
Classes of Lipid Mediators and Their Effects on Vascular Inflammation in Atherosclerosis

Abstract
1. Introduction
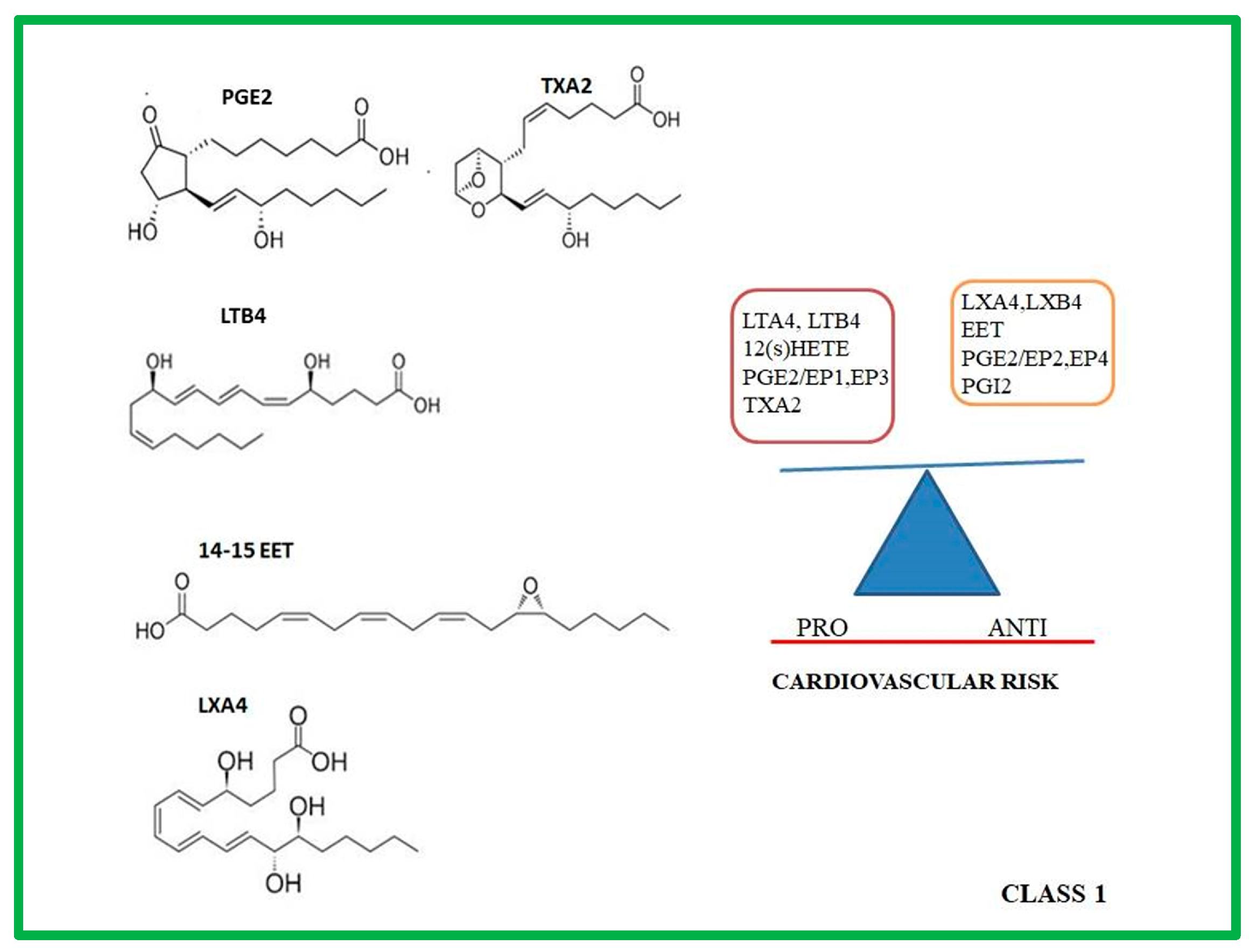
2. Class 1: Eicosanoids
2.1. Molecular and Biochemical Characteristic of PROSTANOIDS
2.2. Molecular and Biochemical Characteristic of LTs and HETEs
2.3. Molecular and Biochemical Characteristic of Lxs
2.4. Molecular and Biochemical Characteristic of EETs
2.5. Role of Prostanoids, LTs, HETEs, LXs, EETs in Atherosclerotic Progression
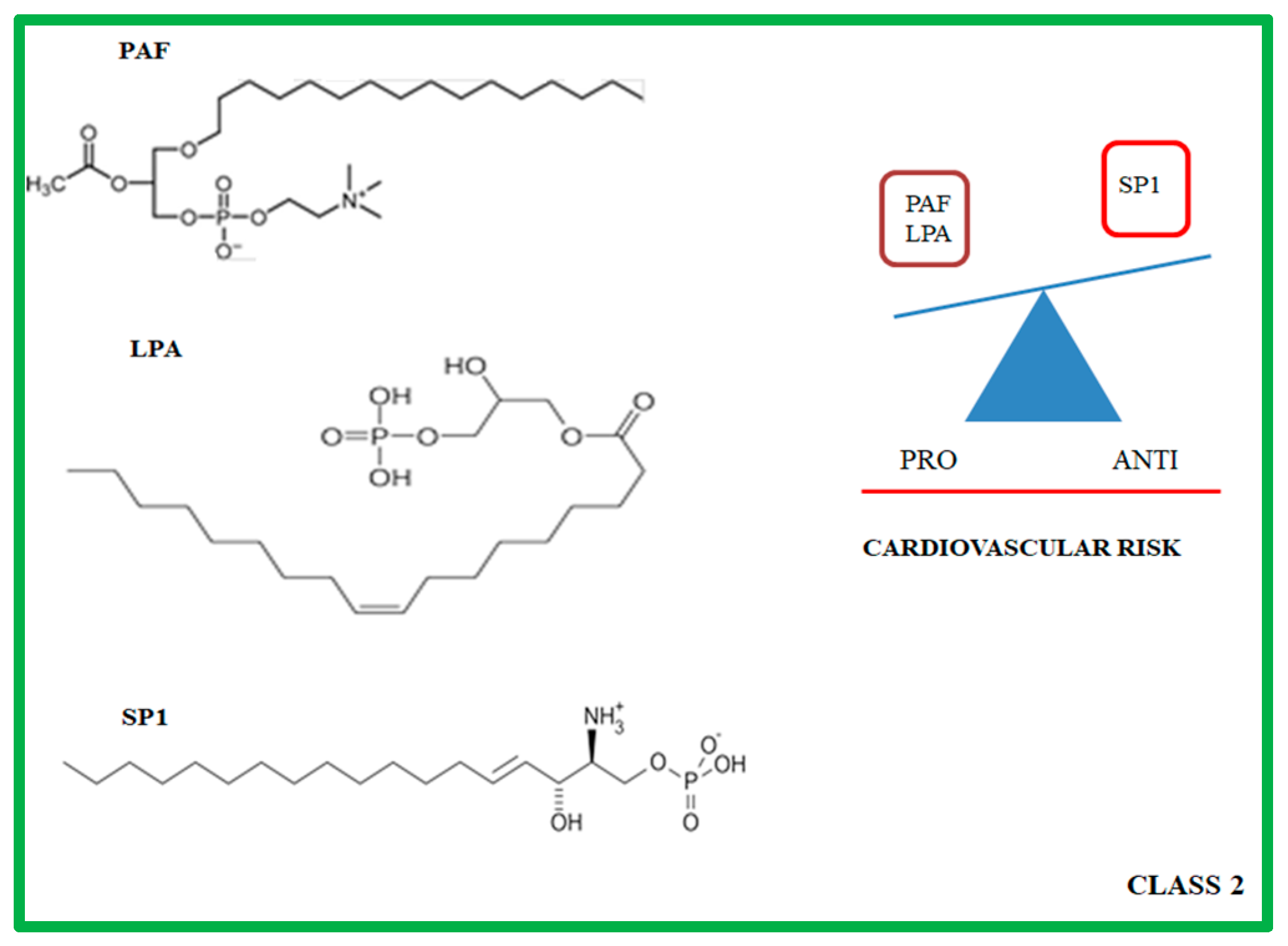
3. Class 2: Lysophospholipids
3.1. Molecular and Biochemical Characteristics of PAF, LPA and SP1
3.2. Role of PAF, LPA, and SP1 in Atherosclerotic Progression
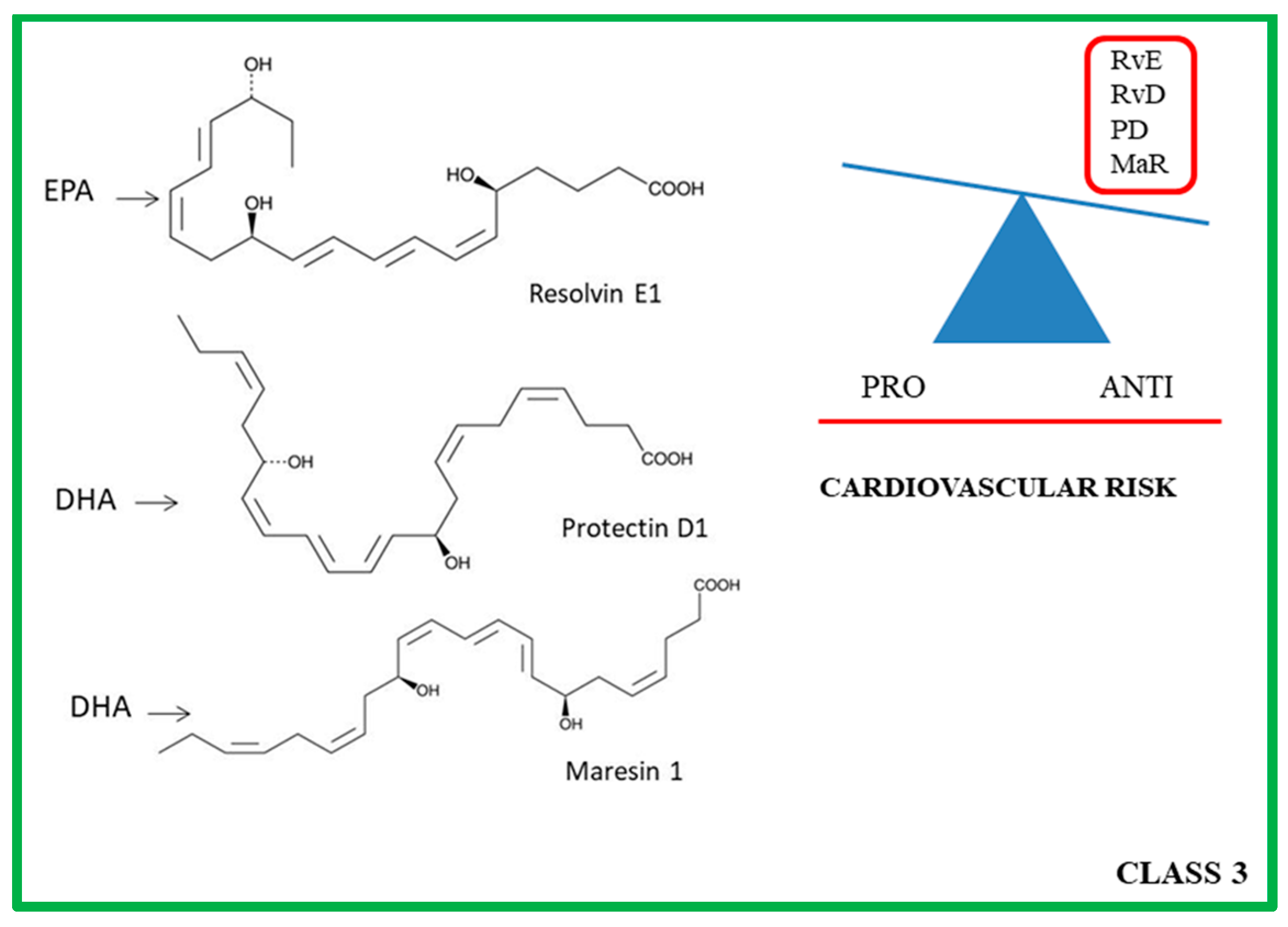
4. Class 3: ω-3 PUFA Derivatives
4.1. Molecular and Biochemical Characteristics of Rvs, PDs and MaRs
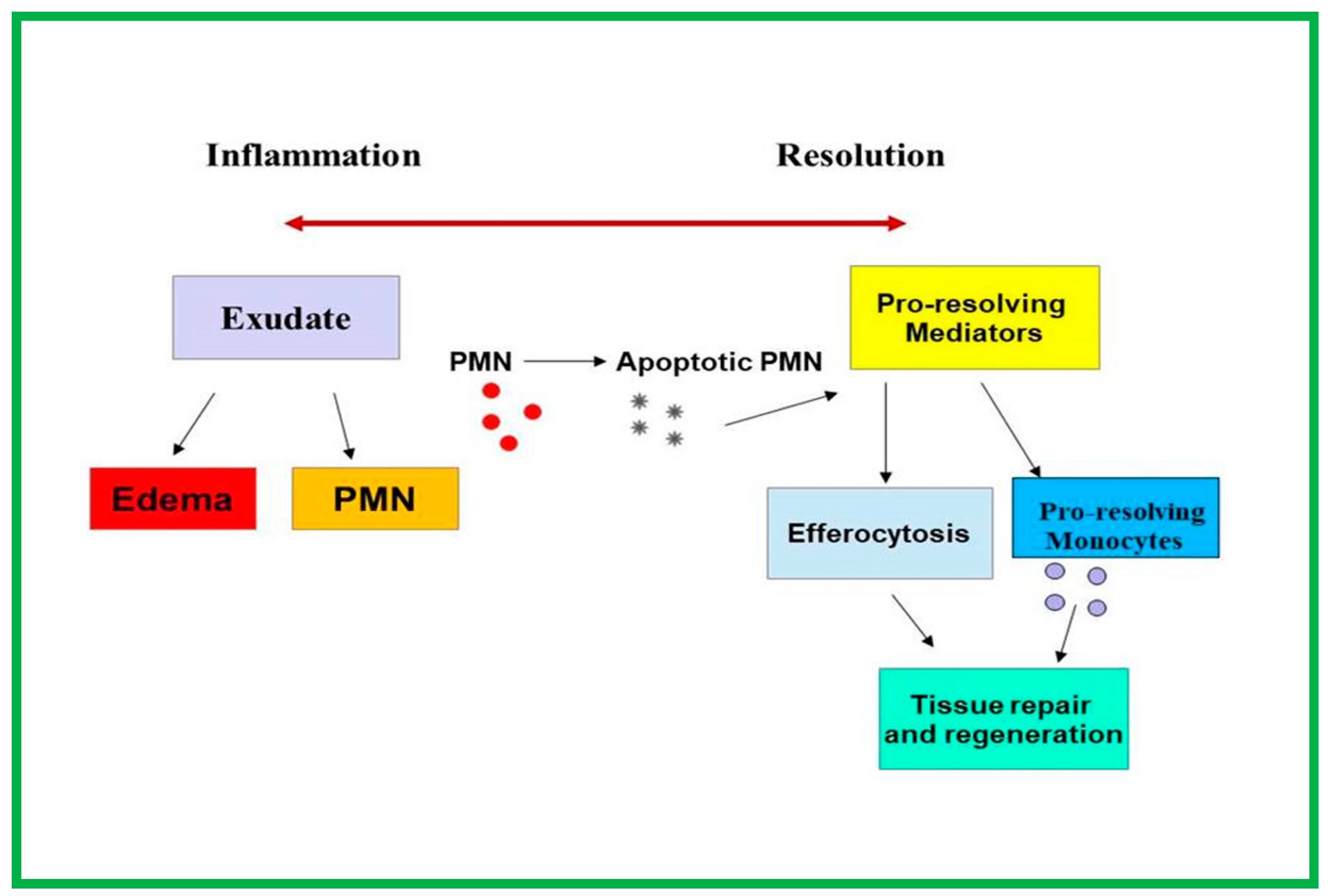
4.2. Role of PRLMs in Atherosclerotic Progression
4.3. Protective Function of ω-3 Fatty Acid Diet in Atherosclerotic Progression
Funding
Acknowledgments
Conflicts of Interest
References
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Doran, A.C.; Cai, B.; Tabas, I. The role of non-resolving inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 2713–2723. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N. The Role of Modified and Dysfunctional Lipoproteins in Atherogenesis. Curr. Med. Chem. 2019, 26, 1509–1511. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Merched, A.J.; Ko, K.; Gotlinger, K.H.; Serhan, C.N.; Chan, L. Atherosclerosis: Evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008, 22, 3595–3606. [Google Scholar] [CrossRef]
- Pullen, A.B.; Jadapalli, J.K.; Rhourri-Frih, B.; Halade, G.V. Re-evaluating the causes and consequences of non-resolving inflammation in chronic cardiovascular disease. Heart Fail. Rev. 2020, 25, 381–391. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Ma, J.; Chen, X. Anti-inflammatory Therapy for Coronary Atherosclerotic Heart Disease: Unanswered questions behind existing successes. Front. Cardiovasc. Med. 2021, 7, 631398. [Google Scholar] [CrossRef]
- Bannenberg, G.L.; Chiang, N.; Ariel, A.; Arita, M.; Tjonahen, E.; Gotlinger, K.H.; Hong, S.; Serhan, C.N. Molecular circuits of resolution: Formation and actions of resolvins and protectins. J. Immunol. 2005, 174, 4345–4355. [Google Scholar] [CrossRef]
- Levy, B.D.; Clish, C.; Schmidt, B.A.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N. Lipid-Derived Mediators in Endogenous Anti-Inflammation and Resolution: Lipoxins and Aspirin-Triggered 15-epi-Lipoxins. Sci. World J. 2002, 2, 169–204. [Google Scholar] [CrossRef]
- Serhan, C.N. Resolution Phase of Inflammation: Novel Endogenous Anti-Inflammatory and Proresolving Lipid Mediators and Pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef]
- Serhan, C.N. Novel Lipid Mediators and Resolution Mechanisms in Acute Inflammation: To Resolve or Not? Am. J. Pathol. 2010, 177, 1576–1591. [Google Scholar] [CrossRef]
- Bennett, M.; Gilroy, D.W. Lipid Mediators in Inflammation. Microbiol. Spectr. 2016, 4, 6. [Google Scholar] [CrossRef]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef]
- Viola, J.R.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Döring, Y.; Drechsler, M.; et al. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ. Res. 2016, 119, 1030–1038. [Google Scholar] [CrossRef]
- Schebb, N.H.; Kühn, H.; Kahnt, A.S.; Rund, K.M.; O’Donnell, V.B.; Flamand, N.; Peters-Golden, M.; Jakobsson, P.-J.; Weylandt, K.H.; Rohwer, N.; et al. Formation, Signaling and Occurrence of Specialized Pro-Resolving Lipid Mediators—What is the Evidence so far? Front. Pharmacol. 2022, 13, 838782. [Google Scholar] [CrossRef]
- Austen, K.F. The cysteinyl leukotrienes: Where do they come from? What are they? Where are they going? Nat. Immunol. 2008, 9, 113–115. [Google Scholar] [CrossRef]
- Narumiya, S.; Fitzgerald, G.A. Genetic and pharmacological analysis of prostanoid receptor function. J. Clin. Investig. 2001, 108, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Botta, E.; Holinstat, M. Eicosanoids in inflammation in the blood and the vessel. Front. Pharmacol. 2022, 13, 997403. [Google Scholar] [CrossRef] [PubMed]
- Idborg, H.; Pawelzik, S.-C. Prostanoid Metabolites as Biomarkers in Human Disease. Metabolites 2022, 12, 721. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, Cellular, and Molecular Biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, Y.; Guo, Z.; Wang, M. Cardiovascular Biology of Prostanoids and Drug Discovery. Arter. Thromb. Vasc. Biol. 2020, 40, 1454–1463. [Google Scholar] [CrossRef]
- Woodward, D.F.; Jones, R.L.; Narumiya, S. International Union of Basic and Clinical Pharmacology. LXXXIII: Classification of Prostanoid Receptors, Updating 15 Years of Progress. Pharmacol. Rev. 2011, 63, 471–538. [Google Scholar] [CrossRef]
- Smith, E.M.; Austin, S.C.; Reilly, M.P.; FitzGerald, G.A. Internalization and sequestration of the human prostacyclin receptor. J. Biol. Chem. 2000, 275, 32037–32045. [Google Scholar] [CrossRef]
- Miller, S.B. Prostaglandins in health and disease: An overview. In Seminars in Arthritis and Rheumatism; WB Saunders: Philadelphia, PA, USA, 2006; Volume 36, pp. 37–49. [Google Scholar]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E Receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, W.; Ge, J.; Zhang, Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMP-PKA/PI3K-Akt signaling pathway. Mol. Med. Rep. 2018, 17, 4702–4712. [Google Scholar] [CrossRef]
- Rucker, D.; Dhamoon, A.S. Physiology, Thromboxane A2. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Chen, H. Role of thromboxane A2 signaling in endothelium-dependent contractions of arteries. Prostaglandins Other Lipid Mediat. 2018, 134, 32–37. [Google Scholar] [CrossRef]
- Funk, C.D.; Chen, X.S.; Johnson, E.N.; Zhao, L. Lipoxygenase genes and their targeted disruption. Prostaglandins Other Lipid Mediat 2002, 68–69, 303–312. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Colazzo, F.; Gelosa, P.; Tremoli, E.; Sironi, L.; Castiglioni, L. Role of the Cysteinyl Leukotrienes in the Pathogenesis and Progression of Cardiovascular Diseases. Mediat. Inflamm. 2017, 2017, 2432958. [Google Scholar] [CrossRef]
- Maekawa, A.; Kanaoka, Y.; Xing, W.; Austen, K.F. Functional recognition of a distinct receptor preferential for leukotriene E 4 in mice lacking the cysteinyl leukotriene 1 and 2 receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 16695–16700. [Google Scholar] [CrossRef]
- Singh, N.K.; Rao, G.N. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog. Lipid Res. 2019, 73, 28–45. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N. Novel endogenous small molecules as the checkpoint controllers in inflammation and resolution: Entrée for resoleomics. Rheum. Dis. Clin. North Am. 2004, 30, 69–95. [Google Scholar] [CrossRef]
- Andrews, D.; Godson, C. Lipoxins and synthetic lipoxin mimetics: Therapeutic potential in renal diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158940. [Google Scholar] [CrossRef]
- Papayianni, A.; Serhan, C.N.; Phillips, M.L.; Rennke, H.G.; Brady, H.R. Transcellular biosynthesis of lipoxin A4 during adhesion of platelets and neutrophils in experimental immune complex glomerulonephritis. Kidney Int. 1995, 47, 1295–1302. [Google Scholar] [CrossRef]
- Bannenberg, G.; Moussignac, R.-L.; Gronert, K.; Devchand, P.R.; Schmidt, B.A.; Guilford, W.J.; Bauman, J.G.; Subramanyam, B.; Perez, H.D.; Parkinson, J.F.; et al. Lipoxins and novel 15-epi-lipoxin analogs display potent anti-inflammatory actions after oral administration. Br. J. Pharmacol. 2004, 143, 43–52. [Google Scholar] [CrossRef]
- Maddox, J.F.; Colgan, S.P.; Clish, C.B.; Petasis, N.A.; Fokin, V.V.; Serhan, C.N. Lipoxin B4regulates human monocyte/neutrophil adherence and motility: Design of stable lipoxin B4analogs with increased biologic activity. FASEB J. 1998, 12, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N. Formyl-Peptide Receptor 2/3/Lipoxin A 4 Receptor Regulates Neutrophil-Platelet Aggregation and Attenuates Cerebral Inflammation: Impact for Therapy in Cardiovascular Disease. Circulation 2016, 133, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.H.; Falck, J.R.; Harris, R.C. Cytochrome P450 and arachidonic acid bioactivation: Molecular and functional properties of the arachidonate monooxygenase. J. Lipid Res. 2000, 41, P163–P181. [Google Scholar] [CrossRef]
- Shi, Z.; He, Z.; Wang, D.W. CYP450 Epoxygenase Metabolites, Epoxyeicosatrienoic Acids, as Novel Anti-Inflammatory Mediators. Molecules 2022, 27, 3873. [Google Scholar] [CrossRef] [PubMed]
- Imig, J. Epoxyeicosanoids in Hypertension. Physiol. Res. 2019, 68, 695–704. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tahara, Y.; Matsumoto, M.; Iguchi, M.; Sano, H.; Murayama, T.; Arai, H.; Oida, H.; Yurugi-Kobayashi, T.; Yamashita, J.K.; et al. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Investig. 2004, 114, 784–794. [Google Scholar] [CrossRef]
- Wang, M.; Zukas, A.M.; Hui, Y.; Ricciotti, E.; Puré, E.; FitzGerald, G.A. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc. Natl. Acad. Sci. 2006, 103, 14507–14512. [Google Scholar] [CrossRef]
- Wang, M.; Lee, E.; Song, W.; Ricciotti, E.; Rader, D.J.; Lawson, J.A.; Puré, E.; FitzGerald, G.A. Microsomal Prostaglandin E Synthase-1 Deletion Suppresses Oxidative Stress and Angiotensin II–Induced Abdominal Aortic Aneurysm Formation. Circulation 2008, 117, 1302–1309. [Google Scholar] [CrossRef]
- Hao, H.; Hu, S.; Wan, Q.; Xu, C.; Chen, H.; Zhu, L.; Xu, Z.; Meng, J.; Breyer, R.M.; Li, N.; et al. Protective Role of mPGES-1 (Microsomal Prostaglandin E Synthase-1)–Derived PGE2 (Prostaglandin E2) and the Endothelial EP4 (Prostaglandin E Receptor) in Vascular Responses to Injury. Arter. Thromb. Vasc. Biol. 2018, 38, 1115–1124. [Google Scholar] [CrossRef]
- Kriska, T.; Cepura, C.; Magier, D.; Siangjong, L.; Gauthier, K.M.; Campbell, W.B. Mice lacking macrophage 12/15-lipoxygenase are resistant to experimental hypertension. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2428–H2438. [Google Scholar] [CrossRef]
- Pang, W.; Li, N.; Ai, D.; Niu, X.-L.; Guan, Y.-F.; Zhu, Y. Activation of peroxisome proliferator-activated receptor-γ downregulates soluble epoxide hydrolase in cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 2011, 38, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Montrucchio, G.; Alloatti, G.; Mariano, F.; De Paulis, R.; Comino, A.; Emanuelli, G.; Camussi, G. Role of platelet-activating factor in the reperfusion injury of rabbit ischemic heart. Am. J. Pathol. 1990, 137, 71–83. [Google Scholar] [PubMed]
- Bot, M.; Bot, I.; Lopez-Vales, R.; van de Lest, C.H.; Saulnier-Blache, J.S.; Helms, J.B.; David, S.; van Berkel, T.J.; Biessen, E.A. Atherosclerotic Lesion Progression Changes Lysophosphatidic Acid Homeostasis to Favor its Accumulation. Am. J. Pathol. 2010, 176, 3073–3084. [Google Scholar] [CrossRef]
- Swendeman, S.L.; Xiong, Y.; Cantalupo, A.; Yuan, H.; Burg, N.; Hisano, Y.; Cartier, A.; Liu, C.H.; Engelbrecht, E.; Blaho, V.; et al. An engineered S1P chaperone attenuates hypertension and ischemic injury. Sci. Signal. 2017, 10, eaal2722. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, K.; Bernier, J.; Godbout, R.; Rousseau, G. Resolvin D1, a Metabolite of Omega-3 Polyunsaturated Fatty Acid, Decreases Post-Myocardial Infarct Depression. Mar. Drugs 2014, 12, 5396–5407. [Google Scholar] [CrossRef]
- Keyes, K.T.; Ye, Y.; Lin, Y.; Zhang, C.; Perez-Polo, J.R.; Gjorstrup, P.; Birnbaum, Y. Resolvin E1 protects the rat heart against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H153–H164. [Google Scholar] [CrossRef]
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.D.; Halade, G.V. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell. Cardiol. 2015, 84, 24–35. [Google Scholar] [CrossRef]
- Joo, M.; Sadikot, R.T. PGD Synthase and PGD2 in Immune Resposne. Mediat. Inflamm. 2012, 2012, 503128. [Google Scholar] [CrossRef]
- Li, K.; Zhao, J.; Wang, M.; Niu, L.; Wang, Y.; Li, Y.; Zheng, Y. The Roles of Various Prostaglandins in Fibrosis: A Review. Biomolecules 2021, 11, 789. [Google Scholar] [CrossRef]
- Smyth, E.M. Thromboxane and the thromboxane receptor in cardiovascular disease. Clin. Lipidol. 2010, 5, 209–219. [Google Scholar] [CrossRef]
- Gutterman, D.D.; Chabowski, D.S.; Kadlec, A.O.; Durand, M.J.; Freed, J.K.; Ait-Aissa, K.; Beyer, A.M. The human microcirculation: Regulation of flow and beyond. Circ. Res. 2016, 118, 157–172. [Google Scholar] [CrossRef]
- Hoxha, M.; Rovati, G.; Cavanillas, A.B. The leukotriene receptor antagonist montelukast and its possible role in the cardiovascular field. Eur. J. Clin. Pharmacol. 2017, 73, 799–809. [Google Scholar] [CrossRef]
- Lotzer, K.; Funk, C.D.; Habenicht, A.J. The 5-lipoxygenase pathway in arterial wall biology and atherosclerosis. Biochim. Biophys. Acta 2005, 1736, 30–37. [Google Scholar] [CrossRef]
- Chawengsub, Y.; Gauthier, K.M.; Campbell, W.B. Role of arachidonic acid lipoxygenase metabolites in the regulation of vascular tone. Am. J. Physiol. Heart Circ Physiol. 2009, 297, H495–H507. [Google Scholar] [CrossRef]
- Oni-Orisan, A.; Edin, M.L.; Lee, J.A.; Wells, M.A.; Christensen, E.S.; Vendrov, K.; Lih, F.B.; Tomer, K.B.; Bai, X.; Taylor, J.; et al. Cytochrome P450-derived epoxyeicosatrienoic acids and coronary artery disease in humans: A targeted metabolomics study. J. Lipid Res. 2016, 57, 109–119. [Google Scholar] [CrossRef]
- Ramakrishnan, A.V.K.P.; Varghese, T.P.; Vanapalli, S.; Nair, N.K.; Mingate, M.D. Platelet activating factor: A potential biomarker in acute coronary syndrome? Cardiovasc. Ther. 2017, 35, 64–70. [Google Scholar] [CrossRef]
- Gabbasov, Z.; Parfyonova, Y.; Popov, E.; Gavrilov, I.; Anuchin, V.; Dubov, P.; Djakonova, Y. Association of platelet function in hypertensive patients with left ventricular hypertrophy, transient myocardial ischemia, and coronary artery disease. Platelets 1998, 9, 191–195. [Google Scholar] [CrossRef]
- Chou, Y.I.; Kong, J.; Song, R.; Yan, L.; Zheng, L.; Zhang, Y. Correlation of platelet derived microparticles with thromboxane B2, platelet activating factor, endothelin 1 and neutrophil to lymphocyte ratio in patients with coronary intermediate lesion. Biomarkers 2014, 8, 684–692. [Google Scholar] [CrossRef]
- Chen, X.; Yang, X.Y.; Wang, N.D.; Ding, C.; Yang, Y.J.; You, Z.J.; Su, Q.; Chen, J.H. Serum lysophosphatidic acid concentrations measured by dot immunogold filtration assay in patients with acute myocardial infarction. Scand. J. Clin. Lab. Investig. 2003, 63, 497–504. [Google Scholar] [CrossRef]
- Dohi, T.; Miyauchi, K.; Ohkawa, R.; Nakamura, K.; Kishimoto, T.; Miyazaki, T.; Nishino, A.; Nakajima, N.; Yaginuma, K.; Tamura, H.; et al. Increased circulating plasma lysophosphatidic acid in patients with acute coronary syndrome. Clin. Chim. Acta 2012, 413, 207–212. [Google Scholar] [CrossRef]
- Rother, E.; Brandl, R.; Baker, D.L.; Goyal, P.; Gebhard, H.; Tigyi, G.; Siess, W. Subtype-Selective Antagonists of Lysophosphatidic Acid Receptors Inhibit Platelet Activation Triggered by the Lipid Core of Atherosclerotic Plaques. Circulation 2003, 108, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.J.; Spite, M.; Owens, C.D.; Lancero, H.; Kroemer, A.H.; Pande, R.; Creager, M.A.; Serhan, C.N.; Conte, M.S. Aspirin-Triggered Lipoxin and Resolvin E1 Modulate Vascular Smooth Muscle Phenotype and Correlate with Peripheral Atherosclerosis. Am. J. Pathol. 2010, 177, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Makriyannis, A.; Nikas, S.P. Aspirin-Triggered Metabolites of EFAs. Chem. Biol. 2011, 18, 1208–1209. [Google Scholar] [CrossRef]
- Conte, M.S.; Desai, T.A.; Wu, B.; Schaller, M.; Werlin, E. Pro-resolving lipid mediators in vascular disease. J. Clin. Investig. 2018, 128, 3727–3735. [Google Scholar] [CrossRef]
- Yang, G.; Chen, L. An Update of Microsomal Prostaglandin E Synthase-1 and PGE2Receptors in Cardiovascular Health and Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 5249086. [Google Scholar] [CrossRef] [PubMed]
- Foudi, N.; Kotelevets, L.; Gomez, I.; Louedec, L.; Longrois, D.; Chastre, E.; Norel, X. Differential reactivity of human mammary artery and saphenous vein to prostaglandin E2: Implication for cardiovascular grafts. Br. J. Pharmacol. 2011, 163, 826–834. [Google Scholar] [CrossRef]
- Chen, L.; Miao, Y.; Zhang, Y.; Dou, D.; Liu, L.; Tian, X.Y.; Yang, G.; Pu, D.; Zhang, X.; Kang, J.; et al. Inactivation of the E-Prostanoid 3 Receptor Attenuates the Angiotensin II Pressor Response via Decreasing Arterial Contractility. Arter. Thromb. Vasc. Biol. 2012, 32, 3024–3032. [Google Scholar] [CrossRef]
- Eskildsen, M.P.; Hansen, P.B.; Stubbe, J.; Toft, A.; Walter, S.; Marcussen, N.; Rasmussen, L.M.; Vanhoutte, P.M.; Jensen, B.L. Prostaglandin I2 and Prostaglandin E2 Modulate Human Intrarenal Artery Contractility Through Prostaglandin E2-EP4, Prostacyclin-IP, and Thromboxane A2-TP Receptors. Hypertension 2014, 64, 551–556. [Google Scholar] [CrossRef]
- Degousee, N.; Fazel, S.; Angoulvant, D.; Stefanski, E.; Pawelzik, S.-C.; Korotkova, M.; Arab, S.; Liu, P.; Lindsay, T.F.; Zhuo, S.; et al. Microsomal Prostaglandin E2 Synthase-1 Deletion Leads to Adverse Left Ventricular Remodeling After Myocardial Infarction. Circulation 2008, 117, 1701–1710. [Google Scholar] [CrossRef]
- Cathcart, M.-C.; Tamosiuniene, R.; Chen, G.; Neilan, B.; Bradford, A.; O’Byrne, K.J.; Fitzgerald, D.J.; Pidgeon, G.P. Cyclooxygenase-2-Linked Attenuation of Hypoxia-Induced Pulmonary Hypertension and Intravascular Thrombosis. J. Pharmacol. Exp. Ther. 2008, 326, 51–58. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Kirkby, N.S.; Ahmetaj-Shala, B.; Armstrong, P.C.; Crescente, M.; Ferreira, P.; Pires, M.E.L.; Vaja, R.; Warner, T.D. Cyclooxygenases and the cardiovascular system. Pharmacol. Ther. 2021, 217, 107624. [Google Scholar] [CrossRef]
- Chen, W.; Yingjie, Z.; Nuan, F.; Zhu, G.; Shuai, W.; Dongming, X. New horizons in the roles and associations of COX-2 and novel natural inhibitors in cardiovascular diseases. Mol. Med. 2021, 27, 123. [Google Scholar] [CrossRef]
- De Caterina, R.; Giannessi, D.; Lazzerini, G.; Bernini, W.; Sicari, R.; Cupelli, F.; Lenzi, S.; Rugolotto, M.M.; Madonna, R.; Maclouf, J. Sulfido-peptide leukotrienes in coronary heart disease—Relationship with disease instability and myocardial ischaemia. Eur. J. Clin. Investig. 2010, 40, 258–272. [Google Scholar] [CrossRef]
- Helgadottir, A.; Manolescu, A.; Thorleifsson, G.; Gretarsdottir, S.; Jonsdottir, H.; Thorsteinsdottir, U.; Samani, N.J.; Gudmundsson, G.; Grant, S.F.; Thorgeirsson, G.; et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat. Genet. 2004, 36, 233–239. [Google Scholar] [CrossRef]
- Dobrian, A.D.; Lieb, D.C.; Cole, B.K.; Taylor-Fishwick, D.A.; Chakrabarti, S.K.; Nadler, J.L. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog. Lipid Res. 2011, 50, 115–131. [Google Scholar] [CrossRef]
- Kayama, Y.; Minamino, T.; Toko, H.; Sakamoto, M.; Shimizu, I.; Takahashi, H.; Okada, S.; Tateno, K.; Moriya, J.; Yokoyama, M. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J. Exp. Med. 2009, 206, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.; Golub, M.; Nozawa, K.; Berger, M.; Knoll, E.; Yanagawa, N.; Natarajan, R.; Nadler, J.L.; Tuck, M.L. Selective inhibition of angiotensin II-mediated vasoconstriction by lipoxygenase blockade. Am. J. Physiol. Content 1989, 257, H434–H443. [Google Scholar] [CrossRef]
- Rubbo, H.; O’Donnell, V. Nitric oxide, peroxynitrite and lipoxygenase in atherogenesis: Mechanistic insights. Toxicology 2008, 208, 305–317. [Google Scholar] [CrossRef]
- Holzhütter, H.G.; Wiesner, R.; Rathmann, J.; Stösser, R.; Kühn, H. A kinetic model for the interaction of nitric oxide with a mammalian lipoxygenase. Eur. J. Biochem. 1997, 245, 608–616. [Google Scholar] [CrossRef]
- Kühn, H. Biosynthesis, metabolization and biological importance of the primary 15-lipoxygenase metabolites 15-hydro(pero)xy-5Z,8Z,11Z,13E-eicosatetraenoic acid and 13-hydro(pero)xy-9Z,11E-octadecadienoic acid. Prog. Lipid Res. 1996, 35, 203–226. [Google Scholar] [CrossRef]
- Kotla, S.; Singh, N.K.; Traylor, J.G.; Orr, A.W.; Rao, G.N. ROS-dependent Syk and Pyk2-mediated STAT1 activation is required for 15(S)-hydroxyeicosatetraenoic acid-induced CD36 expression and foam cell formation. Free Radic. Biol. Med. 2014, 76, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Gertow, K.; Nobili, E.; Folkersen, L.; Newman, J.W.; Pedersen, T.L.; Ekstrand, J.; Swedenborg, J.; Kühn, H.; Wheelock, C.E.; Hansson, G.K.; et al. 12- and 15-lipoxygenases in human carotid atherosclerotic lesions: Associations with cerebrovascular symptoms. Atherosclerosis 2011, 215, 411–416. [Google Scholar] [CrossRef]
- Wittwer, J.; Marti-Jaun, J.; Hersberger, M. Functional polymorphism in ALOX15 results in increased allele-specific transcription in macrophages through binding of the transcription factor SPI1. Hum. Mutat. 2006, 27, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Chen, C. The Role of Epoxyeicosatrienoic Acids in Cardiac Remodeling. Front. Physiol. 2021, 12, 642470. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.B.; Fleming, I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflug. Arch. 2010, 459, 881–895. [Google Scholar] [CrossRef]
- Imig, J.D.; Falck, J.R.; Wei, S.; Capdevila, J.H. Epoxygenase Metabolites Contribute to Nitric Oxide-Independent Afferent Arteriolar Vasodilation in Response to Bradykinin. J. Vasc. Res. 2001, 38, 247–255. [Google Scholar] [CrossRef]
- Capdevila, J.; Falck, J.; Imig, J. Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 2007, 72, 683–689. [Google Scholar] [CrossRef]
- Imig, J.D.; Zhao, X.; Capdevila, J.H.; Morisseau, C.; Hammock, B.D. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 2002, 39, 690–694. [Google Scholar] [CrossRef]
- Lee, J.; Dahl, M.; Grande, P.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Genetically Reduced Soluble Epoxide Hydrolase Activity and Risk of Stroke and Other Cardiovascular Disease. Stroke 2010, 41, 27–33. [Google Scholar] [CrossRef]
- Zhang, L.-N.; Vincelette, J.; Cheng, Y.; Mehra, U.; Chen, D.; Anandan, S.-K.; Gless, R.; Webb, H.K.; Wang, Y.-X. Inhibition of Soluble Epoxide Hydrolase Attenuated Atherosclerosis, Abdominal Aortic Aneurysm Formation, and Dyslipidemia. Arter. Thromb. Vasc. Biol. 2009, 29, 1265–1270. [Google Scholar] [CrossRef]
- Liu, W.; Wang, T.; He, X.; Liu, X.; Wang, B.; Liu, Y.; Li, Z.; Tan, R.; Ding, C.; Wang, H.; et al. CYP2J2 Overexpression Increases EETs and Protects Against HFD-Induced Atherosclerosis in ApoE−/− Mice. J. Cardiovasc. Pharmacol. 2016, 67, 491–502. [Google Scholar] [CrossRef]
- Li, N.; Liu, J.-Y.; Timofeyev, V.; Qiu, H.; Hwang, S.H.; Tuteja, D.; Lu, L.; Yang, J.; Mochida, H.; Low, R.; et al. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J. Mol. Cell. Cardiol. 2009, 47, 835–845. [Google Scholar] [CrossRef]
- Qiu, H.; Li, N.; Liu, J.-Y.; Harris, T.R.; Hammock, B.D.; Chiamvimonvat, N. Soluble Epoxide Hydrolase Inhibitors and Heart Failure. Cardiovasc. Ther. 2011, 29, 99–111. [Google Scholar] [CrossRef]
- Romashko, M.; Schragenheim, J.; Abraham, N.G.; McClung, J.A. Epoxyeicosatrienoic Acid as Therapy for Diabetic and Ischemic Cardiomyopathy. Trends Pharmacol. Sci. 2016, 37, 945–962. [Google Scholar] [CrossRef]
- Xu, D.; Li, N.; He, Y.; Timofeyev, V.; Lu, L.; Tsai, H.-J.; Kim, I.-H.; Tuteja, D.; Mateo, R.K.P.; Singapuri, A.; et al. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. 2006, 103, 18733–18738. [Google Scholar] [CrossRef]
- Althurwi, H.N.; Maayah, Z.H.; Elshenawy, O.H.; El-Kadi, A.O. Early changes in cytochrome P450s and their associated arachidonic acid metabolites play a crucial role in the initiation of cardiac hypertrophy induced by isoproterenol. Drug Metab. Dispos. 2015, 43, 1254–1266. [Google Scholar] [CrossRef]
- Althurwi, H.N.; Tse, M.M.; Abdelhamid, G.; Zordoky, B.N.; Hammock, B.D.; El-Kadi, A.O. Soluble epoxide hydrolase inhibitor, TUPS, protects against isoprenaline-induced cardiac hypertrophy. Br. J. Pharmacol. 2013, 168, 1794–1807. [Google Scholar] [CrossRef]
- Ding, Y.; Tu, P.; Chen, Y.; Huang, Y.; Pan, X.; Chen, W. CYP2J2 and EETs protect against pulmonary arterial hypertension with lung ischemia–reperfusion injury in vivo and in vitro. Respir. Res. 2021, 22, 291. [Google Scholar] [CrossRef]
- Serhan, C.N.; Clish, C.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel Functional Sets of Lipid-Derived Mediators with Antiinflammatory Actions Generated from Omega-3 Fatty Acids via Cyclooxygenase 2–Nonsteroidal Antiinflammatory Drugs and Transcellular Processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef]
- Serhan, C.N.; Maddox, J.F.; Petasis, N.; Akritopoulou-Zanze, I.; Papayianni, A.; Brady, H.R.; Colgan, S.P.; Madara, J.L. Design of Lipoxin A4 Stable Analogs That Block Transmigration and Adhesion of Human Neutrophils. Biochemistry 1995, 34, 14609–14615. [Google Scholar] [CrossRef]
- Tan, S.T.; Ramesh, T.; Toh, X.R.; Nguyen, L.N. Emerging roles of lysophospholipids in health and disease. Prog. Lipid Res. 2020, 80, 101068. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.-W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA Receptors: Subtypes and Biological Actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Nagase, T.; Shimizu, T. Platelet-activating factor receptor. Prostaglandins Other Lipid Mediat. 2002, 68–69, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-Out” Signaling of Sphingosine-1-Phosphate: Therapeutic Targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Shindou, H.; Hishikawa, D.; Nakanishi, H.; Harayama, T.; Ishii, S.; Taguchi, R.; Shimizu, T. A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells: Cloning and characterization of acetyl-coa:lyso-paf acetyltransferase. J. Biol. Chem. 2007, 282, 6532–6539. [Google Scholar] [CrossRef]
- Arai, H. Platelet-activating factor acetylhydrolase. Prostaglandins Other Lipid Mediat. 2002, 69, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Honda, Z.-I.; Ishii, S.; Shimizu, T. Platelet-Activating Factor Receptor. J. Biochem. 2002, 131, 773–779. [Google Scholar] [CrossRef]
- Shimizu, T.; Mutoh, H.; Kato, S. Platelet-activating factor receptor. Gene structure and tissue-specific regulation. Adv. Exp. Med. Biol. 1996, 416, 479–484. [Google Scholar]
- Montrucchio, G.; Lupia, E.; Battaglia, E.; Passerini, G.; Bussolino, F.; Emanuelli, G.; Camussi, G. Tumor necrosis factor alpha-induced angiogenesis depends on in situ platelet-activating factor biosynthesis. J. Exp. Med. 1994, 180, 377–382. [Google Scholar] [CrossRef]
- Reznichenko, A.; Korstanje, R. The role of platelet activating factor in Mesangial Pathophysiology. Am. J. Pathol. 2015, 185, 887–896. [Google Scholar] [CrossRef]
- Braquet, P.; Hosford, D.; Braquet, M.; Bourgain, R.; Bussolino, F. Role of cytokines and platelet activating factor in microvascular immune injury. Int. Arch. Allergy Immunol. 1989, 88, 88–100. [Google Scholar] [CrossRef]
- Yung, Y.C.; Stoddard, N.C.; Mirendil, H.; Chun, J. Lysophosphatidic Acid Signaling in the Nervous System. Neuron 2015, 85, 669–682. [Google Scholar] [CrossRef]
- Knowlden, S.; Georas, S.N. The Autotaxin–LPA Axis Emerges as a Novel Regulator of Lymphocyte Homing and Inflammation. J. Immunol. 2014, 192, 851–857. [Google Scholar] [CrossRef]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Many Ceramides. J. Biol. Chem. 2011, 286, 27855–27862. [Google Scholar] [CrossRef]
- Sandoo, A.; van Zanten, J.J.; Metsios GS: Carrol, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef]
- McIntyre, T.; Zimmerman, G.A.; Satoh, K. Cultured endothelial cells synthesize both PAF and prostacyclin in response to histamine, bradykinin and adenosine tri phosphate. J. Clin. Investig. 1985, 76271–76280. [Google Scholar]
- Zhou, Y.; Little, P.J.; Ta, H.T.; Xu, S.; Kamato, D. Lysophosphatidic acid and its recep tors: Pharmacology and therapeutic potential in atherosclerosis and vascular disease. Pharmacol. Ther. 2019, 204, 107404. [Google Scholar]
- Smyth, S.S.; Cheng, H.-Y.; Miriyala, S.; Panchatcharam, M.; Morris, A.J. Roles of lysophosphatidic acid in cardiovascular physiology and disease. Biochim. Biophys. Acta 2008, 1781, 563–570. [Google Scholar] [CrossRef]
- Tokumura, A.; Yotsumoto, T.; Masuda, Y.; Tanaka, S. Vasopressor effect of lysophosphatidic acid on spontaneously hypertensive rats and Wistar Kyoto rats. Res. Commun. Mol. Pathol. Pharmacol. 1995, 90, 96–102. [Google Scholar] [PubMed]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Baker, D.L.; Yasuda, S.; Makarova, N.; Balazs, L.; Johnson, L.R.; Marathe, G.K.; McIntyre, T.M.; Xu, Y.; Prestwich, G.D.; et al. Lysophosphatidic Acid Induces Neointima Formation Through PPARγ Activation. J. Exp. Med. 2004, 199, 763–774. [Google Scholar] [CrossRef]
- Murakami, M.; Shiraishi, A.; Tabata, K.; Fujita, N. Identification of the orphan GPCR, P2Y10 receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2008, 371, 707–712. [Google Scholar] [CrossRef]
- Aldi, S.; Matic, L.P.; Hamm, G.; van Keulen, D.; Tempel, D.; Holmstrøm, K.; Szwajda, A.; Nielsen, B.S.; Emilsson, V.; Ait-Belkacem, R.; et al. Integrated Human Evaluation of the Lysophosphatidic Acid Pathway as a Novel Therapeutic Target in Atherosclerosis. Mol. Ther.-Methods Clin. Dev. 2018, 10, 17–28. [Google Scholar] [CrossRef]
- Eichholtz, T.; Jalink, K.; Fahrenfort, I.; Moolenaar, W.H. The bioactive phospholipid lysophosphatidic acid is released from activated platelets. Biochem. J. 1993, 291, 677–680. [Google Scholar] [CrossRef]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 2008, 1781, 513–518. [Google Scholar] [CrossRef]
- Yukiura, H.; Kano, K.; Kise, R.; Inoue, A.; Aoki, J. Autotaxin Overexpression Causes Embryonic Lethality and Vascular Defects. PLoS ONE 2015, 10, e0126734. [Google Scholar] [CrossRef]
- Yukiura, H.; Kano, K.; Kise, R.; Inoue, A.; Aoki, J. LPP3 localizes LPA6 signalling to non-contact site in endothelial cells. J. Cell Sci. 2015, 128, 3871–3877. [Google Scholar] [CrossRef]
- Takeda, A.; Kobayashi, D.; Aoi, K.; Sasaki, N.; Sugiura, Y.; Igarashi, H.; Tohya, K.; Inoue, A.; Hata, E.; Akahoshi, N.; et al. Fibroblastic reticular cell-derived lysophosphatidic acid regulates confined intranodal T-cell motility. Elife 2016, 5, e10561. [Google Scholar] [CrossRef] [PubMed]
- Kozian, D.H.; von Haeften, E.; Joho, S.; Czechtizky, W.; Anumala, U.R.; Roux, P.; Dudda, A.; Evers, A.; Nazare, M. Modulation of Hexadecyl-LPA-Mediated Activation of Mast Cells and Microglia by a Chemical Probe for LPA5. Chem. Biochem. 2016, 17, 861–865. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Deng, X.; Liu, Y.; Yang, X.; Wu, Q.; Yu, C. Lysophosphatidic acid directly induces macrophage-derived foam cell formation by blocking the expression of SRBI. Biochem. Biophys. Res. Commun. 2017, 491, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Wang, F.; Zhao, Z.; Wang, H.; Cong, X.; Chen, X. Lysophosphatidic Acid Is Associated with Atherosclerotic Plaque Instability by Regulating NF-κB Dependent Matrix Metalloproteinase-9 Expression via LPA2 in Macrophages. Front. Physiol. 2017, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.J.; Bäck, M. The Role of Matrix Metalloproteinases in Atherothrombosis. Curr. Atheroscler. Rep. 2011, 13, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef] [PubMed]
- Lamour, N.F.; Subramanian, P.; Wijesinghe, D.S.; Stahelin, R.V.; Bonventre, J.V.; Chalfant, C.E. Ceramide 1-Phosphate Is Required for the Translocation of Group IVA Cytosolic Phospholipase A2 and Prostaglandin Synthesis. J. Biol. Chem. 2009, 284, 26897–26907. [Google Scholar] [CrossRef]
- Egom, E.E.; Mamas, M.A.; Soran, H. HDL quality or cholesterol cargo: What really matters—Spotlight on sphingosine-1-phosphate-rich HDL. Curr. Opin. Infect. Dis. 2013, 24, 351–356. [Google Scholar] [CrossRef]
- De Mello, V.D.F.; Lankinen, M.; Schwab, U.; Kolehmainen, M.; Lehto, S.; Seppänen-Laakso, T.; Orešič, M.; Pulkkinen, L.; Uusitupa, M.; Erkkilä, A.T. Link between plasma ceramides, inflammation and insulin resistance: Association with serum IL-6 concentration in patients with coronary heart disease. Diabetologia 2009, 52, 2612–2615. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnaggar, N.; Zdanyte, M.; Müller, I.; Borst, O.; Geisler, T.; et al. Regulation of oxidized platelet lipidome: Implications for coronary artery disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef]
- Cheng, J.M.; Suoniemi, M.; Kardys, I.; Vihervaara, T.; de Boer, S.P.M.; Akkerhuis, K.M.; Sysi-Aho, M.; Ekroos, K.; Garcia-Garcia, H.M.; Oemrawsingh, R.M.; et al. Plasma concentrations of molecular lipid species in relation to coronary plaque characteristics and cardiovascular outcome: Results of the ATHEROREMO-IVUS study. Atherosclerosis 2015, 243, 560–566. [Google Scholar] [CrossRef]
- Ariel, A.; Serhan, C.N. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 2007, 28, 176–183. [Google Scholar] [CrossRef]
- Seki, H.; Tani, Y.; Arita, M. Omega-3 PUFA derived anti-inflammatory lipid mediator resolvin E1. Prostaglandins Other Lipid Mediat. 2009, 89, 126–130. [Google Scholar] [CrossRef]
- Vik, A.; Hansen, T.V. Stereoselective syntheses and biological activities of E-series resolvins. Org. Biomol. Chem. 2021, 19, 705–721. [Google Scholar] [CrossRef]
- Freire, M.O.; Van Dyke, T.E. Natural resolution of inflammation. Periodontology 2000 2013, 63, 149–164. [Google Scholar] [CrossRef]
- Isobe, Y.; Arita, M.; Matsueda, S.; Iwamoto, R.; Fujihara, T.; Nakanishi, H.; Taguchi, R.; Masuda, K.; Sasaki, K.; Urabe, D.; et al. Identification and structure determination of novel anti-inflammatory mediator resolvin E3, 17,18-dihydroxyeicosapentaenoic acid. J. Biol. Chem. 2012, 287, 10525–10534. [Google Scholar] [CrossRef]
- Libreros, S.; Shay, A.E.; Nshimiyimana, R.; Fichtner, D.; Martin, M.J.; Wourms, N.; Serhan, C.N. A New E-Series Resolvin: RvE4 Stereochemistry and Function in Efferocytosis of Inflammation-Resolution. Front. Immunol. 2021, 11, 631319. [Google Scholar] [CrossRef]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Asp. Med. 2019, 58, 114–129. [Google Scholar] [CrossRef]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef]
- Arita, M.; Ohira, T.; Sun, Y.-P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 Selectively Interacts with Leukotriene B4 Receptor BLT1 and ChemR23 to Regulate Inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar] [CrossRef]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef]
- Serhan, C.N.; Gotlinger, K.; Hong, S.; Lu, Y.; Siegelman, J.; Baer, T.; Yang, R.; Colgan, S.P.; Petasis, N.A. Anti-Inflammatory Actions of Neuroprotectin D1/Protectin D1 and Its Natural Stereoisomers: Assignments of Dihydroxy-Containing Docosatrienes. J. Immunol. 2006, 176, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.F.; Claudino, R.F.; Dutra, R.C.; Marcon, R.; Calixto, J.B. Omega-3 Fatty Acid-Derived Mediators 17(R)-Hydroxy Docosahexaenoic Acid, Aspirin-Triggered Resolvin D1 and Resolvin D2 Prevent Experimental Colitis in Mice. J. Immunol. 2011, 187, 1957–1969. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Cui, J.-G.; Marcheselli, V.L.; Bodker, M.; Botkjaer, A.; Gotlinger, K.; Serhan, C.N.; Bazan, N.G. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Investig. 2005, 115, 2774–2783. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Marcheselli, V.L.; Serhan, C.N.; Bazan, N.G. Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. USA 2004, 101, 8491–8496. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Delgado, M. Endogenous anti-inflammatory neuropeptides and pro-resolving lipid mediators: A new therapeutic approach for immune disorders. J. Cell. Mol. Med. 2008, 12, 1830–1847. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N. Endogenous pro-resolving and anti-inflammatory lipid mediators: A new pharmacologic genus. Br. J. Pharmacol. 2008, 153, S200–S215. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Chiang, N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta 2015, 1851, 397–413. [Google Scholar] [CrossRef]
- Shinohara, M.; Mirakaj, V.; Serhan, C.N. Functional Metabolomics Reveals Novel Active Products in the DHA Metabolome. Front. Immunol. 2012, 3, 81. [Google Scholar] [CrossRef]
- Li, Q.F.; Hao, H.; Tu, W.S.; Guo, N.; Zhou, X.Y. Maresins: Anti-inflammatory pro-resolving mediators with therapeutic potential. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7442–7453. [Google Scholar] [CrossRef]
- Hasturk, H.; Kantarci, A.; Goguet-Surmenian, E.; Blackwood, A.; Andry, C.; Serhan, C.N.; Van Dyke, T.E. Resolvin E1 Regulates Inflammation at the Cellular and Tissue Level and Restores Tissue Homeostasis In Vivo. J. Immunol. 2007, 179, 7021–7029. [Google Scholar] [CrossRef]
- Reátegui, E.; Jalali, F.; Khankhel, A.H.; Wong, E.; Cho, H.; Lee, J.; Serhan, C.N.; Dalli, J.; Elliott, H.; Irimia, D. Microscale arrays for the profiling of start and stop signals coordinating human-neutrophil swarming. Nat. Biomed. Eng. 2017, 1, 0094. [Google Scholar] [CrossRef]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Investig. 2011, 121, 569–581. [Google Scholar] [CrossRef]
- Norling, L.V.; Serhan, C.N. Profiling in resolving inflammatory exudates identifies novel anti-inflammatory and pro-resolving mediators and signals for termination. J. Intern. Med. 2010, 268, 15–24. [Google Scholar] [CrossRef]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef]
- Werz, O.; Gerstmeier, J.; Libreros, S.; De la Rosa, X.; Werner, M.; Norris, P.C.; Chiang, N.; Serhan, C.N. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun. 2018, 9, 59. [Google Scholar] [CrossRef]
- Motwani, M.P.; Colas, R.A.; George, M.J.; Flint, J.D.; Dalli, J.; Richard-Loendt, A.; De Maeyer, R.P.; Serhan, C.N.; Gilroy, D.W. Pro-resolving mediators promote resolution in a human skin model of UV-killed Escherichia coli–driven acute inflammation. J. Clin. Investig. 2018, 3, e94463. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400. [Google Scholar] [CrossRef]
- Fredman, G.; Van Dyke, T.E.; Serhan, C.N. Resolvin E1 Regulates Adenosine Diphosphate Activation of Human Platelets. Arter. Thromb. Vasc. Biol. 2010, 30, 2005–2013. [Google Scholar] [CrossRef]
- Petri, M.H.; Laguna-Fernandez, A.; Arnardottir, H.; Wheelock, C.E.; Perretti, M.; Hansson, G.K.; Bäck, M. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E−/−mice. Br. J. Pharmacol. 2017, 174, 4043–4054. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol. Asp. Med. 2018, 64, 1–17. [Google Scholar] [CrossRef]
- Kozłowska, H.; Baranowska-Kuczko, M.; Schlicker, E.; Kozłowski, M.; Zakrzeska, A.; Grzęda, E.; Malinowska, B. EP3 receptor-mediated contraction of human pulmonary arteries and inhibition of neurogenic tachycardia in pithed rats. Pharmacol. Rep. 2012, 64, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Lannan, K.L.; Spinelli, S.L.; Blumberg, N.; Phipps, R.P. Maresin 1 induces a novel pro-resolving phenotype in human platelets. J. Thromb. Haemost. 2017, 15, 802–813. [Google Scholar] [CrossRef]
- Abdulnour, R.-E.E.; Dalli, J.; Colby, J.K.; Krishnamoorthy, N.; Timmons, J.Y.; Tan, S.H.; Colas, R.A.; Petasis, N.A.; Serhan, C.N.; Levy, B.D. Maresin 1 biosynthesis during platelet–neutrophil interactions is organ-protective. Proc. Natl. Acad. Sci. USA 2014, 111, 16526–16531. [Google Scholar] [CrossRef] [PubMed]
- Dona, M.; Fredman, G.; Schwab, J.M.; Chiang, N.; Arita, M.; Goodarzi, A.; Cheng, G.; von Andrian, U.H.; Serhan, C.N. Resolvin E1, an EPA-derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood 2008, 112, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Elajami, T.K.; Colas, R.A.; Dalli, J.; Chiang, N.; Serhan, C.N.; Welty, F.K. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J. 2016, 30, 2792–2801. [Google Scholar] [CrossRef]
- Sansbury, B.E.; Spite, M. Resolution of Acute Inflammation and the Role of Resolvins in Immunity, Thrombosis, and Vascular Biology. Circ. Res. 2016, 119, 113–130. [Google Scholar] [CrossRef]
- Filep, J.G.; Zouki, C.; Petasis, N.A.; Hachicha, M.; Serhan, C.N. Anti-inflammatory actions of lipoxin A(4) stable analogs are demonstrable in human whole blood: Modulation of leukocyte adhesion molecules and inhibition of neutrophil-endothelial interactions. Blood 1999, 94, 4132–4142. [Google Scholar] [CrossRef]
- Chattopadhyay, R.; Mani, A.M.; Singh, N.K.; Rao, G.N. Resolvin D1 blocks H2O2-mediated inhibitory crosstalk between SHP2 and PP2A and suppresses endothelial-monocyte interactions. Free Radic. Biol. Med. 2018, 117, 119–131. [Google Scholar] [CrossRef]
- Chatterjee, A.; Sharma, A.; Chen, M.; Toy, R.; Mottola, G.; Conte, M.S. The Pro-Resolving Lipid Mediator Maresin 1 (MaR1) Attenuates Inflammatory Signaling Pathways in Vascular Smooth Muscle and Endothelial Cells. PLoS ONE 2014, 9, e113480. [Google Scholar] [CrossRef]
- Chattopadhyay, R.; Raghavan, S.; Rao, G.N. Resolvin D1 via prevention of ROS-mediated SHP2 inactivation protects endothelial adherens junction integrity and barrier function. Redox Biol. 2017, 12, 438–455. [Google Scholar] [CrossRef]
- Miyahara, T.; Runge, S.; Chatterjee, A.; Chen, M.; Mottola, G.; Fitzgerald, J.M.; Serhan, C.N.; Conte, M.S. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J. 2013, 27, 2220–2232. [Google Scholar] [CrossRef]
- Wu, B.; Mottola, G.; Chatterjee, A.; Lance, K.D.; Chen, M.; Siguenza, I.O.; Desai, T.A.; Conte, M.S. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rat model of arterial injury. J. Vasc. Surg. 2017, 65, 207–217.e3. [Google Scholar] [CrossRef]
- Egert, S.; Somoza, V.; Kannenberg, F.; Fobker, M.; Krome, K.; Erbersdobler, H.F.; Wahrburg, U. Influence of three rapeseed oil-rich diets, fortified with α-linolenic acid, eicosapentaenoic acid or docosahexaenoic acid on the composition and oxidizability of low-density lipoproteins: Results of a controlled study in healthy volunteers. Eur. J. Clin. Nutr. 2007, 61, 314–325. [Google Scholar] [CrossRef]
- Calder, P.C. Polyunsaturated fatty acids, inflammation, and immunity. Lipids 2000, 36, 1007–1024. [Google Scholar] [CrossRef]
- Zárate, R.; El Jaber-Vazdekis, N.; Tejera, N.; Pérez, J.A.P.; Rodríguez, C. Significance of long chain polyunsaturated fatty acids in human health. Clin. Transl. Med. 2017, 6, 25. [Google Scholar] [CrossRef]
- Brenna, J.T.; Salem, N.; Sinclair, A.J.; Cunnane, S.C. International Society for the Study of Fatty Acids and Lipids, ISSFAL. α-Linolenic acid supplementation and conversion to n-3 long-chain polyunsaturated fatty acids in humans. Prostaglandins Leukot. Essent. Fat. Acids 2009, 80, 85–91. [Google Scholar] [CrossRef]
- Bazan, H.A.; Lu, Y.; Jun, B.; Fang, Z.; Woods, T.C.; Hong, S. Circulating inflammation-resolving lipid mediators RvD1 and DHA are decreased in patients with acutely symptomatic carotid disease. Prostaglandins, Leukot. Essent. Fat. Acids 2017, 125, 43–47. [Google Scholar] [CrossRef]
- Tan, A.; Sullenbarger, B.; Prakash, R.; McDaniel, J.C. Supplementation with eicosapentaenoic acid and docosahexaenoic acid reduces high levels of circulating proinflammatory cytokines in aging adults: A randomized, controlled study. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 23–29. [Google Scholar] [CrossRef]
- Jialal, I.; Devaraj, S.; Huet, B.A.; Traber, M. GISSI-Prevenzione trial. GISSI-Prevenzione trial. Lancet 1999, 354, 1554. [Google Scholar] [CrossRef]
- Shen, S.; Gong, C.; Jin, K.; Zhou, L.; Xiao, Y.; Ma, L. Omega-3 Fatty Acid Supplementation and Coronary Heart Disease Risks: A Meta-Analysis of Randomized Controlled Clinical Trials. Front. Nutr. 2022, 9, 809311. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. REDUCE-IT Investigators. Effects of icosapent ethyl on total ischemic events: From REDUCE-IT. J. Am. Coll. Cardiol. 2019, 73, 2791–2802. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Gencer, B.; Djousse, L.; Al-Ramady, O.T.; Cook, N.R.; Manson, J.E.; Albert, C.M. Effect of Long-Term Marine ɷ-3 Fatty Acids Supplementation on the Risk of Atrial Fibrillation in Randomized Controlled Trials of Cardiovascular Outcomes: A Systematic Review and Meta-Analysis. Circulation 2021, 144, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhen, P.; Wei, Q.; Yu, F.; Song, S.; Tong, J. Effects of omega-3 polyunsaturated fatty acids supplementation for patients with cardiovascular disease risks: A dose-response meta-analysis. Am. J. Transl. Res. 2021, 13, 8526–8539. [Google Scholar] [PubMed]
- Hu, X.F.; Kenny, T.-A.; Chan, H.M. Inuit Country Food Diet Pattern Is Associated with Lower Risk of Coronary Heart Disease. J. Acad. Nutr. Diet. 2018, 118, 1237–1248.e1. [Google Scholar] [CrossRef] [PubMed]
- Sales-Campos, H.; Souza, P.R.; Peghini, B.C.; da Silva, J.S.; Cardoso, C.R. An overview of the modulatory effects of oleic acid in health and disease. Mini Rev. Med. Chem. 2013, 13, 201–210. [Google Scholar]
- Widmer, R.J.; Flammer, A.J.; Lerman, L.O.; Lerman, A. The Mediterranean Diet, its Components, and Cardiovascular Disease. Am. J. Med. 2015, 128, 229–238. [Google Scholar] [CrossRef]
- Liyanage, T.; Ninomiya, T.; Wang, A.; Neal, B.; Jun, M.; Wong, M.G.; Jardine, M.; Hillis, G.S.; Perkovic, V. Effects of the Mediterranean Diet on Cardiovascular Outcomes—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0159252. [Google Scholar] [CrossRef]
- Román, G.C.; Jackson, R.E.; Gadhia, R.; Román, A.N.; Reis, J. Mediterranean diet: The role of long-chain ω-3 fatty acids in fish; polyphenols in fruits, vegetables, cereals, coffee, tea, cacao and wine; probiotics and vitamins in prevention of stroke, age-related cognitive decline, and Alzheimer disease. Rev. Neurol. 2019, 175, 724–741. [Google Scholar] [CrossRef]
- Lavie, C.J.; Milani, R.V.; Mehra, M.R.; Ventura, H.O. Omega-3 Polyunsaturated Fatty Acids and Cardiovascular Diseases. J. Am. Coll. Cardiol. 2009, 54, 585–594. [Google Scholar] [CrossRef]
- Mozaffarian, D. Fish and n−3 fatty acids for the prevention of fatal coronary heart disease and sudden cardiac death. Am. J. Clin. Nutr. 2008, 87, 1991S–1996S. [Google Scholar] [CrossRef]
- Rauch, B.; Schiele, R.; Schneider, S.; Diller, F.; Victor, N.; Gohlke, H.; Gottwik, M.; Steinbeck, G.; Del Castillo, U.; Sack, R.; et al. OMEGA Study Group. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation 2010, 122, 2152–2159. [Google Scholar] [CrossRef]
- Martino, A.; Pezzi, L.; Magnano, R.; Salustri, E.; Penco, M.; Calo’, L. Omega 3 and atrial fibrillation: Where are we? World J. Cardiol. 2016, 8, 114–119. [Google Scholar] [CrossRef]
- Rizos, E.C.; Ntzani, E.E.; Bika, E.; Kostapanos, M.S.; Elisaf, M.S. Association Between Omega-3 Fatty Acid Supplementation and Risk of Major Cardiovascular Disease Events: A systematic review and meta-analysis. JAMA 2012, 308, 1024–1033. [Google Scholar] [CrossRef]
- He, K.; Song, Y.; Daviglus, M.L.; Liu, K.; Van Horn, L.; Dyer, A.R.; Greenland, P. Accumulated Evidence on Fish Consumption and Coronary Heart Disease Mortality. Circulation 2004, 109, 2705–2711. [Google Scholar] [CrossRef]
- Bosch, J.; Gerstein, H.C.; Dagenais, G.R.; Diaz, R.; Dyal, L.; Jung, H.; Maggiono, A.P.; Probstfield, J.; Ramachandran, A.; Riddle, M.C.; et al. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N. Engl. J. Med. 2012, 367, 309–318. [Google Scholar]
- Alexander, D.D.; Miller, P.E.; Van Elswyk, M.E.; Kuratko, C.N.; Bylsma, L.C. A meta-analysis of randomized controlled trials and prospective cohort studies of eicosapentaenoic and docosahexaenoic long-chain omega-3 fatty acids and coronary heart disease risk. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2017; Volume 92, pp. 15–29. [Google Scholar]
- Aung, T.; Halsey, J.; Kromhout, D.; Gerstein, H.C.; Marchioli, R.; Tavazzi, L.; Geleijnse, J.M.; Rauch, B.; Ness, A.; Galan, P.; et al. Omega-3 Treatment Trialists’ Collaboration. Associations of omega-3 fatty acid supplement use with cardiovascular disease risks: Meta-analysis of 10 trials involving 77917 individuals. JAMA Cardiol. 2018, 3, 225–234. [Google Scholar] [CrossRef]
- Poorani, R.; Bhatt, A.N.; Dwarakanath, B.; Das, U.N. COX-2, aspirin and metabolism of arachidonic, eicosapentaenoic and docosahexaenoic acids and their physiological and clinical significance. Eur. J. Pharmacol. 2016, 785, 116–132. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studies in Animal Models | Lipid Mediators | Effects | Ref. |
|---|---|---|---|
| ApoE-deficient mice | ↑ TXA2 | ↑ Atherogenesis | [48] |
| ↓ PGI2 | ↑ Atherogenesis | [48] | |
| Mice deficient in LDL receptor/mPGEs 1 | ↑ PGI2 | ↓ Atherosclerosis | [49,50] |
| Mice COX-2 knockout | ↑ HBP, Trombosis | [51] | |
| ALOX15-deficient mice | ↓ HETES | ↑ resistance L-NAME-induced hypertension | [52] |
| Mice Ang II-induced hypertension | ↓EETS↑ sEH | ↑ AngII-induced cardiac hypertrophy | [53] |
| Administration of PAF receptor antagonist (SDZ 63.675) before reperfusion of the ischemic-isolated rabbit heart | ↓ PAF | ↓ Myocardial injury during reperfusion | [54] |
| Male LDLr−/− mice (two weeks cholesterol + cacao butter diet) before surgery of carotid artery | ↑ LPA in atherosclerotic tissue | Atherosclerotic Lesion Progression | [55] |
| WT mice administered with soluble carrier for S1P (ApoM-Fc) after I/R injury in heart | ↑ SP1 Receptor | ↓ Myocardial damage after I/R injury | [56] |
| Myocardial I/R in mouse | ↑ RvE, RvD, PD, MaR | ↓ ROS, Inflammation | [57,58,59] |
| Lipid Mediators | Effects | Ref |
|---|---|---|
| ↑ TXA2 | Systemic inflammation, myocardial ischemia | [61] |
| ↑ LTs | Inflammation, CV risk, MI | [64,65] |
| ↑ 12(S)-HETE | VSMC relaxation | [66] |
| ↓ EETs | Obstructed CAD | [67] |
| ↑ PAF | Ischemic events | [68,69,70] |
| ↑ LPA | Acute heart attacks and coronary syndrome | [71,72,73] |
| ↓ RvE1 | Vascular inflammation | [74,75] |
| ↓ 15-epi-LXA 4 | Disease severity | [74,76] |
| Study | Design | Supplementation | End Point | Ref. |
|---|---|---|---|---|
| GISSI-Prevenzione Trials | Clinical trial of 11,324 post-MI patients | Fish oil 850–882 mg EPA and DHA | Reduction Death Risk | [201] |
| Meta analysis of 14 clinical Trials | 135,291 subjects | 0.8-1.2 g omega-3 FA(EPA, DHA) | MACE, CVD, and MI | [202] |
| Trial | 8179 participants and 5 years follow-up | 4 g/d ω-3 PUFA | 30% reduction CVE/ control | [203] |
| JELIS trial in Japan | 18,645 patients with a total cholesterol of 6.5 mmol/L | 1800 mg of EPA daily | 19% reduction of coronary events | [204] |
| Trial | Patients suffering of cardiovascular diseases | >1 g/d marine ɷ-3 fatty | Increased AF Risk | [205] |
| Trial | Partecipants suffering major CVE and death | PUFA daily dose × period > 8 grams/day × years | More benefit/1 g/d for 1-year ω-3 PUFA | [206] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lubrano, V.; Ndreu, R.; Balzan, S. Classes of Lipid Mediators and Their Effects on Vascular Inflammation in Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 1637. https://doi.org/10.3390/ijms24021637
Lubrano V, Ndreu R, Balzan S. Classes of Lipid Mediators and Their Effects on Vascular Inflammation in Atherosclerosis. International Journal of Molecular Sciences. 2023; 24(2):1637. https://doi.org/10.3390/ijms24021637
Chicago/Turabian StyleLubrano, Valter, Rudina Ndreu, and Silvana Balzan. 2023. "Classes of Lipid Mediators and Their Effects on Vascular Inflammation in Atherosclerosis" International Journal of Molecular Sciences 24, no. 2: 1637. https://doi.org/10.3390/ijms24021637
APA StyleLubrano, V., Ndreu, R., & Balzan, S. (2023). Classes of Lipid Mediators and Their Effects on Vascular Inflammation in Atherosclerosis. International Journal of Molecular Sciences, 24(2), 1637. https://doi.org/10.3390/ijms24021637