
Epigenetic Modulators as Therapeutic Agents in Cancer
Abstract
:1. Introduction
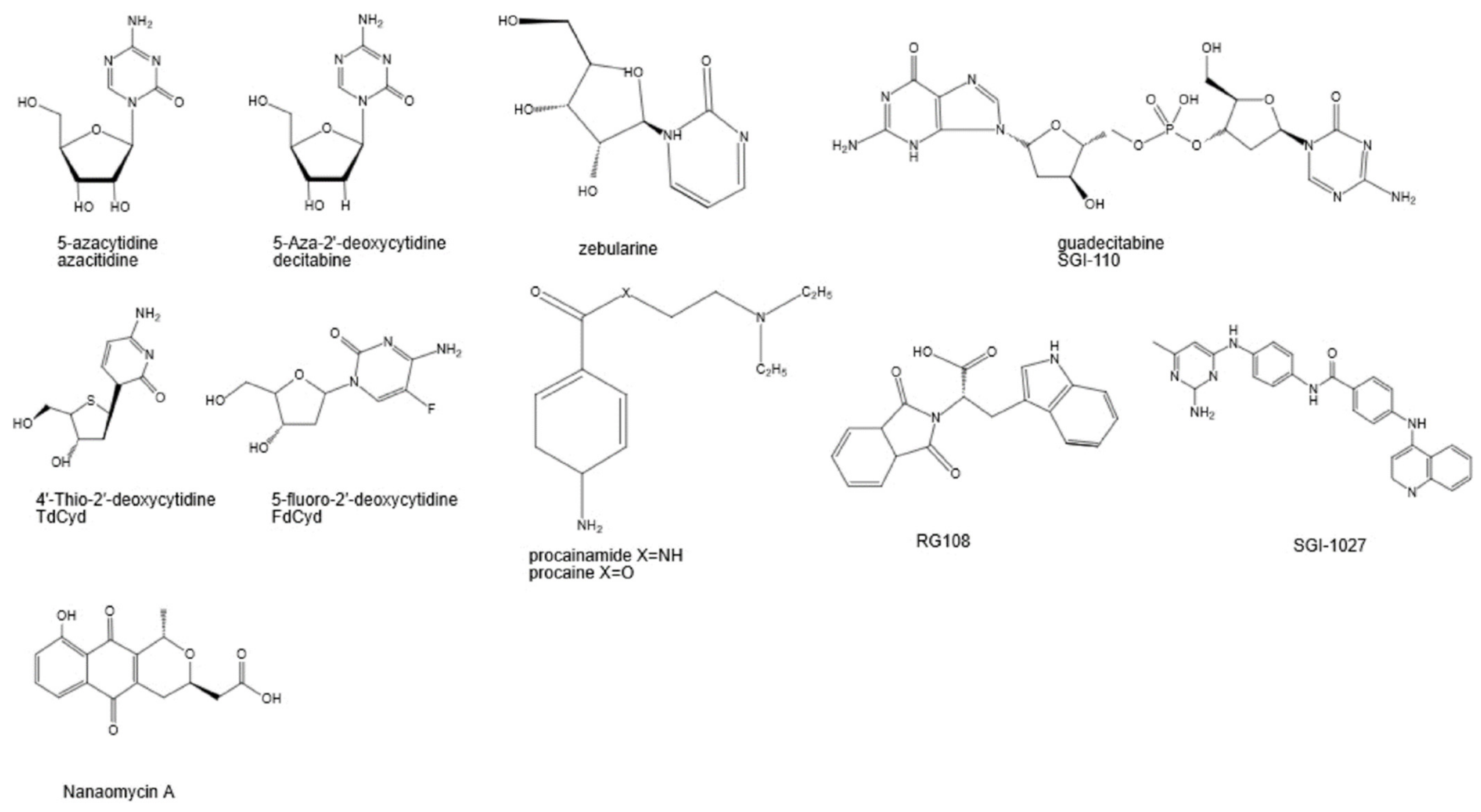
2. DNA Methylation Inhibitors
3. Histone Deacetylase Inhibitors
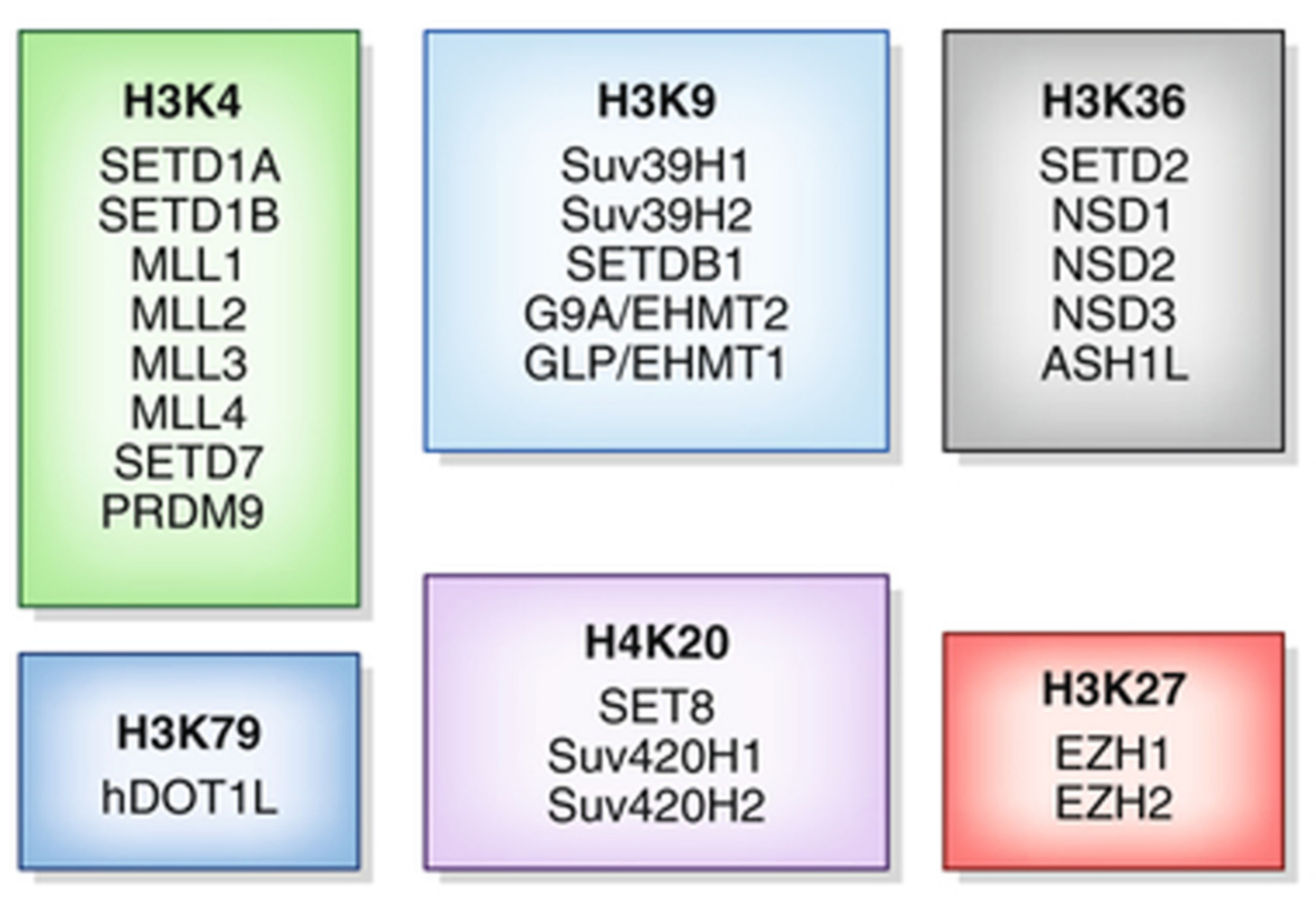
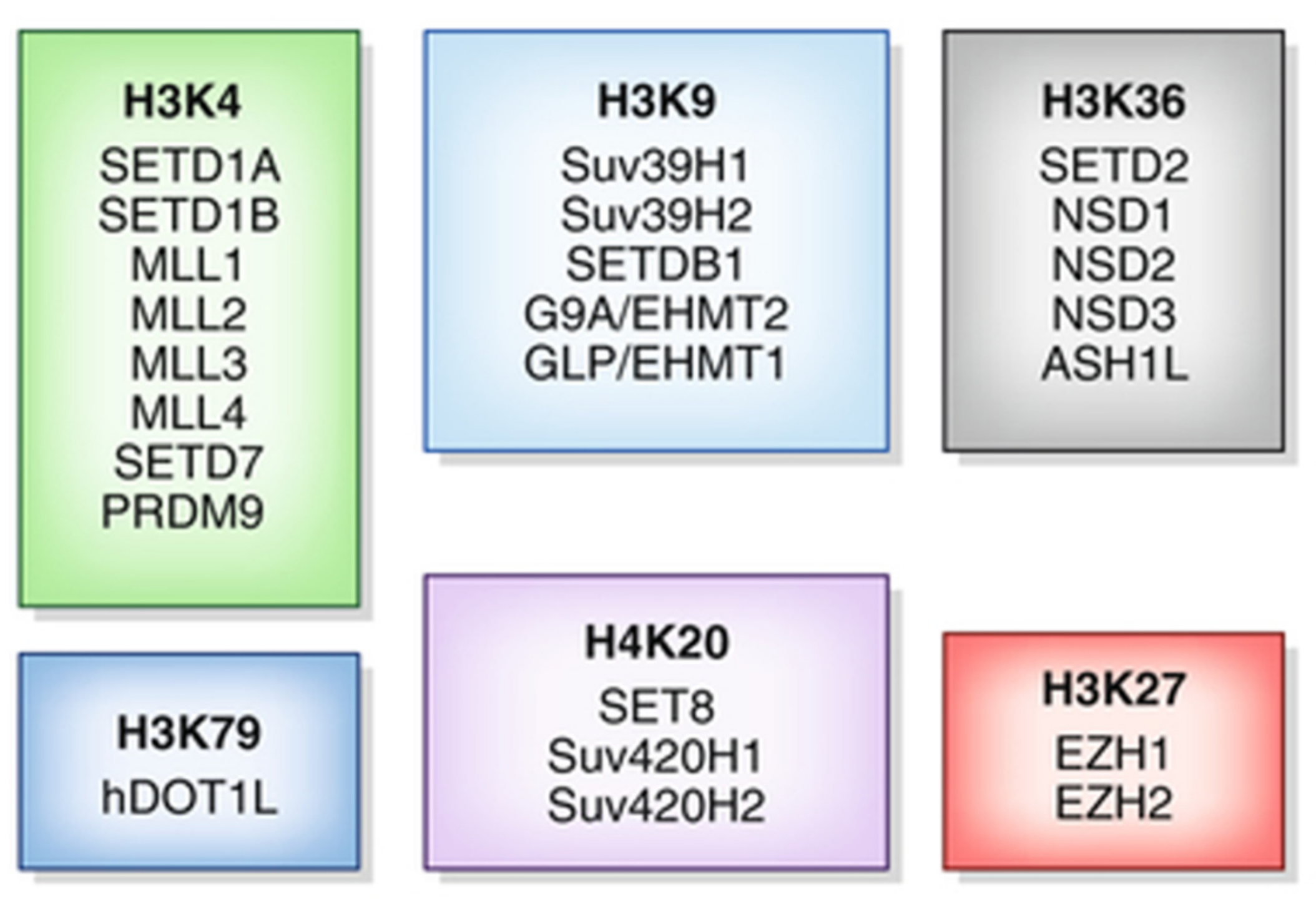
4. Histone Methylation Inhibitors
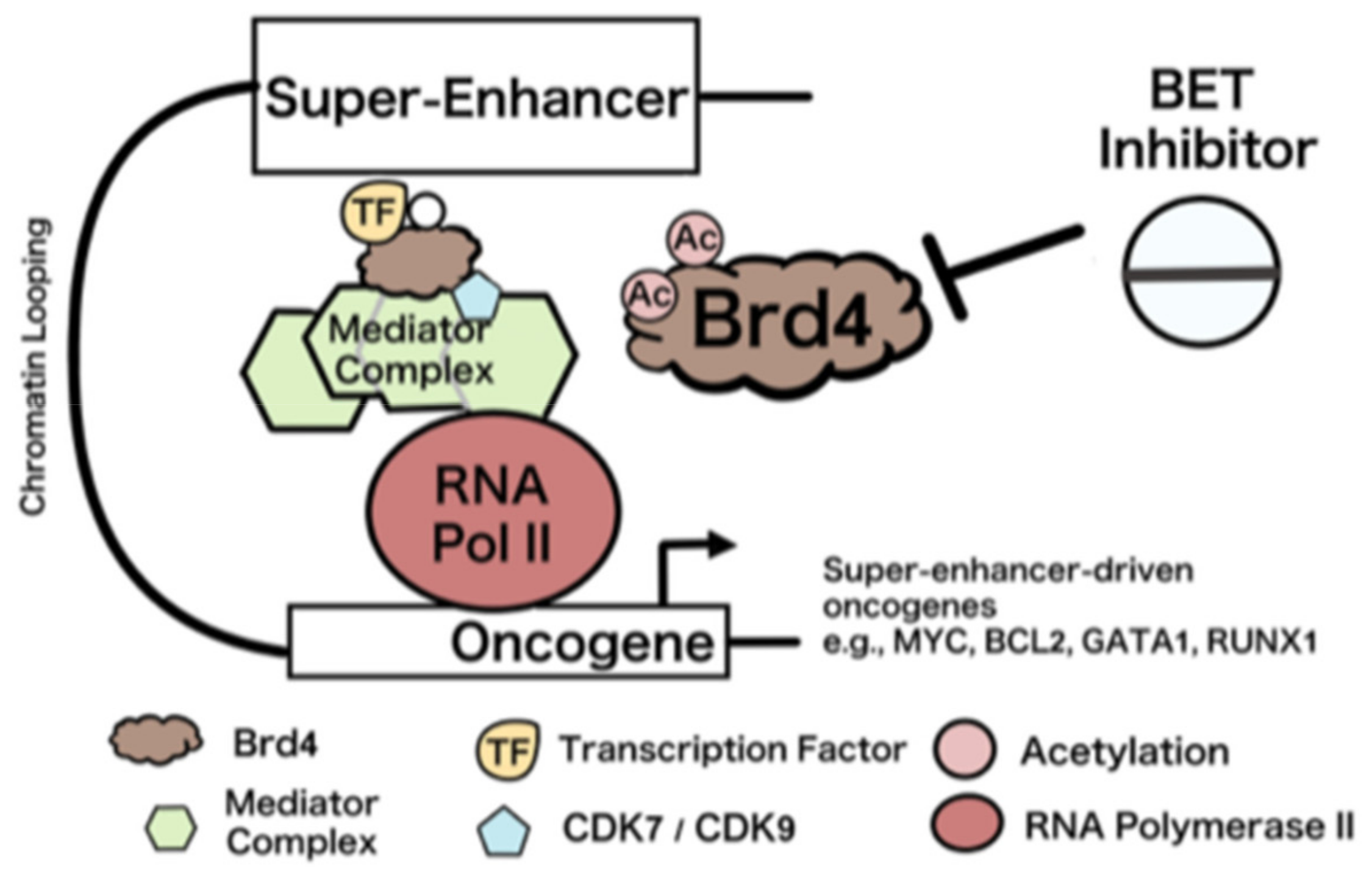
5. Bromodomain and Bet Inhibitors
6. Current Challenges and Discoveries
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ma, T.; Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Sig Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.H.; El-Fawal, H.A.N.; Abdelnaser, A. Hepigenetics: A Review of Epigenetic Modulators and Potential Therapies in Hepatocellular Carcinoma. Biomed. Res. Int. 2020, 2020, 9593254. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Zykovich, A.; Hubbard, A.; Flynn, J.M.; Tarnopolsky, M.; Fraga, M.F.; Kerksick, C.; Ogborn, D.; MacNeil, L.; Mooney, S.D.; Melov, S. Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell 2014, 13, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef]
- Morgan, A.E.; Davies, T.J.; Mc Auley, M.T. The role of DNA methylation in ageing and cancer. Proc. Nutr. Soc. 2018, 77, 412–422. [Google Scholar] [CrossRef]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA methyltransferases: A novel target for prevention and therapy. Front. Oncol. 2014, 4, 80. [Google Scholar] [CrossRef] [PubMed]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer. Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Loaeza-Loaeza, J.; Beltran, A.S.; Hernández-Sotelo, D. DNMTs and Impact of CpG Content, Transcription Factors, Consensus Motifs, lncRNAs, and Histone Marks on DNA Methylation. Genes 2020, 11, 1336. [Google Scholar] [CrossRef] [PubMed]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.M.; Ausseil, F.; Vispé, S.; Arimondo, P.B. DNA methylation inhibitors in cancer: Recent and future approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.; Zhang, S.; Zhou, Y.; Guan, Q. DNA Methyltransferase Inhibitors: Catalysts for Antitumour Immune Responses. OncoTargets Ther. 2019, 12, 10903–10916. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. Epigenetic therapies in MDS and AML. Adv. Exp. Med. Biol. 2013, 754, 253–283. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Yang, X.; Lay, F.; Han, H.; Jones, P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol. Sci. 2010, 31, 536–546. [Google Scholar] [CrossRef]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef]
- Li, N.; Yang, L.; Qi, X.K.; Lin, Y.X.; Xie, X.; He, G.P.; Feng, Q.S.; Liu, L.R.; Xie, X.; Zeng, Y.X.; et al. BET bromodomain inhibitor JQ1 preferentially suppresses EBV-positive nasopharyngeal carcinoma cells partially through repressing c-Myc. Cell Death Dis. 2018, 9, 761. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Coletta, C.; Asimakopoulou, A.; Szczesny, B.; Chao, C.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide 2014, 41, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Musch, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Marampon, F.; Popov, V.M.; Pestell, R.G.; Zani, B.M.; Tombolini, V. Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol. Cancer 2010, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Vidal, L.; Griffin, M.; Lesley, M.; de Bono, J.; Coulthard, S.; Sludden, J.; Siu, L.L.; Chen, E.X.; Oza, A.M.; et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 3177–3183. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.; Sterner, D.E.; Berger, S.L. Histone modifications: Now summoning sumoylation. Proc. Natl. Acad. Sci. USA 2003, 100, 13118–13120. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005, 6, 227. [Google Scholar] [CrossRef] [PubMed]
- Poulin, M.B.; Schneck, J.L.; Matico, R.E.; Hou, W.; McDevitt, P.J.; Holbert, M.; Schramm, V.L. Nucleosome Binding Alters the Substrate Bonding Environment of Histone H3 Lysine 36 Methyltransferase NSD2. J. Am. Chem. Soc. 2016, 138, 6699–6702. [Google Scholar] [CrossRef] [PubMed]
- Del Rizzo, P.A.; Trievel, R.C. Substrate and product specificities of SET domain methyltransferases. Epigenetics 2011, 6, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.J.; Arnaudo, A.M.; Garcia, B.A. Large-scale global identification of protein lysine methylation in vivo. Epigenetics 2013, 8, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.N.; Gasteiger, E.; Wilkins, M.R. Identification of arginine- and lysine-methylation in the proteome of Saccharomyces cerevisiae and its functional implications. BMC Genomics 2010, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Milite, C.; Feoli, A.; Horton, J.R.; Rescigno, D.; Cipriano, A.; Pisapia, V.; Viviano, M.; Pepe, G.; Amendola, G.; Novellino, E.; et al. Discovery of a Novel Chemotype of Histone Lysine Methyltransferase EHMT1/2 (GLP/G9a) Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Co-crystal Structure. J. Med. Chem. 2019, 62, 2666–2689. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Di Croce, L. Emerging roles for Polycomb proteins in cancer. Curr. Opin. Genet. Dev. 2016, 36, 50–58. [Google Scholar] [CrossRef]
- Julia, E.; Salles, G. EZH2 inhibition by tazemetostat: Mechanisms of action, safety and efficacy in relapsed/refractory follicular lymphoma. Future Oncol. 2021, 17, 2127–2140. [Google Scholar] [CrossRef]
- Kim, S.K.; Jung, I.; Lee, H.; Kang, K.; Kim, M.; Jeong, K.; Kwon, C.S.; Han, Y.M.; Kim, Y.S.; Kim, D.; et al. Human histone H3K79 methyltransferase DOT1L protein [corrected] binds actively transcribing RNA polymerase II to regulate gene expression. J. Biol. Chem. 2012, 287, 39698–39709. [Google Scholar] [CrossRef]
- Campagna, R.; Vignini, A. NAD+ Homeostasis and NAD+-Consuming Enzymes: Implications for Vascular Health. Antioxidants 2023, 12, 376. [Google Scholar] [CrossRef] [PubMed]
- Togni, L.; Mascitti, M.; Sartini, D.; Campagna, R.; Pozzi, V.; Salvolini, E.; Offidani, A.; Santarelli, A.; Emanuelli, M. Nicotinamide N-Methyltransferase in Head and Neck Tumors: A Comprehensive Review. Biomolecules 2021, 11, 1594. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Wellen, K.E. Metabolism and epigenetics: A link cancer cells exploit. Curr. Opin. Biotechnol. 2015, 34, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Campagna, R.; Pozzi, V.; Spinelli, G.; Sartini, D.; Milanese, G.; Galosi, A.B.; Emanuelli, M. The Utility of Nicotinamide N-Methyltransferase as a Potential Biomarker to Predict the Oncological Outcomes for Urological Cancers: An Update. Biomolecules 2021, 11, 1214. [Google Scholar] [CrossRef] [PubMed]
- Van Haren, M.J.; Gao, Y.; Buijs, N.; Campagna, R.; Sartini, D.; Emanuelli, M.; Mateuszuk, L.; Kij, A.; Chlopicki, S.; Escudé Martinez de Castilla, P.; et al. Esterase-Sensitive Prodrugs of a Potent Bisubstrate Inhibitor of Nicotinamide N-Methyltransferase (NNMT) Display Cellular Activity. Biomolecules 2021, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Rajagopal, S.; Kadnur, S.V.; Hallur, M.S.; Rani, S.; Kristam, R.; Swaminathan, S.; Zope, B.R.; Gondrala, P.K.; Swamy, I.; et al. Novel tricyclic small molecule inhibitors of Nicotinamide N-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 2022, 12, 15440. [Google Scholar] [CrossRef] [PubMed]
- Zaware, N.; Zhou, M.M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Pérez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef]
- Zeng, L.; Yap, K.L.; Ivanov, A.V.; Wang, X.; Mujtaba, S.; Plotnikova, O.; Rauscher, F.J., 3rd; Zhou, M.M. Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat. Struct. Mol. Biol. 2008, 15, 626–633. [Google Scholar] [CrossRef]
- Jung, M.; Philpott, M.; Müller, S.; Schulze, J.; Badock, V.; Eberspächer, U.; Moosmayer, D.; Bader, B.; Schmees, N.; Fernández-Montalván, A.; et al. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014, 289, 9304–9319. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Umehara, T.; Inoue, M.; Saito, K.; Kigawa, T.; Jang, M.K.; Ozato, K.; Yokoyama, S.; Padmanabhan, B.; Güntert, P. Solution structure of the extraterminal domain of the bromodomain-containing protein BRD4. Protein Sci. 2008, 17, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yao, S.; Tang, J.; Liu, S.; Chen, J.; Deng, D.; Zeng, C. Integrative analysis of super enhancer SNPs for type 2 diabetes. PLoS ONE 2018, 13, e0192105. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Jaganathan, A.; Sun, Y.; Ma, N.; Li, N.; Han, X.; Sun, X.; Yi, H.; Fu, S.; et al. BRD4-PRC2 represses transcription of T-helper 2-specific negative regulators during T-cell differentiation. EMBO J. 2023, 42, e111473. [Google Scholar] [CrossRef] [PubMed]
- French, C.A.; Miyoshi, I.; Aster, J.C.; Kubonishi, I.; Kroll, T.G.; Dal Cin, P.; Vargas, S.O.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19). Am. J. Pathol. 2001, 159, 1987–1992. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef]
- LeRoy, G.; Rickards, B.; Flint, S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef]
- Hsu, P.L.; Chien, C.W.; Tang, Y.A.; Lin, B.W.; Lin, S.C.; Lin, Y.S.; Chen, S.Y.; Sun, H.S.; Tsai, S.J. Targeting BRD3 eradicates nuclear TYRO3-induced colorectal cancer metastasis. Sci. Adv. 2023, 9, eade3422. [Google Scholar] [CrossRef] [PubMed]
- French, C.A. NUT midline carcinoma. Cancer Genet. Cytogenet. 2010, 203, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak-Celińska, A.; Warych, A.; Szoszkiewicz, M. Novel Approaches to Epigenetic Therapies: From Drug Combinations to Epigenetic Editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET family in immunity and disease. Signal Transduct. Target. Ther. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenet. 2021, 13, 166. [Google Scholar] [CrossRef]
- To, K.K.W.; Xing, E.; Larue, R.C.; Li, P.-K. BET Bromodomain Inhibitors: Novel Design Strategies and Therapeutic Applications. Molecules 2023, 28, 3043. [Google Scholar] [CrossRef]
- Hnilicová, J.; Hozeifi, S.; Stejskalová, E.; Dušková, E.; Poser, I.; Humpolíčková, J.; Hof, M.; Staněk, D. The C-terminal domain of Brd2 is important for chromatin interaction and regulation of transcription and alternative splicing. Mol. Biol. Cell 2013, 24, 3557–3568. [Google Scholar] [CrossRef]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Da Costa, D.; Thanasopoulou, A.; Filippakopoulos, P.; Fish, P.V.; Philpott, M.; Fedorov, O.; Brennan, P.; Bunnage, M.E.; Owen, D.R.; et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013, 73, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, M.; Guo, C.; Wu, Y.; Zhao, L.; Shi, Q.; Song, J.; Song, B. Therapeutic Targeting of Cancer: Epigenetic Homeostasis. Front. Oncol. 2021, 11, 747022. [Google Scholar] [CrossRef] [PubMed]
- Borah, N.A.; Sradhanjali, S.; Barik, M.R.; Jha, A.; Tripathy, D.; Kaliki, S.; Rath, S.; Raghav, S.K.; Patnaik, S.; Mittal, R.; et al. Aurora Kinase B Expression, Its Regulation and Therapeutic Targeting in Human Retinoblastoma. Investig. Ophthalmol. Vis. Sci. 2021, 62, 16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Bilbao, M.; Sookram, J.; Linden, K.M.; Morgan, A.B.; Ostrovsky, O. Epigenetic drugs induce the potency of classic chemotherapy, suppress post-treatment re-growth of breast cancer, but preserve the wound healing ability of stem cells. Cancer Biol. Ther. 2022, 23, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y. Predictive biomarkers and potential drug combinations of epi-drugs in cancer therapy. Clin. Epigenet. 2021, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Liou, J.P. Recent developments in epigenetic cancer therapeutics: Clinical advancement and emerging trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Brueckner, B.; Musch, T.; Stopper, H.; Lyko, F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006, 66, 2794–2800. [Google Scholar] [CrossRef]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef]
- Adhikari, S.; Bhattacharya, A.; Adhikary, S.; Singh, V.; Gadad, S.S.; Roy, S.; Das, C. The paradigm of drug resistance in cancer: An epigenetic perspective. Biosci. Rep. 2022, 42, BSR20211812. [Google Scholar] [CrossRef]
- Musolino, E.; Pagiatakis, C.; Serio, S.; Borgese, M.; Gamberoni, F.; Gornati, R.; Bernardini, G.; Papait, R. The Yin and Yang of epigenetics in the field of nanoparticles. Nanoscale Adv. 2022, 4, 979–994. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Lutz, B.M.; Tao, Y.X. The CRISPR/Cas9 system for gene editing and its potential application in pain research. Transl. Perioper. Pain. Med. 2016, 1, 22–33. [Google Scholar] [PubMed]
- Rodríguez-Rodríguez, D.R.; Ramírez-Solís, R.; Garza-Elizondo, M.A.; Garza-Rodríguez, M.L.; Barrera-Saldaña, H.A. Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases (Review). Int. J. Mol. Med. 2019, 43, 1559–1574. [Google Scholar] [CrossRef] [PubMed]
- Enríquez, P. CRISPR-Mediated Epigenome Editing. Yale J. Biol. Med. 2016, 89, 471–486. [Google Scholar] [PubMed]
- Zhou, W.; Jia, Y.; Liu, Y.; Chen, Y.; Zhao, P. Tumor Microenvironment-Based Stimuli-Responsive Nanoparticles for Controlled Release of Drugs in Cancer Therapy. Pharmaceutics 2022, 14, 2346. [Google Scholar] [CrossRef] [PubMed]
- Zolnik, B.S.; González-Fernández, A.; Sadrieh, N.; Dobrovolskaia, M.A. Nanoparticles and the immune system. Endocrinology 2010, 151, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Hartwig, F.P.; Davey Smith, G. From stem cells to the law courts: DNA methylation, the forensic epigenome and the possibility of a biosocial archive. Int. J. Epidemiol. 2015, 44, 1083–1093. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Seco-Cervera, M.; Tollefsbol, T.O.; Romá-Mateo, C.; Peiró-Chova, L.; Lapunzina, P.; Pallardó, F.V. Epigenetic biomarkers: Current strategies and future challenges for their use in the clinical laboratory. Crit. Rev. Clin. Lab. Sci. 2017, 54, 529–550. [Google Scholar] [CrossRef]
- Lelièvre, S.A. Can the epigenome contribute to risk stratification for cancer onset? NAR Cancer 2021, 3, zcab043. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Drug | Formula | Target | Status | Uses/Concerns/Other |
|---|---|---|---|---|
| Azacytidine | C8H12N4O5 | DNMT inhibitor | FDA-approved | Myelodysplastic syndromes |
| Decitabine | C8H12N4O4 | DNMT inhibitor | FDA-approved | Myelodysplastic syndromes |
| Pseudoisocytidine | C9H13N3O5 | DNMT inhibitor | Discontinued | Hepatotoxicity concerns |
| DHAC | C8H14N4O5 | DNMT inhibitor | Discontinued | Cardiotoxicity concerns |
| Guadecitabine | C18H23N9O10P | DNMT inhibitor | Discontinued | Lack of Phase 3 efficacy |
| Vorinostat | C14H20N2O3 | HDAC inhibitor | FDA-approved | Cutaneous T-cell lymphoma |
| Romidepsin | C24H36N4O6S2 | HDAC inhibitor | FDA-approved | Cutaneous T-cell lymphoma |
| Butyric acid | C4H8O2 | HDAC inhibitor | Awaiting | Exploring inhibition in models |
| Belinostat | C15H14N2O4S | HDAC inhibitor | FDA-approved | Peripheral T-cell lymphoma |
| Panobinostat | C21H23N3O2 | HDAC inhibitor | FDA approval withdrawn | Peripheral T-cell lymphoma |
| Entinostat | C21H20N4O3 | HDAC inhibitor | Paused | Lack of Phase 3 efficacy |
| Tucidinostat | C22H19FN4O2 | HDAC inhibitor | CFDA-approved | Peripheral T-cell lymphoma |
| Pinometostat | C30H42N8O3 | HMT inhibitor | Discontinued | Lack of efficacy |
| Tazemetostat | C34H44N4O4 | HMT inhibitor | FDA-approved | Relapsed/refractory follicular lymphoma |
| Pemrametostat | C24H32N6O3 | HMT inhibitor | Paused | Clinical trial paused |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patnaik, E.; Madu, C.; Lu, Y. Epigenetic Modulators as Therapeutic Agents in Cancer. Int. J. Mol. Sci. 2023, 24, 14964. https://doi.org/10.3390/ijms241914964
Patnaik E, Madu C, Lu Y. Epigenetic Modulators as Therapeutic Agents in Cancer. International Journal of Molecular Sciences. 2023; 24(19):14964. https://doi.org/10.3390/ijms241914964
Chicago/Turabian StylePatnaik, Eshaan, Chikezie Madu, and Yi Lu. 2023. "Epigenetic Modulators as Therapeutic Agents in Cancer" International Journal of Molecular Sciences 24, no. 19: 14964. https://doi.org/10.3390/ijms241914964
APA StylePatnaik, E., Madu, C., & Lu, Y. (2023). Epigenetic Modulators as Therapeutic Agents in Cancer. International Journal of Molecular Sciences, 24(19), 14964. https://doi.org/10.3390/ijms241914964