
Stereoselective Synthesis and Antiproliferative Activities of Tetrafunctional Diterpene Steviol Derivatives

 ,
,  , , ,
, , , 

Abstract
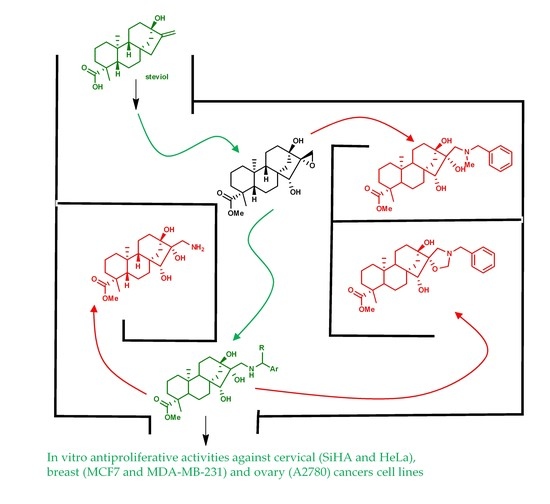
1. Introduction
2. Results and Discussion
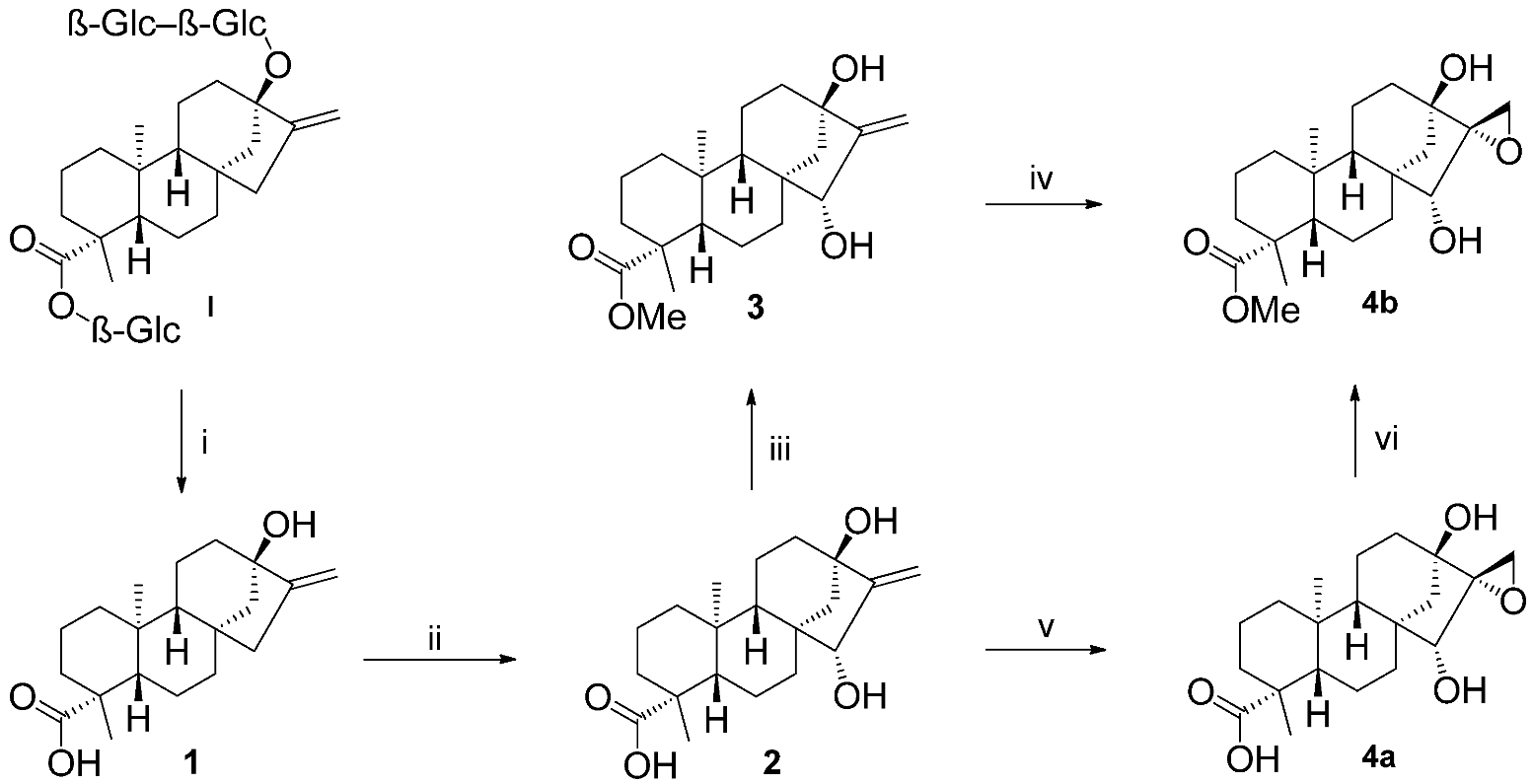
2.1. Synthesis of Key Intermediate Spiro-Epoxide 4b
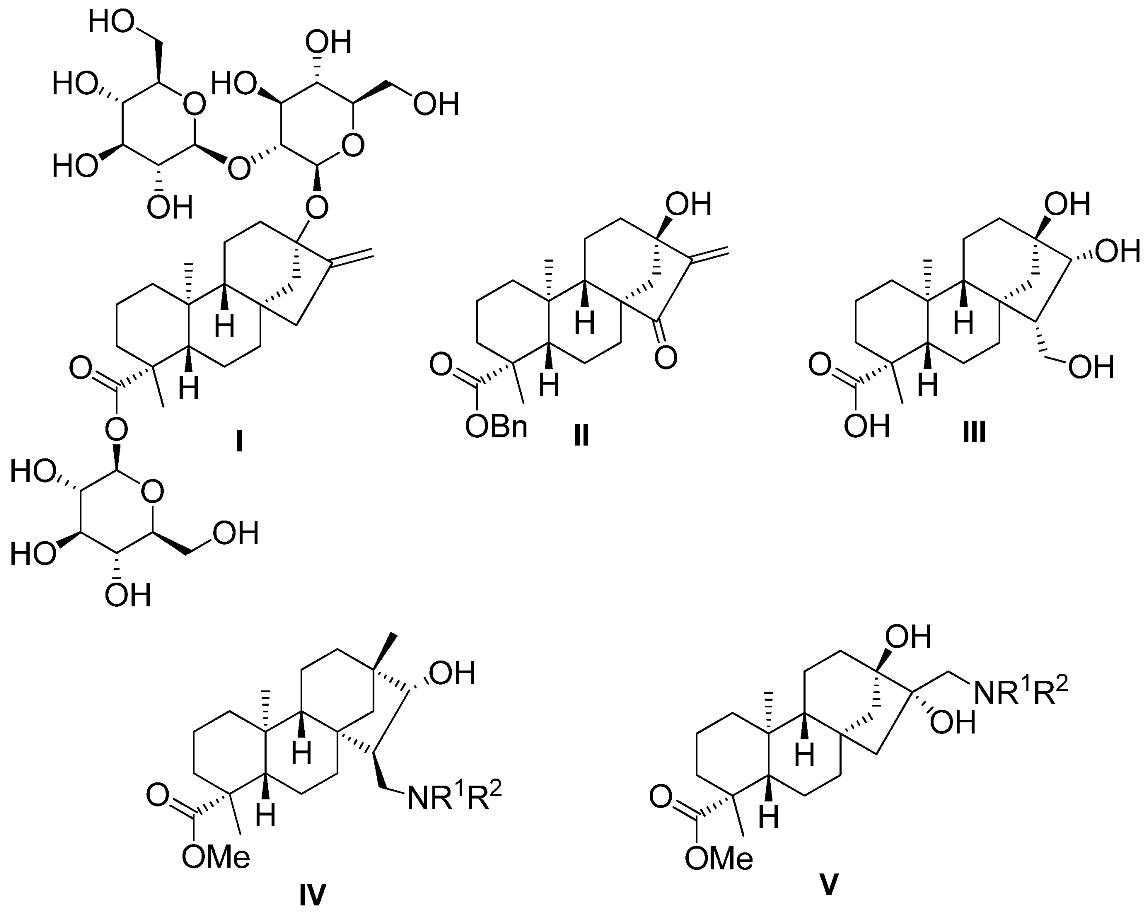
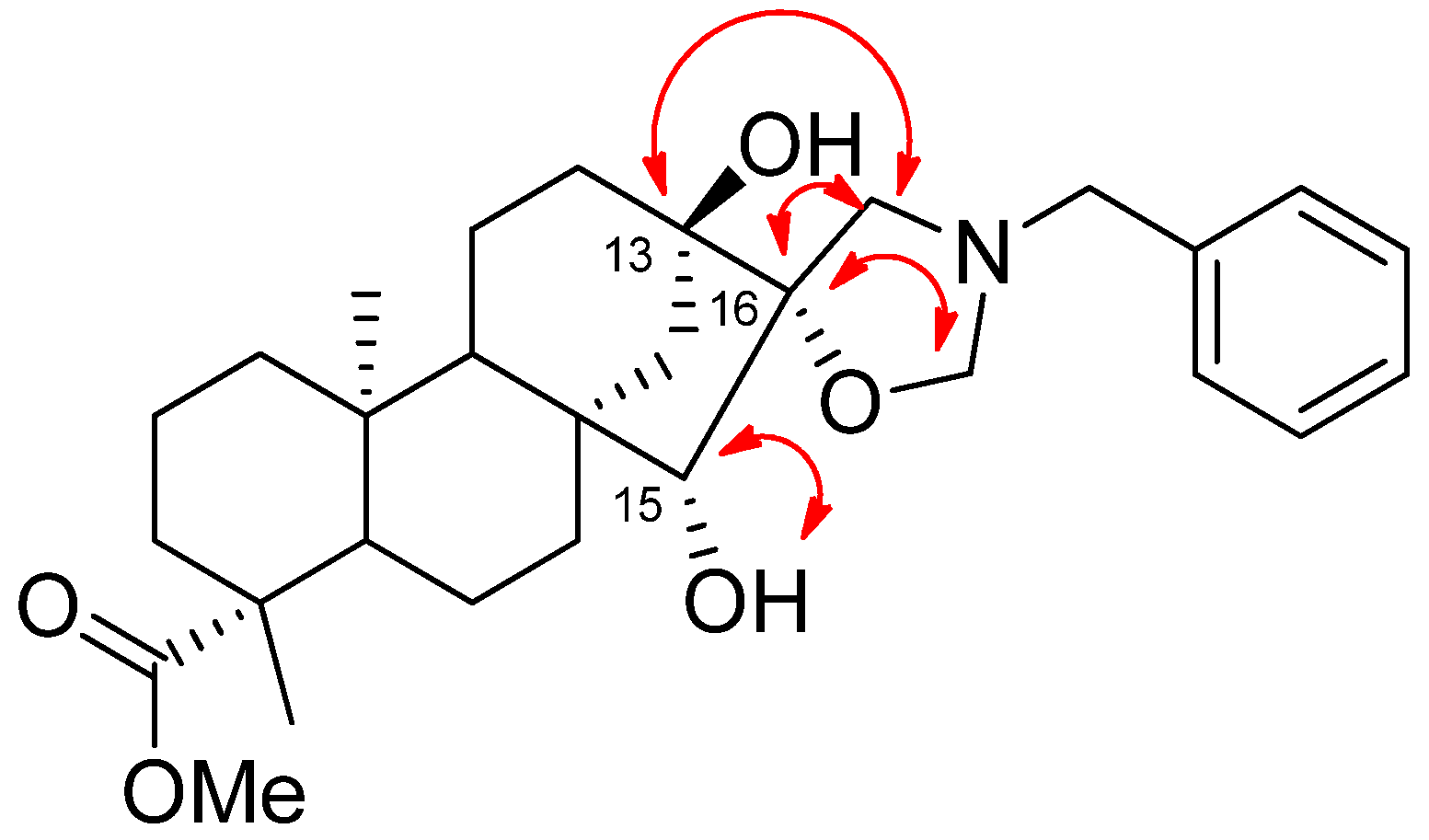
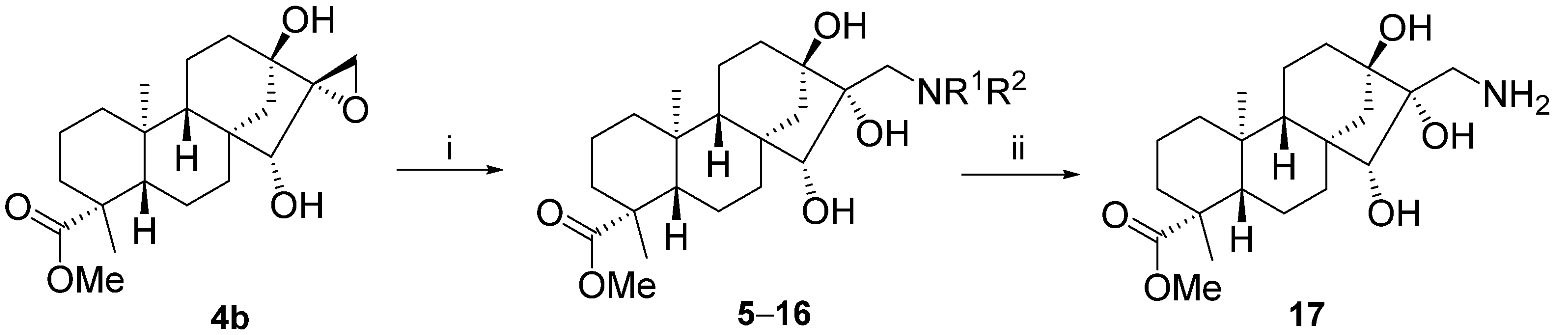
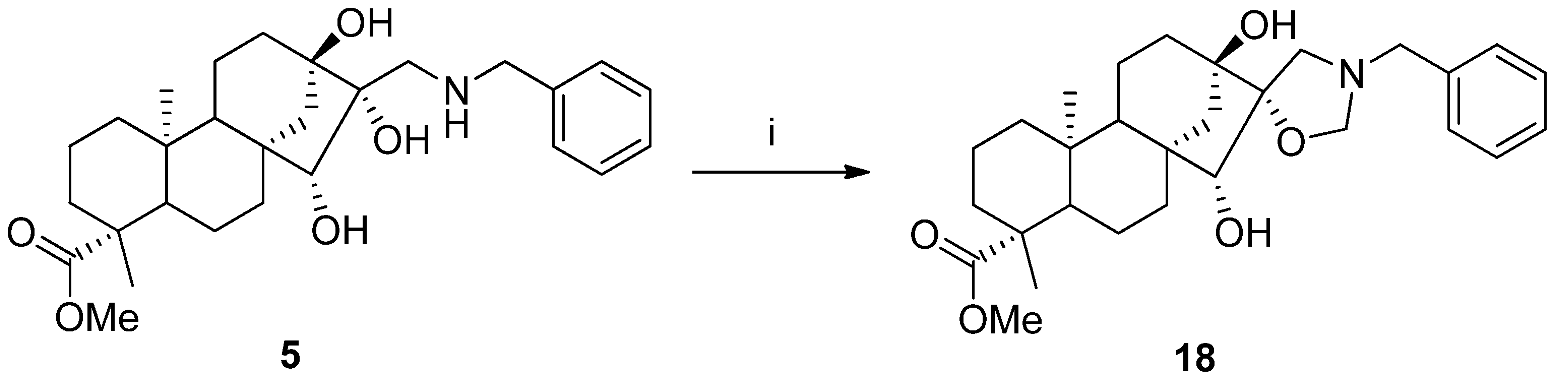
2.2. Synthesis of Aminotriol Derivatives 5–17
2.3. Drug-Likeness Properties of Steviol-Based Aminotriols Using in Silico and Experimental Physicochemical Parameters
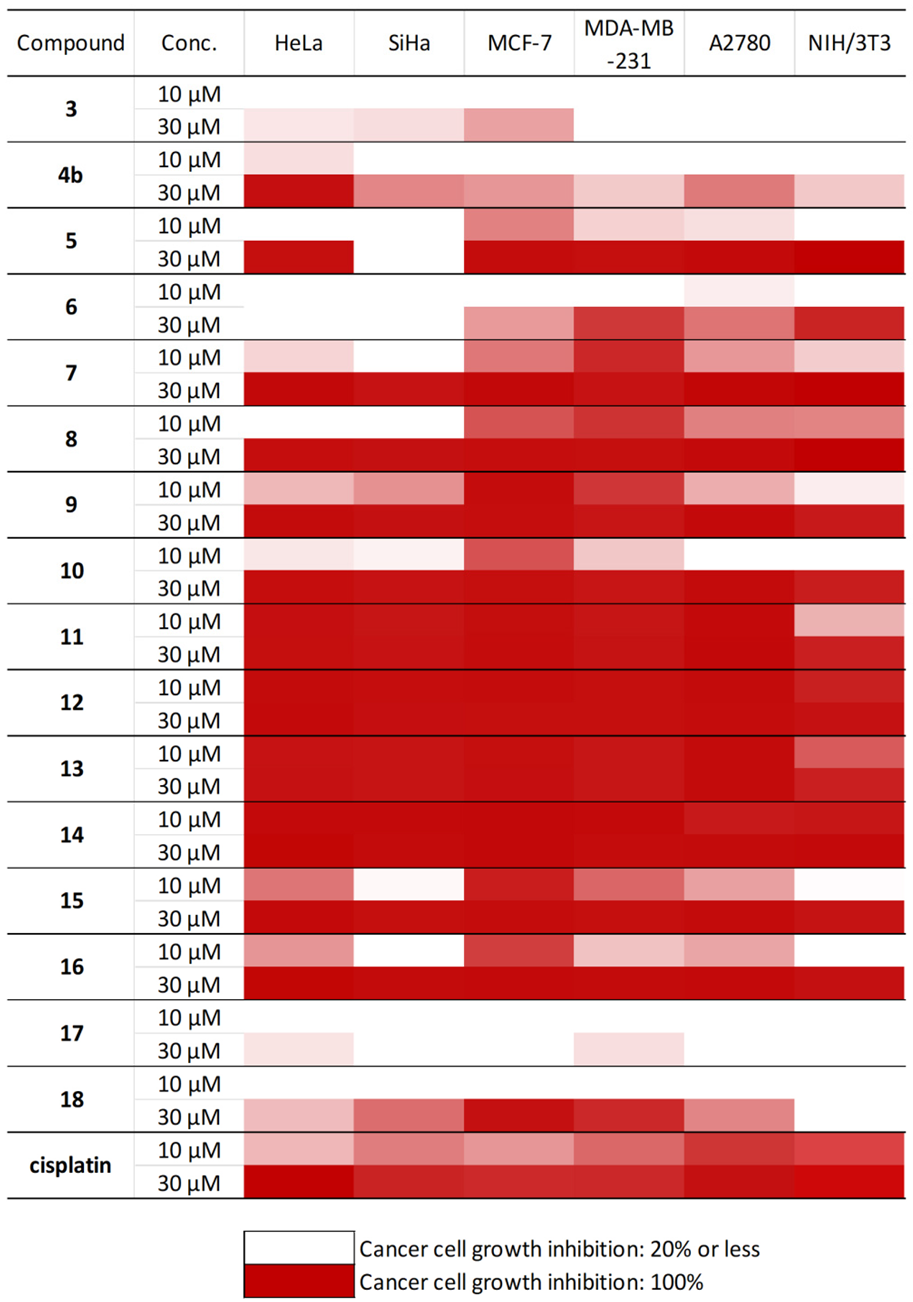
2.4. In Vitro Antiproliferative Studies of Steviol-Based Aminotriols and Structure-Activity Relationship
3. Materials and Methods
3.1. General Methods
3.2. Starting Materials
3.2.1. (4R,6aR,7R,9S,11bS)-Methyl 7,9-Dihydroxy-4,11b-dimethyl-8-methylenetetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (3)
3.2.2. (2’R,4R,6aR,7R,9S,11bS)-Methyl 7,9-Dihydroxy-4,11b-dimethyldodecahydro-1H-spiro[6a,9-methanocyclohepta[a]naphthalene-8,2’-oxirane]-4-carboxylate (4b)
3.3. General Procedure for Preparation of Aminotriols with Primary and Secondary Amines
3.3.1. (4R,6aR,7R,8R,9S,11bS)-Methyl 8-((Benzylamino)methyl)-7,8,9-trihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (5)
3.3.2. (4R,6aR,7R,8R,9S,11bS)-Methyl 8-((Benzyl(methyl)amino)methyl)-7,8,9-trihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (6)
3.3.3. (4R,6aR,7R,8R,9S,11bS)-Methyl 7,8,9-Trihydroxy-4,11b-dimethyl-8-((((R)-1-phenylethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (7)
3.3.4. (4R,6aR,7R,8R,9S,11bS)-Methyl 7,8,9-Trihydroxy-4,11b-dimethyl-8-((((S)-1-phenylethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (8)
3.3.5. (4R,6aR,7R,8R,9S,11bS)-Methyl 8-(((4-Fluorobenzyl)amino)methyl)-7,8,9-trihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (9)
3.3.6. (4R,6aR,7R,8R,9S,11bS)-Methyl 7,8,9-Trihydroxy-8-(((4-methoxybenzyl)amino)methyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (10)
3.3.7. (4R,4aS,6aR,7R,8R,11aS,11bS)-Methyl 8-((((R)-1-(4-Fluorophenyl)ethyl)amino)methyl)-7,8,9-trihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (11)
3.3.8. (4R,6aR,7R,8R,9S,11bS)-Methyl 7,8,9-Trihydroxy-4,11b-dimethyl-8-((((R)-1-(naphthalen-2-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (12)
3.3.9. (4R,6aR,7R,8R,9S,11bS)-Methyl 7,8,9-Trihydroxy-4,11b-dimethyl-8-((((S)-1-(naphthalen-2-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (13)
3.3.10. (4R,6aR,7R,8R,9S,11bS)-Methyl 7,8,9-Trihydroxy-4,11b-dimethyl-8-(((naphthalen-1-ylmethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (14)
3.3.11. (4R,6aR,7R,8R,9S,11bS)-Methyl 7,8,9-Trihydroxy-4,11b-dimethyl-8-((((R)-1-phenylpropyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (15)
3.3.12. (4R,6aR,7R,8R,9S,11bS)-Methyl 7,8,9-Trihydroxy-4,11b-dimethyl-8-((((S)-1-phenylpropyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (16)
3.3.13. (4R,6aR,7R,8R,9S,11bS)-Methyl 8-(Aminomethyl)-7,8,9-trihydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (17)
3.3.14. (4R,5’R,6aR,7R,9S,11bS)-Methyl 3’-Benzyl-7,9-dihydroxy-4,11b-dimethyldodecahydro-1H-spiro [6a,9-methanocyclohepta[a]naphthalene-8,5’-oxazolidine]-4-carboxylate (18)
3.4. Analytical Method for Physicochemical Investigations
3.5. Kinetic Solubility and PAMPA-GI Measurements
3.6. Determination of the Antiproliferative Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawash, M. Recent Advances of Tubulin Inhibitors Targeting the Colchicine Binding Site for Cancer Therapy. Biomolecules 2022, 12, 1843. [Google Scholar] [CrossRef] [PubMed]
- Hawash, M.M.A.; Kahraman, D.C.; Eren, F.; Cetin Atalay, R.; Baytas, S.N. Synthesis and Biological Evaluation of Novel Pyrazolic Chalcone Derivatives as Novel Hepatocellular Carcinoma Therapeutics. Eur. J. Med. Chem. 2017, 129, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Caprio, V.; Williams, J.M.J. Catalysis in Asymmetric Synthesis; John Wiley & Sons: Oxford, UK, 2009; ISBN 1-4051-9091-4. [Google Scholar]
- Satyanarayana, T.; Kagan, H.B. The Multi-Substrate Screening of Asymmetric Catalysts. Adv. Synth. Catal. 2005, 347, 737–748. [Google Scholar] [CrossRef]
- Gaunt, M.J.; Johansson, C.C.C.; McNally, A.; Vo, N.T. Enantioselective Organocatalysis. Drug Discov. Today 2007, 12, 8–27. [Google Scholar] [CrossRef] [PubMed]
- El Alami, M.S.I.; El Amrani, M.A.; Agbossou-Niedercorn, F.; Suisse, I.; Mortreux, A. Chiral Ligands Derived from Monoterpenes: Application in the Synthesis of Optically Pure Secondary Alcohols via Asymmetric Catalysis. Chem.-Eur. J. 2015, 21, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, J.T.; Sharpe, R.J.; Johnson, J.S. Enantioselective Synthesis of Pactamycin, a Complex Antitumor Antibiotic. Science 2013, 340, 180–182. [Google Scholar] [CrossRef]
- Luo, L.; Yamamoto, H. Synthesis of Virtually Enantiopure Aminodiols with Three Adjacent Stereogenic Centers by Epoxidation and Ring-Opening. Org. Biomol. Chem. 2015, 13, 10466–10470. [Google Scholar] [CrossRef]
- Miyagawa, T.; Inuki, S.; Oishi, S.; Ohno, H. Construction of Quaternary Carbon Stereocenter of α-Tertiary Amine through Remote C–H Functionalization of Tris Derivatives: Enantioselective Total Synthesis of Myriocin. Org. Lett. 2019, 21, 5485–5490. [Google Scholar] [CrossRef]
- Raschmanová, J.Š.; Martinková, M.; Pilátová, M.B.; Nosálová, N.; Kuchár, J.; Bodnár, G. Synthesis and in Vitro Anticancer Activity of Penaresidin-Related Stereoisomeric Analogues. Carbohydr. Res. 2021, 508, 108419. [Google Scholar] [CrossRef]
- Bamou, F.Z.; Le, T.M.; Tayeb, B.A.; Tahaei, S.A.S.; Minorics, R.; Zupkó, I.; Szakonyi, Z. Antiproliferative Activity of (−)-Isopulegol-based 1,3-Oxazine, 1,3-Thiazine and 2,4-Diaminopyrimidine Derivatives. ChemistryOpen 2022, 11, e202200169. [Google Scholar] [CrossRef]
- Sarwar, M.S.; Xia, Y.-X.; Liang, Z.-M.; Tsang, S.W.; Zhang, H.-J. Mechanistic Pathways and Molecular Targets of Plant-Derived Anticancer Ent-Kaurane Diterpenes. Biomolecules 2020, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.; Gomes, D.; Lyra, F.; dos Santos, G.; Martins, L. The Remarkable Structural Diversity Achieved in Ent-Kaurane Diterpenes by Fungal Biotransformations. Molecules 2014, 19, 1856–1886. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Iriti, M.; Vitalini, S.; Antolak, H.; Pawlikowska, E.; Kręgiel, D.; Sharifi-Rad, J.; Oyeleye, S.I.; Ademiluyi, A.O.; Czopek, K.; et al. Euphorbia-Derived Natural Products with Potential for Use in Health Maintenance. Biomolecules 2019, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-S.; Luo, L.-P.; Xu, G.; Xu, X.-J.; Xu, C.; Ou, E.; Zhang, H.-Y.; Yuan, Z.-Q.; Zhao, Y. Synthesis and Biological Evaluation of Steviol Derivatives with Improved Cytotoxic Activity and Selectivity. J. Nat. Prod. 2022, 85, 1945–1958. [Google Scholar] [CrossRef] [PubMed]
- Lohoelter, C.; Weckbecker, M.; Waldvogel, S.R. (-)-Isosteviol as a Versatile Ex-Chiral-Pool Building Block for Organic Chemistry: (-)-Isosteviol as a Building Block for Organic Chemistry. Eur. J. Org. Chem. 2013, 2013, 5539–5554. [Google Scholar] [CrossRef]
- Wang, M.; Li, H.; Xu, F.; Gao, X.; Li, J.; Xu, S.; Zhang, D.; Wu, X.; Xu, J.; Hua, H.; et al. Diterpenoid Lead Stevioside and Its Hydrolysis Products Steviol and Isosteviol: Biological Activity and Structural Modification. Eur. J. Med. Chem. 2018, 156, 885–906. [Google Scholar] [CrossRef]
- Ullah, A.; Munir, S.; Mabkhot, Y.; Badshah, S. Bioactivity Profile of the Diterpene Isosteviol and Its Derivatives. Molecules 2019, 24, 678. [Google Scholar] [CrossRef]
- Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Synthesis and Biological Application of Isosteviol-Based 1,3-Aminoalcohols. Int. J. Mol. Sci. 2021, 22, 11232. [Google Scholar] [CrossRef]
- Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols. Int. J. Mol. Sci. 2019, 21, 184. [Google Scholar] [CrossRef]
- Ukiya, M.; Sawada, S.; Kikuchi, T.; Kushi, Y.; Fukatsu, M.; Akihisa, T. Cytotoxic and Apoptosis-Inducing Activities of Steviol and Isosteviol Derivatives against Human Cancer Cell Lines. Chem. Biodivers. 2013, 10, 177–188. [Google Scholar] [CrossRef]
- Shi, L.-Y.; Wu, J.-Q.; Zhang, D.-Y.; Wu, Y.-C.; Hua, W.-Y.; Wu, X.-M. Efficient Synthesis of Novel Jolkinolides and Related Derivatives Starting from Stevioside. Synthesis 2011, 2011, 3807–3814. [Google Scholar] [CrossRef]
- Li, J.; Zhang, D.; Wu, X. Synthesis and Biological Evaluation of Novel Exo-Methylene Cyclopentanone Tetracyclic Diterpenoids as Antitumor Agents. Bioorg. Med. Chem. Lett. 2011, 21, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Peña, A.; Alarcón, L.; Aparicio, R.; Rojas, J.; Usubillaga, A. On the Allylic Hydroxylation of Ent-Kaurenic Acid with SeO2. Av. En Quím. 2014, 9, 7–13. [Google Scholar]
- Avent, A.G.; Hanson, J.R.; Hitchcock, P.B.; De Oliveira, B.H. The Influence of a 15-Hydroxy Group on the Rearrangement Reactions of Steviol and Its 16,17-Epoxide. J. Chem. Soc. Perkin Trans. 1 1990, 22, 2661–2665. [Google Scholar] [CrossRef]
- Vandichel, M.; Leus, K.; Van Der Voort, P.; Waroquier, M.; Van Speybroeck, V. Mechanistic Insight into the Cyclohexene Epoxidation with VO(Acac)2 and Tert-Butyl Hydroperoxide. J. Catal. 2012, 294, 1–18. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Csillag, K.; Fülöp, F. Stereoselective Synthesis of Carane-Based Aminodiols as Chiral Ligands for the Catalytic Addition of Diethylzinc to Aldehydes. Tetrahedron Asymmetry 2011, 22, 1021–1027. [Google Scholar] [CrossRef]
- Le, T.M.; Csámpai, A.; Fülöp, F.; Szakonyi, Z. Regio- and Stereoselective Synthesis of Bicyclic Limonene-Based Chiral Aminodiols and Spirooxazolidines. Chem.-Eur. J. 2018, 24, 13607–13615. [Google Scholar] [CrossRef]
- Le, T.M.; Szilasi, T.; Volford, B.; Szekeres, A.; Fülöp, F.; Szakonyi, Z. Stereoselective Synthesis and Investigation of Isopulegol-Based Chiral Ligands. Int. J. Mol. Sci. 2019, 20, 4050. [Google Scholar] [CrossRef]
- Azizi, N.; Mirmashhori, B.; Saidi, M.R. Lithium Perchlorate Promoted Highly Regioselective Ring Opening of Epoxides under Solvent-Free Conditions. Catal. Commun. 2007, 8, 2198–2203. [Google Scholar] [CrossRef]
- Saddique, F.A.; Zahoor, A.F.; Faiz, S.; Naqvi, S.A.R.; Usman, M.; Ahmad, M. Recent Trends in Ring Opening of Epoxides by Amines as Nucleophiles. Synth. Commun. 2016, 46, 831–868. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Zupkó, I.; Sillanpää, R.; Fülöp, F. Stereoselective Synthesis and Cytoselective Toxicity of Monoterpene-Fused 2-Imino-1,3-Thiazines. Molecules 2014, 19, 15918–15937. [Google Scholar] [CrossRef] [PubMed]
- Szakonyi, Z.; Zupkó, I.; Fülöp, F. Stereoselective Synthesis and Antiproliferative Activity of Monoterpene-Fused 2- Imino-1,3-Oxazines. Curr. Org. Synth. 2017, 14, 612–619. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Pethő, B.; Zwillinger, M.; Csenki, J.T.; Káncz, A.E.; Krámos, B.; Müller, J.; Balogh, G.T.; Novák, Z. Palladium-Catalyzed 2,2,2-Trifluoroethoxylation of Aromatic and Heteroaromatic Chlorides Utilizing Borate Salt and the Synthesis of a Trifluoro Analogue of Sildenafil. Chem.-Eur. J. 2017, 23, 15628–15632. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Springthorpe, B. The Influence of Drug-like Concepts on Decision-Making in Medicinal Chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J. Lipophilicity in Drug Discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Subrahmanyam, C.; Rao, B.V.; Ward, R.S.; Hursthouse, M.B.; Hibbs, D.E. Diterpenes from the Marine Mangrove Bruguieragymnorhiza. Phytochemistry 1999, 51, 83–90. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, J.-H.; Dai, G.-F.; Liu, C.-J.; Tian, G.-Q.; Ma, W.-Y.; Tao, J.-C. Stereoselective Synthesis of Bioactive Isosteviol Derivatives as α-Glucosidase Inhibitors. Bioorg. Med. Chem. 2009, 17, 1464–1473. [Google Scholar] [CrossRef]
- Coates, R.M.; Kang, H.Y. Synthesis and Evaluation of Cyclobutylcarbinyl Derivatives as Potential Intermediates in Diterpene Biosynthesis. J. Org. Chem. 1987, 52, 2065–2074. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Product | R1 | R2 | Yield [%] |
|---|---|---|---|---|
| 1 | 5 | H | benzyl | 93 |
| 2 | 6 | Me | benzyl | 45 |
| 3 | 7 | H | (R)-α-methylbenzyl | 59 |
| 4 | 8 | H | (S)-α-methylbenzyl | 60 |
| 5 | 9 | H | 4-fluorobenzyl | 95 |
| 6 | 10 | H | 4-methoxybenzyl | 96 |
| 7 | 11 | H | (R)-4-fluoro-α-methylbenzyl | 50 |
| 8 | 12 | H | (R)-1-(2-naphthyl)ethyl | 60 |
| 9 | 13 | H | (S)-1-(2-naphthyl)ethyl | 55 |
| 10 | 14 | H | 1-naphthylmethyl | 60 |
| 11 | 15 | H | (R)-(+)-α-ethylbenzyl | 80 |
| 12 | 16 | H | (S)-(-)-α-ethylbenzyl | 65 |
| Product | MW a | pKa,base/pKa,acid | logPa/logD7.4 | TPSA | Lipinski Ro5 Violation a |
|---|---|---|---|---|---|
| 2 | 334 | -/4.6 | 3.2/0.4 | 77.8 | No |
| 3 | 348 | -/- | 3.8/3.8 | 66.8 | No |
| 4b | 364 | -/- | 2.6/2.6 | 79.3 | No |
| 5 | 472 | 9.5/- | 4.5/2.8 | 99 | No |
| 6 | 486 | 8.6/- | 4.8/4.0 | 90.2 | No |
| 7 | 486 | 9.6/- | 4.8/3.1 | 99 | No |
| 8 | 486 | 9.6/- | 4.8/3.1 | 99 | No |
| 9 | 490 | 9.5/- | 4.4/2.8 | 99 | No |
| 10 | 502 | 9.9/- | 4.3/2.3 | 108.3 | Moderate: Mw |
| 11 | 504 | 9.6/- | 4.8/3.2 | 99 | Moderate: Mw |
| 12 | 536 | 9.3/- | 5.7/4.3 | 99 | High: Mw, logP |
| 13 | 536 | 9.3/- | 5.7/4.3 | 99 | High: Mw, logP |
| 14 | 522 | 9.2/- | 5.5/4.2 | 99 | High: Mw, logP |
| 15 | 500 | 9.6/- | 5.2/3.5 | 99 | Moderate: logP |
| 16 | 500 | 9.6/- | 5.2/3.5 | 99 | Moderate: logP |
| 17 | 382 | 10.2/- | 2.3/-0.1 | 113 | No |
| 18 | 484 | 5.2/- | 4.1/4.1 | 79.2 | No |
| Products | Kinetic Solubility a () | PAMPA-GI | |
|---|---|---|---|
| Pe b (10−7 cm/s) | MR c (%) | ||
| 2 | 443.4 ± 35.7 | 5.2 ± 0.2 | 6.5 ± 0.7 |
| 3 | ND d | ND d | ND d |
| 4b | ND d | ND d | ND d |
| 5 | 290.8 ± 35.5 | 6.4 ± 0.3 | 29.8 ± 0.3 |
| 6 | 114.3 ± 16.0 | 2.3 ± 0.8 | 67.3 ± 8.6 |
| 7 | 181.6 ± 3.2 | 6.4 ± 0.7 | 28.8 ± 5.8 |
| 8 | 131.3 ± 2.2 | 5.8 ± 0.1 | 42.6 ± 3.9 |
| 9 | 42.1 ± 10.8 | 4.0 ± 0.4 | 63.8 ± 4.9 |
| 10 | 142.9 ± 26.8 | 3.7 ± 1.1 | 6.1 ± 1.1 |
| 11 | 145.4 ± 14.1 | 4.8 ± 0.5 | 22.4 ± 2.0 |
| 12 | 13.3 ± 0.9 | ND e | 92.6 ± 2.1 |
| 13 | 11.9 ± 1.8 | ND e | 92.6 ± 1.1 |
| 14 | 18.9 ± 2.4 | ND e | 94.2 ± 2.0 |
| 15 | 145.0 ± 28.2 | 1.4 ± 0.4 | 46.8 ± 3.3 |
| 16 | 89.9 ± 8.5 | 2.5 ± 0.9 | 44.6 ± 4.2 |
| 17 | 535.4 ± 38.4 | ND e | 1.5 ± 0.3 |
| 18 | 321.1 ± 88.5 | ND e | 2.7 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, D.; Schelz, Z.; Erdős, D.; Kis, A.K.; Nagy, V.; Zupkó, I.; Balogh, G.T.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activities of Tetrafunctional Diterpene Steviol Derivatives. Int. J. Mol. Sci. 2023, 24, 1121. https://doi.org/10.3390/ijms24021121
Bai D, Schelz Z, Erdős D, Kis AK, Nagy V, Zupkó I, Balogh GT, Szakonyi Z. Stereoselective Synthesis and Antiproliferative Activities of Tetrafunctional Diterpene Steviol Derivatives. International Journal of Molecular Sciences. 2023; 24(2):1121. https://doi.org/10.3390/ijms24021121
Chicago/Turabian StyleBai, Dorottya, Zsuzsanna Schelz, Dóra Erdős, Anna K. Kis, Viktória Nagy, István Zupkó, György T. Balogh, and Zsolt Szakonyi. 2023. "Stereoselective Synthesis and Antiproliferative Activities of Tetrafunctional Diterpene Steviol Derivatives" International Journal of Molecular Sciences 24, no. 2: 1121. https://doi.org/10.3390/ijms24021121
APA StyleBai, D., Schelz, Z., Erdős, D., Kis, A. K., Nagy, V., Zupkó, I., Balogh, G. T., & Szakonyi, Z. (2023). Stereoselective Synthesis and Antiproliferative Activities of Tetrafunctional Diterpene Steviol Derivatives. International Journal of Molecular Sciences, 24(2), 1121. https://doi.org/10.3390/ijms24021121