
Biology and Management of High-Grade Chondrosarcoma: An Update on Targets and Treatment Options
 , , and
, , and
Abstract
1. Introduction
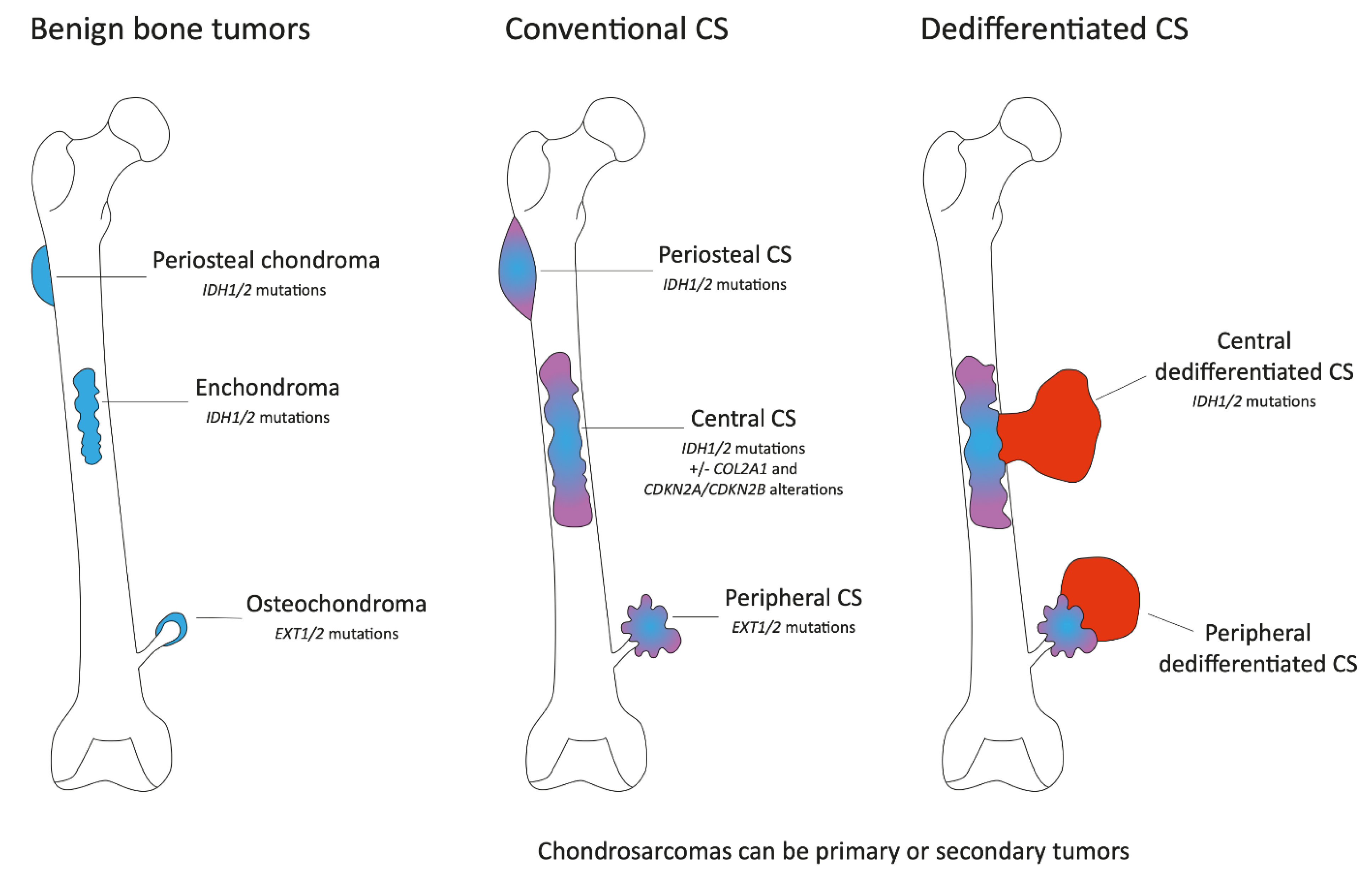
2. Clinical, Pathological and Molecular Characteristics
2.1. Conventional Chondrosarcoma
2.2. Dedifferentiated Chondrosarcoma
2.3. Mesenchymal Chondrosarcoma
3. Current Treatment Management
3.1. Conventional and Dedifferentiated Chondrosarcoma
3.2. Mesenchymal Chondrosarcoma
4. Targets and Novel Treatments Options in Chondrosarcoma
4.1. Angiogenesis Pathway
4.2. Isocitrate Dehydrogenase (IDH) Mutations
4.3. Immunotherapy Strategies
4.4. Cell Cycle Pathway
4.5. PI3K-AKT-mTOR Pathway
4.6. Epigenetic Strategies
4.7. Anti DR5
4.8. Others
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Pinieux, G.; Karanian, M.; Le Loarer, F.; Le Guellec, S.; Chabaud, S.; Terrier, P.; Bouvier, C.; Batistella, M.; Neuville, A.; Robin, Y.M.; et al. Nationwide incidence of sarcomas and connective tissue tumors of intermediate malignancy over four years using an expert pathology review network. PLoS ONE 2021, 16, e0246958. [Google Scholar] [CrossRef]
- Brandolini, F.; Bacchini, P.; Moscato, M.; Bertoni, F. Chondrosarcoma as a complicating factor in Paget’s disease of bone. Skelet. Radiol. 1997, 26, 497–500. [Google Scholar] [CrossRef]
- Wu, L.C.; Kleinerman, R.A.; Curtis, R.E.; Savage, S.A.; Berrington de Gonzalez, A. Patterns of bone sarcomas as a second malignancy in relation to radiotherapy in adulthood and histologic type. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1993–1999. [Google Scholar] [CrossRef]
- Herget, G.W.; Strohm, P.; Rottenburger, C.; Kontny, U.; Krauss, T.; Bohm, J.; Sudkamp, N.; Uhl, M. Insights into Enchondroma, Enchondromatosis and the risk of secondary Chondrosarcoma. Review of the literature with an emphasis on the clinical behaviour, radiology, malignant transformation and the follow up. Neoplasma 2014, 61, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.; Hogendoorn, P.C.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.; Bovée, J.V. The clinical approach towards chondrosarcoma. Oncologist 2008, 13, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Arjen, H.G.; Cleven, J.L.B.R.T. Periosteal Chondrosarcoma; WHO Classification of Tumours; © International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- de Andrea, C.E.; Reijnders, C.M.; Kroon, H.M.; de Jong, D.; Hogendoorn, P.C.; Szuhai, K.; Bovee, J.V. Secondary peripheral chondrosarcoma evolving from osteochondroma as a result of outgrowth of cells with functional EXT. Oncogene 2012, 31, 1095–1104. [Google Scholar] [CrossRef]
- Cleven, A.H.; Zwartkruis, E.; Hogendoorn, P.C.; Kroon, H.M.; Briaire-de Bruijn, I.; Bovee, J.V. Periosteal chondrosarcoma: A histopathological and molecular analysis of a rare chondrosarcoma subtype. Histopathology 2015, 67, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Ye, H.; Forbes, G.; Damato, S.; Maggiani, F.; Pollock, R.; Tirabosco, R.; Flanagan, A.M. Isocitrate dehydrogenase 1 mutations (IDH1) and p16/CDKN2A copy number change in conventional chondrosarcomas. Virchows Arch. 2015, 466, 217–222. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. World Health Organization Classification of Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Van Praag Veroniek, V.M.; Rueten-Budde, A.J.; Ho, V.; Dijkstra, P.D.S.; Study group Bone and Soft tissue tumours, (WeBot); Fiocco, M.; van de Sande, M.A.J. Incidence, outcomes and prognostic factors during 25 years of treatment of chondrosarcomas. Surg. Oncol. 2018, 27, 402–408. [Google Scholar] [CrossRef]
- Bjornsson, J.; McLeod, R.A.; Unni, K.K.; Ilstrup, D.M.; Pritchard, D.J. Primary chondrosarcoma of long bones and limb girdles. Cancer 1998, 83, 2105–2119. [Google Scholar] [CrossRef]
- Cleven, A.H.G.; Suijker, J.; Agrogiannis, G.; Briaire-de Bruijn, I.H.; Frizzell, N.; Hoekstra, A.S.; Wijers-Koster, P.M.; Cleton-Jansen, A.M.; Bovee, J. IDH1 or -2 mutations do not predict outcome and do not cause loss of 5-hydroxymethylcytosine or altered histone modifications in central chondrosarcomas. Clin. Sarcoma Res. 2017, 7, 8. [Google Scholar] [CrossRef]
- Lugowska, I.; Teterycz, P.; Mikula, M.; Kulecka, M.; Kluska, A.; Balabas, A.; Piatkowska, M.; Wagrodzki, M.; Pienkowski, A.; Rutkowski, P.; et al. IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma. J. Cancer 2018, 9, 998–1005. [Google Scholar] [CrossRef]
- Kostine, M.; Cleven, A.H.; de Miranda, N.F.; Italiano, A.; Cleton-Jansen, A.M.; Bovee, J.V. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Grimer, R.J.; Gosheger, G.; Taminiau, A.; Biau, D.; Matejovsky, Z.; Kollender, Y.; San-Julian, M.; Gherlinzoni, F.; Ferrari, C. Dedifferentiated chondrosarcoma: Prognostic factors and outcome from a European group. Eur. J. Cancer 2007, 43, 2060–2065. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Mir, O.; Cioffi, A.; Palmerini, E.; Piperno-Neumann, S.; Perrin, C.; Chaigneau, L.; Penel, N.; Duffaud, F.; Kurtz, J.E.; et al. Advanced chondrosarcomas: Role of chemotherapy and survival. Ann. Oncol. 2013, 24, 2916–2922. [Google Scholar] [CrossRef]
- Van Maldegem, A.M.; Bovee, J.V.; Gelderblom, H. Comprehensive analysis of published studies involving systemic treatment for chondrosarcoma of bone between 2000 and 2013. Clin. Sarcoma Res. 2014, 4, 11. [Google Scholar] [CrossRef]
- Yoshida, K.I.; Machado, I.; Motoi, T.; Parafioriti, A.; Lacambra, M.; Ichikawa, H.; Kawai, A.; Antonescu, C.R.; Yoshida, A. NKX3-1 Is a Useful Immunohistochemical Marker of EWSR1-NFATC2 Sarcoma and Mesenchymal Chondrosarcoma. Am. J. Surg. Pathol. 2020, 44, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Sainati, L.; Scapinello, A.; Montaldi, A.; Bolcato, S.; Ninfo, V.; Carli, M.; Basso, G. A mesenchymal chondrosarcoma of a child with the reciprocal translocation (11;22)(q24;q12). Cancer Genet. Cytogenet. 1993, 71, 144–147. [Google Scholar] [CrossRef]
- Nyquist, K.B.; Panagopoulos, I.; Thorsen, J.; Haugom, L.; Gorunova, L.; Bjerkehagen, B.; Fossa, A.; Guriby, M.; Nome, T.; Lothe, R.A.; et al. Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS ONE 2012, 7, e49705. [Google Scholar] [CrossRef]
- Wang, L.; Motoi, T.; Khanin, R.; Olshen, A.; Mertens, F.; Bridge, J.; Dal Cin, P.; Antonescu, C.R.; Singer, S.; Hameed, M.; et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012, 51, 127–139. [Google Scholar] [CrossRef]
- Mitchell, A.D.; Ayoub, K.; Mangham, D.C.; Grimer, R.J.; Carter, S.R.; Tillman, R.M. Experience in the treatment of dedifferentiated chondrosarcoma. J. Bone Joint Surg. Br. 2000, 82, 55–61. [Google Scholar] [CrossRef]
- Dickey, I.D.; Rose, P.S.; Fuchs, B.; Wold, L.E.; Okuno, S.H.; Sim, F.H.; Scully, S.P. Dedifferentiated chondrosarcoma: The role of chemotherapy with updated outcomes. J. Bone Joint Surg. Am. 2004, 86, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- van Maldegem, A.M.; Gelderblom, H.; Palmerini, E.; Dijkstra, S.D.; Gambarotti, M.; Ruggieri, P.; Nout, R.A.; van de Sande, M.A.; Ferrari, C.; Ferrari, S.; et al. Outcome of advanced, unresectable conventional central chondrosarcoma. Cancer 2014, 120, 3159–3164. [Google Scholar] [CrossRef] [PubMed]
- DeLaney, T.F.; Liebsch, N.J.; Pedlow, F.X.; Adams, J.; Weyman, E.A.; Yeap, B.Y.; Depauw, N.; Nielsen, G.P.; Harmon, D.C.; Yoon, S.S.; et al. Long-term results of Phase II study of high dose photon/proton radiotherapy in the management of spine chordomas, chondrosarcomas, and other sarcomas. J. Surg. Oncol. 2014, 110, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Morioka, H.; Takahashi, S.; Araki, N.; Sugiura, H.; Ueda, T.; Takahashi, M.; Yonemoto, T.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Results of sub-analysis of a phase 2 study on trabectedin treatment for extraskeletal myxoid chondrosarcoma and mesenchymal chondrosarcoma. BMC Cancer 2016, 16, 479. [Google Scholar] [CrossRef]
- Nacev, B.A.; Smith, S.A.; Antonescu, C.R.; Rosenbaum, E.; Shi, H.; Tang, C.; Socci, N.D.; Rana, S.; Gularte-Mérida, R.; Zehir, A.; et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. medRxiv 2022. [Google Scholar] [CrossRef]
- McGough, R.L.; Aswad, B.I.; Terek, R.M. Pathologic neovascularization in cartilage tumors. Clin. Orthop. Relat. Res. 2002, 397, 76–82. [Google Scholar] [CrossRef]
- Ayala, G.; Liu, C.; Nicosia, R.; Horowitz, S.; Lackman, R. Microvasculature and VEGF expression in cartilaginous tumors. Hum. Pathol. 2000, 31, 341–346. [Google Scholar] [CrossRef]
- van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schoffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Stacchiotti, S.; Boglione, A.; Ferraresi, V.; Frustaci, S.; Comandone, A.; Casali, P.G.; Ferrari, S.; Aglietta, M. A phase 2 trial of imatinib mesylate in patients with recurrent nonresectable chondrosarcomas expressing platelet-derived growth factor receptor-alpha or -beta: An Italian Sarcoma Group study. Cancer 2011, 117, 826–831. [Google Scholar] [CrossRef]
- Chow, W.; Frankel, P.; Ruel, C.; Araujo, D.M.; Milhem, M.; Okuno, S.; Hartner, L.; Undevia, S.; Staddon, A. Results of a prospective phase 2 study of pazopanib in patients with surgically unresectable or metastatic chondrosarcoma. Cancer 2020, 126, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Villalobos, V.M.; Cote, G.M.; Burris, H.; Janku, F.; Mir, O.; Beeram, M.; Wagner, A.J.; Jiang, L.; Wu, B.; et al. Phase I Study of the Mutant IDH1 Inhibitor Ivosidenib: Safety and Clinical Activity in Patients With Advanced Chondrosarcoma. J. Clin. Oncol. 2020, 38, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gao, S.; Zhu, L.; Wang, J.; Zhang, P.; Li, P.; Zhang, F.; Yao, W. Efficacy and safety of anlotinib in patients with unresectable or metastatic bone sarcoma: A retrospective multiple institution study. Cancer Med. 2021, 10, 7593–7600. [Google Scholar] [CrossRef]
- Duffaud, F.; Italiano, A.; Bompas, E.; Rios, M.; Penel, N.; Mir, O.; Piperno-Neumann, S.; Chevreau, C.; Delcambre, C.; Bertucci, F.; et al. Efficacy and safety of regorafenib in patients with metastatic or locally advanced chondrosarcoma: Results of a non-comparative, randomised, double-blind, placebo controlled, multicentre phase II study. Eur. J. Cancer 2021, 150, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Tap, W.D.; Qin, L.X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: A multicentre, open-label, phase 2 trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Schaap, F.G.; French, P.J.; Bovee, J.V. Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 in tumors. Adv. Anat. Pathol. 2013, 20, 32–38. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, G.; Yang, Z.; Zeng, K.; Liu, F.; Sun, J. Expression of PD-L1/PD-L2 is associated with high proliferation index of Ki-67 but not with TP53 overexpression in chondrosarcoma. Int. J. Biol. Markers 2018, 33, 507–513. [Google Scholar] [CrossRef]
- Iseulys, R.; Anne, G.B.; Corinne, B.; Gonzague, D.B.P.; Marie, K.; Jean-Yves, B.; Aurelie, D. The immune landscape of chondrosarcoma reveals an immunosuppressive environment in the dedifferentiated subtypes and exposes CSFR1+ macrophages as a promising therapeutic target. J. Bone Oncol. 2020, 20, 100271. [Google Scholar]
- Cote, G.M.; He, J.; Choy, E. Next-Generation Sequencing for Patients with Sarcoma: A Single Center Experience. Oncologist 2018, 23, 234–242. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Behjati, S.; Cooke, S.L.; Van Loo, P.; Wedge, D.C.; Pillay, N.; Marshall, J.; O’Meara, S.; Davies, H.; Nik-Zainal, S.; et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat. Genet. 2013, 45, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Pollack, S.M.; Redman, M.W.; Baker, K.K.; Wagner, M.J.; Schroeder, B.A.; Loggers, E.T.; Trieselmann, K.; Copeland, V.C.; Zhang, S.; Black, G.; et al. Assessment of Doxorubicin and Pembrolizumab in Patients with Advanced Anthracycline-Naive Sarcoma: A Phase 1/2 Nonrandomized Clinical Trial. JAMA Oncol. 2020, 6, 1778–1782. [Google Scholar] [CrossRef]
- Al Baghdadi, T.; Halabi, S.; Garrett-Mayer, E.; Mangat, P.K.; Ahn, E.R.; Sahai, V.; Alvarez, R.H.; Kim, E.S.; Yost, K.J.; Rygiel, A.L.; et al. Palbociclib in Patients with Pancreatic and Biliary Cancer with CDKN2A Alterations: Results from the Targeted Agent and Profiling Utilization Registry Study. JCO Precis. Oncol. 2019, 3, 1–8. [Google Scholar]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef]
- Zhang, Y.X.; van Oosterwijk, J.G.; Sicinska, E.; Moss, S.; Remillard, S.P.; van Wezel, T.; Buhnemann, C.; Hassan, A.B.; Demetri, G.D.; Bovee, J.V.; et al. Functional profiling of receptor tyrosine kinases and downstream signaling in human chondrosarcomas identifies pathways for rational targeted therapy. Clin. Cancer Res. 2013, 19, 3796–3807. [Google Scholar] [CrossRef]
- Bernstein-Molho, R.; Kollender, Y.; Issakov, J.; Bickels, J.; Dadia, S.; Flusser, G.; Meller, I.; Sagi-Eisenberg, R.; Merimsky, O. Clinical activity of mTOR inhibition in combination with cyclophosphamide in the treatment of recurrent unresectable chondrosarcomas. Cancer Chemother. Pharm. 2012, 70, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Thornton, K.A.; Chen, A.R.; Trucco, M.M.; Shah, P.; Wilky, B.A.; Gul, N.; Carrera-Haro, M.A.; Ferreira, M.F.; Shafique, U.; Powell, J.D.; et al. A dose-finding study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcoma. Int. J. Cancer 2013, 133, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Gagné, L.M.; Boulay, K.; Topisirovic, I.; Huot, M.E.; Mallette, F.A. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar] [CrossRef]
- Venneker, S.; Kruisselbrink, A.B.; Baranski, Z.; Palubeckaite, I.; Briaire-de Bruijn, I.H.; Oosting, J.; French, P.J.; Danen, E.H.J.; Bovee, J. Beyond the Influence of IDH Mutations: Exploring Epigenetic Vulnerabilities in Chondrosarcoma. Cancers 2020, 12, 3589. [Google Scholar] [CrossRef]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef]
- Fervienza, A.; Moya, I.; Martinez-Camacho, A.; Sala-Blanch, X. Median nerve block in the forearm and persistent median artery. Rev. Esp. Anestesiol. Reanim. 2019, 66, 494–495. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Hwang, E.S.; McManus, M.T.; Amsterdam, A.; Tian, Y.; Kalmukova, R.; Mueller, E.; Benjamin, T.; Spiegelman, B.M.; Sharp, P.A.; et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 2005, 309, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wu, A.; Li, P.; Li, G.; Qin, L.; Song, H.; Mak, K.K. Yap1 Regulates Multiple Steps of Chondrocyte Differentiation during Skeletal Development and Bone Repair. Cell Rep. 2016, 14, 2224–2237. [Google Scholar] [CrossRef]
- Fullenkamp, C.A.; Hall, S.L.; Jaber, O.I.; Pakalniskis, B.L.; Savage, E.C.; Savage, J.M.; Ofori-Amanfo, G.K.; Lambertz, A.M.; Ivins, S.D.; Stipp, C.S.; et al. TAZ and YAP are frequently activated oncoproteins in sarcomas. Oncotarget 2016, 7, 30094–30108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Gui, T.; Sang, Y.; Yang, J.; Li, Y.H.; Liang, G.H.; Li, T.; He, Q.Y.; Zha, Z.G. The BET Bromodomain Inhibitor JQ1 Suppresses Chondrosarcoma Cell Growth via Regulation of YAP/p21/c-Myc Signaling. J. Cell Biochem. 2017, 118, 2182–2192. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Bolejack, V.; Choy, E.; Ganjoo, K.N.; Staddon, A.P.; Chow, W.A.; Tawbi, H.A.; Samuels, B.L.; Patel, S.R.; von Mehren, M.; et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer 2017, 123, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef]
- Campbell, V.T.; Nadesan, P.; Ali, S.A.; Wang, C.Y.; Whetstone, H.; Poon, R.; Wei, Q.; Keilty, J.; Proctor, J.; Wang, L.W.; et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Mol. Cancer Ther. 2014, 13, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type (WHO 2020) | Biologic Marker | Incidence | O’Neal & Ackerman Grade | Prognosis | |
|---|---|---|---|---|---|
| Conventional CS | Central CS | IDH1/2 mutation | 72% (mainly in flat bones and long bones) | 1 (central ACT) 2 or 3 | No metastasis Potential for metastasize |
| Peripheral CS | No IDH1/2 mutation | 13% | 1 (secondary peripheral ACT) 2 or 3 | No metastasis Potential for metastasize | |
| Periosteal CS | IDH1/2 mutation | 1.5% | Not applicable | Good | |
| Dedifferentiated CS | IDH1/2 mutation (except in dedifferentiated peripheral CS) | 10% | Not applicable | High grade Poor prognosis | |
| Mesenchymal CS | Recurrent fusion transcript (HEY1-NCOA2, IRF2BP2-CDX1) | 1% | Not applicable | High grade Poor prognosis | |
| Clinical Trial | NCT Number | Conditions | Drugs | Drug Classes |
|---|---|---|---|---|
| Trial of Sunitinib and/or Nivolumab Plus Chemotherapy in Advanced Soft Tissue and Bone Sarcomas | NCT03277924 | All sarcoma subtypes | Sunitinib Nivolumab Epirubicin Ifosfamide Doxorubicin Dacarbazine Cisplatin Methotrexate | Antiangiogenic PD-L1 inhibitor Chemotherapy |
| Study of LY3410738 Administered to Patients with Advanced Solid Tumors with IDH1 or IDH2 Mutations | NCT04521686 | Basket trial with IDH1/2 mutations | LY3410738 | IDH1 and IDH2 inhibitor |
| A Study of HMPL-306 in Advanced Solid Tumors with IDH Mutations | NCT04762602 | Basket trial with IDH1/2 mutations | HMPL-306 | IDH1 and IDH2 inhibitor |
| AG-120 in People with IDH1 Mutant Chondrosarcoma | NCT04278781 | Chondrosarcoma with IDH1 Gene Mutation | AG-120 | IDH1 inhibitor |
| A Study of FT 2102 in Participants with Advanced Solid Tumors and Gliomas with an IDH1 Mutation | NCT03684811 | Basket trial with IDH1 mutation | FT-2102 Azacitidine Nivolumab Gemcitabine and Cisplatin | IDH1 inhibitor DNA methyltransferase inhibitor PD-L1 inhibitor Chemotherapy |
| Safety, Tolerability, and Pharmacokinetics of an Anti-PD-1 Monoclonal Antibody in Subjects with Advanced Malignancies | NCT03474640 | All tumors (Phase 1) | Toripalimab | PD-1 inhibitor |
| A Phase II of Nivolumab Plus Ipilimumab in Non-resectable Sarcoma and Endometrial Carcinoma | NCT02982486 | Soft Tissue Sarcoma Bone Sarcoma Chondrosarcoma Gastrointestinal Stromal Sarcoma Ewing’s Tumor Metastatic Ewing’s Tumor Recurrent Osteosarcoma Desmoplastic Small Round Cell Tumor | Ipilimumab Nivolumab | CTLA-4 inhibitor PD-L1 inhibitor |
| IACS-6274 with or without Pembrolizumab for the Treatment of Advanced Solid Tumors | NCT05039801 | Basket trial | IPN60090 Pembrolizumab | Glutaminase Inhibitor PD-1 inhibitor |
| LN-145 or LN-145-S1 in Treating Patients with Relapsed or Refractory Ovarian Cancer, Anaplastic Thyroid Cancer, Osteosarcoma, or Other Bone and Soft Tissue Sarcomas | NCT03449108 | Basket trial | Aldesleukin Autologous Tumor Infiltrating Lymphocytes LN-145 Autologous Tumor Infiltrating Lymphocytes LN-145-S1 Cyclophosphamide Fludarabine Ipilimumab Nivolumab | Recombinant IL-2 Cell therapy Chemotherapy CTLA-4 inhibitor PD-L1 inhibitor |
| Autologous Dendritic Cell Vaccine in Patients with Soft Tissue Sarcoma | NCT01883518 | All sarcoma subtypes | Autologous dendritic cell vaccine | Cell therapy |
| Study of INBRX-109 in Conventional Chondrosarcoma | NCT04950075 | CS | INBRX-109 | Tetravalent DR5 agonistic antibody |
| Phase 1 Study of INBRX-109 in Subjects with Locally Advanced or Metastatic Solid Tumors Including Sarcomas | NCT03715933 | Basket trial | INBRX-109 Carboplatin Cisplatin Pemetrexed 5-fluorouracil Irinotecan Temozolomide | Antibody targeting Death Receptor 5 (DR5) Chemotherapy |
| Phase I Study of IGM-8444 as a Single Agent and in Combination with Subjects with Relapsed and/or Refractory Solid Cancers | NCT04553692 | Basket trial | IGM-8444 FOLFIRI Bevacizumab (and approved biosimilars) Birinapant Venetoclax | Antibody targeting Death Receptor 5 (DR5) Chemotherapy Antiangiogenic Second mitochondrial-derived activator of caspases (SMAC) and inhibitor of IAP (Inhibitor of Apoptosis Protein) BCL2 inhibitor |
| Abemaciclib for Bone and Soft Tissue Sarcoma with Cyclin- Dependent Kinase (CDK) Pathway Alteration | NCT04040205 | Chondrosarcoma Osteosarcoma Soft Tissue Sarcoma | Abemaciclib | CDK4/6 inhibitor |
| Sirolimus and Cyclophosphamide in Metastatic or Unresectable Myxoid Liposarcoma and Chondrosarcoma | NCT02821507 | CS Myxoid Liposarcoma Mesenchymal Chondrosarcoma Dedifferentiated Chondrosarcoma | Sirolimus and cyclophosphamide | mTOR inhibitor and chemotherapy |
| TQB3525 for Advanced Bone Sarcomas with PI3KA Mutations or PTEN Loss | NCT04690725 | All tumors (Phase 1) | TQB3525 | PI3Ka inhibitor |
| Multicohort Trial of Trabectedin and Low-dose Radiation Therapy in Advanced/Metastatic Sarcomas | NCT05131386 | Soft Tissue Sarcoma Bone Tumors Small Round-cell Sarcomas | Trabectedin | Chemotherapy |
| Testing the Combination of Belinostat and SGI-110 (Guadecitabine) or ASTX727 for the Treatment of Unresectable and Metastatic Conventional Chondrosarcoma | NCT04340843 | Locally Advanced Unresectable Primary Central Chondrosarcoma Metastatic Primary Central Chondrosarcoma Unresectable Primary Central Chondrosarcoma | Belinostat Decitabine and Cedazuridine Guadecitabine | HDAC inhibitor Antimetabolites Anitmetabolite |
| Vismodegib in Treating Patients with Advanced Chondrosarcomas | NCT01267955 | All chondrosarcoma subtypes | Vismodegib | Hedgehog pathway inhibitor |
| Itacitinib in Treating Patients with Refractory Metastatic/Advanced Sarcomas | NCT03670069 | All sarcoma subtypes | Itacitinib | JAK-1 inhibitor |
| Trial | Trial Phase | Intervention | Type of Tumor | Years of Inclusion | Geographic Distribution | Median Age | N | Previous Lines of Treatment | CR (N) | PR (N) | SD (N) | PD (N) | RR (%) | DCR (%) | Median PFS (Months) | Median OS (Months) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grignani G. et al. Cancer 2011 [32] | II | Imatinib | CS | 2007–2009 | Multicenter Italy | 61 | 26 | At least 1: 100% | 0 | 0 | 8 | 18 | 0 | 30 | 3 | 11 |
| Chow W. et al. Cancer 2020 [33] | II (1 arm) | Pazopanib | CS | 2011–2015 | Multicenter USA/UK | 58 | 47 | At least 1: 32% | 0 | 1 | 30 | 11 | 2 | 65 | 7.9 | 17.6 |
| Tap W. D. et al. J Clin Oncol 2020 [34] | I | Ivosidenib | CS | March 2014 | Multicenter | 55 | 21 | >1: 50% <1: 50% | 0 | 1 | 11 | 6 | 4 | 57 | 5.6 | NA |
| Liu Z. L. et al.Cancer Med 2021 [35] | Restrospective | Anlotinib | Bone sarcoma | 2018–2020 | China | 24 | 9 | 1: 37 2: 31 3: 12 | 0 | 0 | 7 | 2 | 0 | 77 | 4.2 | NA |
| Duffaud F. et al.Eur J Cancer 2021 [36] | II | Regorafenib | CS | 2014–2019 | Multicenter France | 64 | 24 | 1: 17 2: 7 | 0 | 2 | 11 | 10 | 8 | 54 | 5 | 11.7 |
| Tawbi H.A. et al. Lancet Oncol 2017 [37] | II (1 arm) | Pembrolizumab | Soft Tissue/bone sarcoma | 2015–2016 | Multicenter | 33 (bone sarcoma) | 5 (CS) | 1: 19% 2: 38% 3: 43% | 0 | 1 | 1 | 3 | 20 | 32 | 8 | 12 |
| Schwartz G.K. et al. Lancet Oncol 2013 [38] | II | Cixutumumab + temsirolimus | Bone sarcoma | 2009–2012 | Multicenter USA | 47 | 17 | <2: 50% >2: 50% | 0 | 1 | NA | NA | 6 | NA | 5.2 | 13.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tlemsani, C.; Larousserie, F.; De Percin, S.; Audard, V.; Hadjadj, D.; Chen, J.; Biau, D.; Anract, P.; Terris, B.; Goldwasser, F.; et al. Biology and Management of High-Grade Chondrosarcoma: An Update on Targets and Treatment Options. Int. J. Mol. Sci. 2023, 24, 1361. https://doi.org/10.3390/ijms24021361
Tlemsani C, Larousserie F, De Percin S, Audard V, Hadjadj D, Chen J, Biau D, Anract P, Terris B, Goldwasser F, et al. Biology and Management of High-Grade Chondrosarcoma: An Update on Targets and Treatment Options. International Journal of Molecular Sciences. 2023; 24(2):1361. https://doi.org/10.3390/ijms24021361
Chicago/Turabian StyleTlemsani, Camille, Frédérique Larousserie, Sixtine De Percin, Virginie Audard, Djihad Hadjadj, Jeanne Chen, David Biau, Philippe Anract, Benoit Terris, François Goldwasser, and et al. 2023. "Biology and Management of High-Grade Chondrosarcoma: An Update on Targets and Treatment Options" International Journal of Molecular Sciences 24, no. 2: 1361. https://doi.org/10.3390/ijms24021361
APA StyleTlemsani, C., Larousserie, F., De Percin, S., Audard, V., Hadjadj, D., Chen, J., Biau, D., Anract, P., Terris, B., Goldwasser, F., Pasmant, E., & Boudou-Rouquette, P. (2023). Biology and Management of High-Grade Chondrosarcoma: An Update on Targets and Treatment Options. International Journal of Molecular Sciences, 24(2), 1361. https://doi.org/10.3390/ijms24021361