.E. Cardiac Ion Channels. Annu. Rev. Physiol. 2002, 64, 431–447. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, E.; Pappone, C.; Boneschi, F.M.; Di Resta, C.; Carbone, M.R.; Salvi, E.; Vergara, P.; Sala, S.; Cusi, D.; Ferrari, M.; et al. Genetics can contribute to the prognosis of Brugada syndrome: A pilot model for risk stratification. Eur. J. Hum. Genet. 2013, 21, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Nasilli, G.; Yiangou, L.; Palandri, C.; Cerbai, E.; Davis, R.P.; O Verkerk, A.; Casini, S.; Remme, C.A. Beneficial effects of chronic mexiletine treatment in a human model of SCN5A overlap syndrome. Europace 2023, 25, euad154. [Google Scholar] [CrossRef]
- Li, Z.; Jin, X.; Wu, T.; Huang, G.; Wu, K.; Lei, J.; Pan, X.; Yan, N. Structural Basis for Pore Blockade of the Human Cardiac Sodium Channel Nav1.5 by the Antiarrhythmic Drug Quinidine. Angew. Chem. Int. Ed. 2021, 60, 11474–11480. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jin, X.; Wu, T.; Zhao, X.; Wang, W.; Lei, J.; Pan, X.; Yan, N. Structure of human Nav1.5 reveals the fast inactivation-related segments as a mutational hotspot for the long QT syndrome. Proc. Natl. Acad. Sci. USA 2021, 18, e2100069118. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Jin, X.; Zhao, Y.; Huang, G.; Huang, X.; Shen, Z.; Cao, Y.; Dong, M.; Lei, J.; et al. Comparative structural analysis of human Nav1.1 and Nav1.5 reveals mutational hotspots for sodium channelopathies. Proc. Natl. Acad. Sci. USA 2021, 118, e2100066118. [Google Scholar] [CrossRef]
- Vatta, M.; Dumaine, R.; Antzelevitch, C.; Brugada, R.; Li, H.; Bowles, N.E.; Nademanee, K.; Brugada, J.; Brugada, P.; Towbin, J.A. Novel mutations in domain I of SCN5A cause Brugada syndrome. Mol. Genet. Metab. 2002, 75, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Ciconte, G.; Monasky, M.M.; Santinelli, V.; Micaglio, E.; Vicedomini, G.; Anastasia, L.; Negro, G.; Borrelli, V.; Giannelli, L.; Santini, F.; et al. Brugada syndrome genetics is associated with phenotype severity. Eur. Heart J. 2021, 42, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, J.M.; Barajas-Martinez, H.; Hong, K.; Burashnikov, E.; Pfeiffer, R.; Orsino, A.-M.; Wu, Y.S.; Hu, D.; Brugada, J.; Brugada, P.; et al. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation 2006, 114, 2026–2033. [Google Scholar] [CrossRef]
- Pfahnl, A.E.; Viswanathan, P.C.; Weiss, R.; Shang, L.L.; Sanyal, S.; Shusterman, V.; Kornblit, C.; London, B.; Dudley, S.C. A sodium channel pore mutation causing Brugada syndrome. Heart Rhythm 2007, 4, 46–53. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Yang, J.; Xu, B.; Wen, Y.; Xiang, G.; Wei, G.; Zhu, C.; Xing, Y.; Li, Y. Electrophysiological and trafficking defects of the SCN5A T353I mutation in Brugada syndrome are rescued by alpha-allocryptopine. Eur. J. Pharmacol. 2015, 746, 333–343. [Google Scholar] [CrossRef]
- Makiyama, T.; Akao, M.; Tsuji, K.; Doi, T.; Ohno, S.; Takenaka, K.; Kobori, A.; Ninomiya, T.; Yoshida, H.; Takano, M.; et al. High risk for bradyarrhythmic complications in patients with Brugada syndrome caused by SCN5A gene mutations. J. Am. Coll. Cardiol. 2005, 46, 2100–2106. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Rebs, S.; El-Battrawy, I.; Chasan, S.; Krause, T.; Haas, J.; Zhong, R.; Liao, Z.; Xu, Q.; Zhou, X.; et al. Identification of SCN5A p.C335R Variant in a Large Family with Dilated Cardiomyopathy and Conduction Disease. Int. J. Mol. Sci. 2021, 22, 12990. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.D.; Zhu, W.; Mangold, K.; Chung, W.; Silva, J.R. A Molecularly Detailed NaV1.5 Model Reveals a New Class I Antiarrhythmic Target. JACC Basic. Transl. Sci. 2019, 4, 736–751. [Google Scholar] [CrossRef]
- Wible, B.A.; Hawryluk, P.; Ficker, E.; Kuryshev, Y.A.; Kirsch, G.; Brown, A.M. HERG-Lite: A novel comprehensive high-throughput screen for drug-induced hERG risk. J. Pharmacol. Toxicol. Methods 2005, 52, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.M.; McEwen, D.P.; Zhang, L.; Arendt, K.L.; Van Genderen, K.M.; Martens, J.R. Antiarrhythmic drug-induced internalization of the atrial-specific K+ channel kv1.5. Circ. Res. 2009, 104, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Keller, D.I.; Huang, H.; Fressart, V.; Schmied, C.; Timour, Q.; Chahine, M. Mexiletine differentially restores the trafficking defects caused by two brugada syndrome mutations. Front. Pharmacol. 2012, 3, 62. [Google Scholar] [CrossRef]
- Ruan, Y.; Denegri, M.; Liu, N.; Bachetti, T.; Seregni, M.; Morotti, S.; Severi, S.; Napolitano, C.; Priori, S.G. Trafficking defects and gating abnormalities of a novel SCN5A mutation question gene-specific therapy in long QT syndrome type 3. Circ. Res. 2010, 106, 1374–1383. [Google Scholar] [CrossRef]
- Hu, R.M.; Tester, D.J.; Li, R.; Sun, T.; Peterson, B.Z.; Ackerman, M.J.; Makielski, J.C.; Tan, B.-H. Mexiletine rescues a mixed biophysical phenotype of the cardiac sodium channel arising from the SCN5A mutation, N406K, found in LQT3 patients. Channels 2018, 12, 176–186. [Google Scholar] [CrossRef]
- Valdivia, C.R.; Tester, D.J.; Rok, B.A.; Porter, C.-B.J.; Munger, T.M.; Jahangir, A.; Makielski, J.C.; Ackerman, M.J. A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 2004, 62, 53–62. [Google Scholar] [CrossRef]
- Rivolta, I.; Abriel, H.; Tateyama, M.; Liu, H.; Memmi, M.; Vardas, P.; Napolitano, C.; Priori, S.G.; Kass, R.S. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J. Biol. Chem. 2001, 276, 30623–30630. [Google Scholar] [CrossRef] [PubMed]
- Bugiardini, E.; Rivolta, I.; Binda, A.; Caminero, A.S.; Cirillo, F.; Cinti, A.; Giovannoni, R.; Botta, A.; Cardani, R.; Wicklund, M.; et al. SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul. Disord. 2015, 25, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Tadros, R.; Tan, H.L.; el Mathari, S.; A Kors, J.; Postema, P.G.; Lahrouchi, N.; Beekman, L.; Radivojkov-Blagojevic, M.; Amin, A.S.; Meitinger, T.; et al. Predicting cardiac electrical response to sodium-channel blockade and Brugada syndrome using polygenic risk scores. Eur. Heart J. 2019, 40, 3097–3107. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||||||
| WT | p.A344S/WT | p.A344S | p.N347K/WT | p.D349N/WT | |||
| n | 17 | 20 | 12 | 27 | 24 | ||
| Current density @ −20 mV | (pA/pF) | −134.5 ± 14 | −77.8 ± 9 * | −38.8 ± 6 *,# | −63.3 ± 7 * | −69.5 ± 8 * | |
| Activation | V1/2 (mV) | −33.7 ± 1 | −29.3 ± 5 * | −29.2 ± 5 * | −29.7 ± 1* | −31.1 ± 1 | |
| k | 7.8 ± 0.5 | 8.9 ± 0.3 | 8.3 ± 0.5 | 8.4 ± 0.3 | 7.8 ± 0.6 | ||
| Inactivation | V1/2 (mV) | −82.5 ± 2 | −82.8 ± 2 | −77.5 ± 1 * | −83.2 ± 2 | −77.6 ± 1 * | |
| k | 5.4 ± 0.3 | 6.5 ± 0.3 | 5.3 ± 0.3 | 6.1 ± 0.3 | 5.2 ± 0.2 | ||
| Fast inactivation @ −20 mV | 0.74 ± 0.06 | 0.85 ± 0.09 | 1.03 ± 0.09 * | 0.95 ± 0.08 | 0.74 ± 0.04 | ||
| n | 12 | 7 | 4 | 23 | 17 | ||
| Recovery from inactivation | 8.8 ± 0.6 | 10.6 ± 4 | 7.8 ± 0.8 | 7.4 ± 0.7 | 8.3 ± 0.7 | ||
| 210.4 ± 26 | 450.5 ± 191 | 396.7 ± 187 | 171.9 ± 32 | 304.4 ± 44 | |||
| (B) | |||||||
| +Mexiletine 0.1 mM | |||||||
| p.A344S | Change | p.N347K | Change | p.D349N | Change | ||
| Current density @ −20 mV | (pA/pF) | −5.72 ± 0.4 | −85% * | −28.15 ± 6 | +21% * | −7.45 ± 1 | +5% * |
| Activation | V1/2 (mV) | −14.5 ± 0.5 | +19 # | −24.7 ± 0.5 | +9 # | −19.1 ± 0.8 | +14.6 # |
| k | 8.9 ± 0.5 | 8.1 ± 0.4 | 7.8 ± 0.7 | ||||
| n | 7 | 13 | 6 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frosio, A.; Micaglio, E.; Polsinelli, I.; Calamaio, S.; Melgari, D.; Prevostini, R.; Ghiroldi, A.; Binda, A.; Carrera, P.; Villa, M.; et al. Unravelling Novel SCN5A Mutations Linked to Brugada Syndrome: Functional, Structural, and Genetic Insights. Int. J. Mol. Sci. 2023, 24, 15089. https://doi.org/10.3390/ijms242015089
Frosio A, Micaglio E, Polsinelli I, Calamaio S, Melgari D, Prevostini R, Ghiroldi A, Binda A, Carrera P, Villa M, et al. Unravelling Novel SCN5A Mutations Linked to Brugada Syndrome: Functional, Structural, and Genetic Insights. International Journal of Molecular Sciences. 2023; 24(20):15089. https://doi.org/10.3390/ijms242015089
Chicago/Turabian StyleFrosio, Anthony, Emanuele Micaglio, Ivan Polsinelli, Serena Calamaio, Dario Melgari, Rachele Prevostini, Andrea Ghiroldi, Anna Binda, Paola Carrera, Marco Villa, and et al. 2023. "Unravelling Novel SCN5A Mutations Linked to Brugada Syndrome: Functional, Structural, and Genetic Insights" International Journal of Molecular Sciences 24, no. 20: 15089. https://doi.org/10.3390/ijms242015089
APA StyleFrosio, A., Micaglio, E., Polsinelli, I., Calamaio, S., Melgari, D., Prevostini, R., Ghiroldi, A., Binda, A., Carrera, P., Villa, M., Mastrocinque, F., Presi, S., Salerno, R., Boccellino, A., Anastasia, L., Ciconte, G., Ricagno, S., Pappone, C., & Rivolta, I. (2023). Unravelling Novel SCN5A Mutations Linked to Brugada Syndrome: Functional, Structural, and Genetic Insights. International Journal of Molecular Sciences, 24(20), 15089. https://doi.org/10.3390/ijms242015089