
The Mechanism of Interleukin 33-Induced Stimulation of Interleukin 6 in MLO-Y4 Cells
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
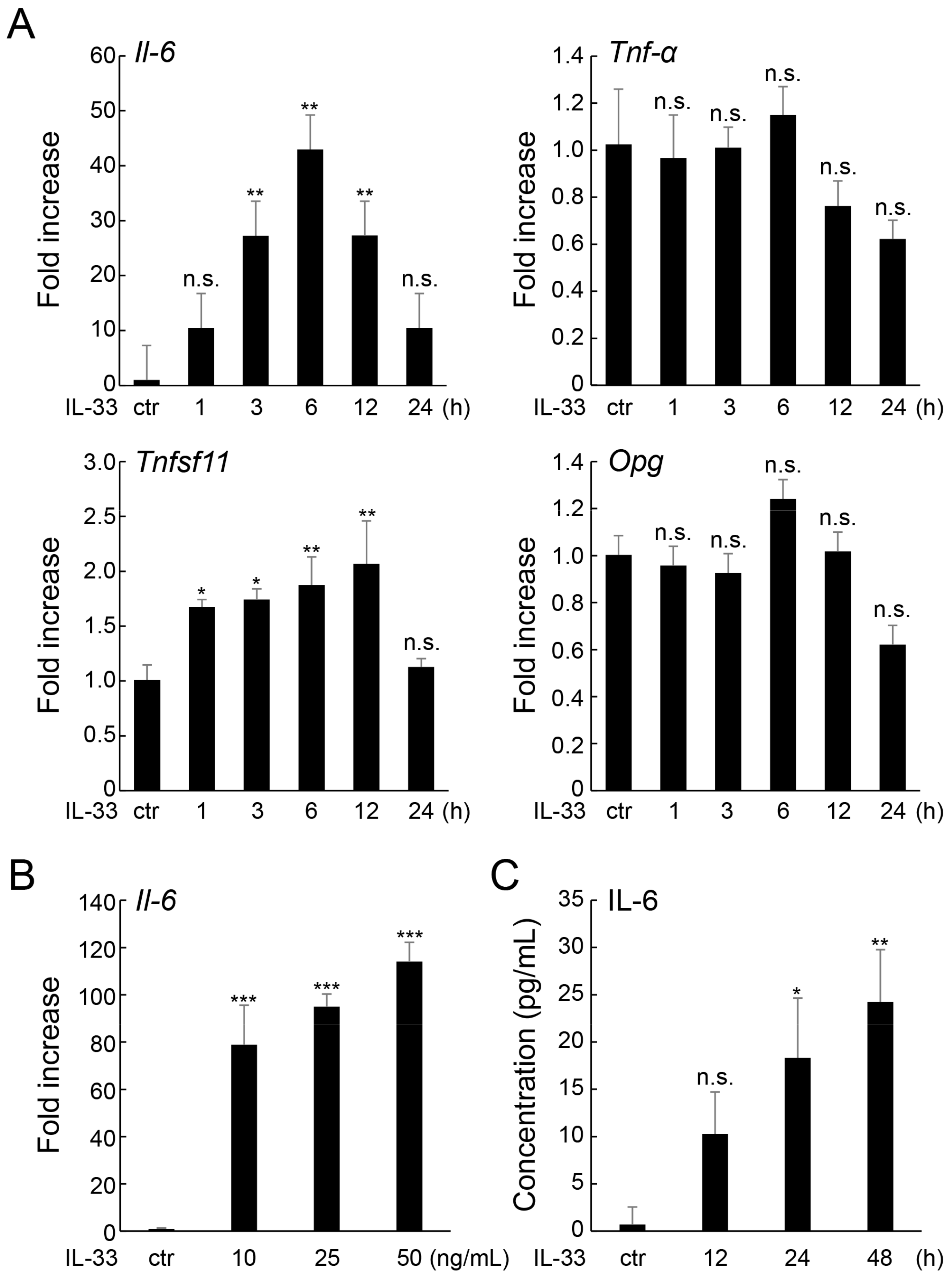
2.1. IL-33 Increases IL-6 Expression in MLO-Y4 Cells
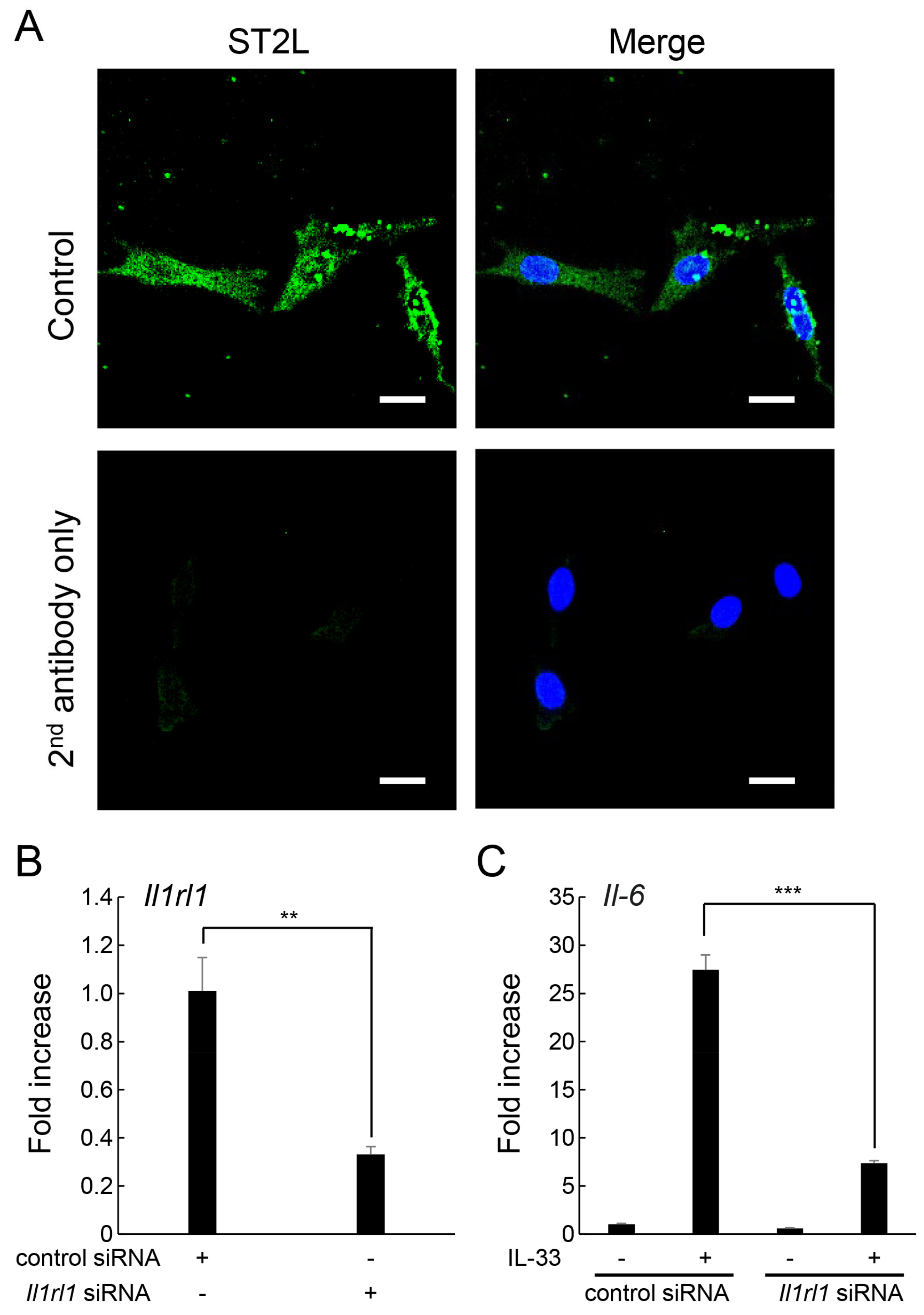
2.2. ST2L on the Cell Surface of MLO-Y4 Cells Is Involved in the Induction of IL-6 Expression by IL-33
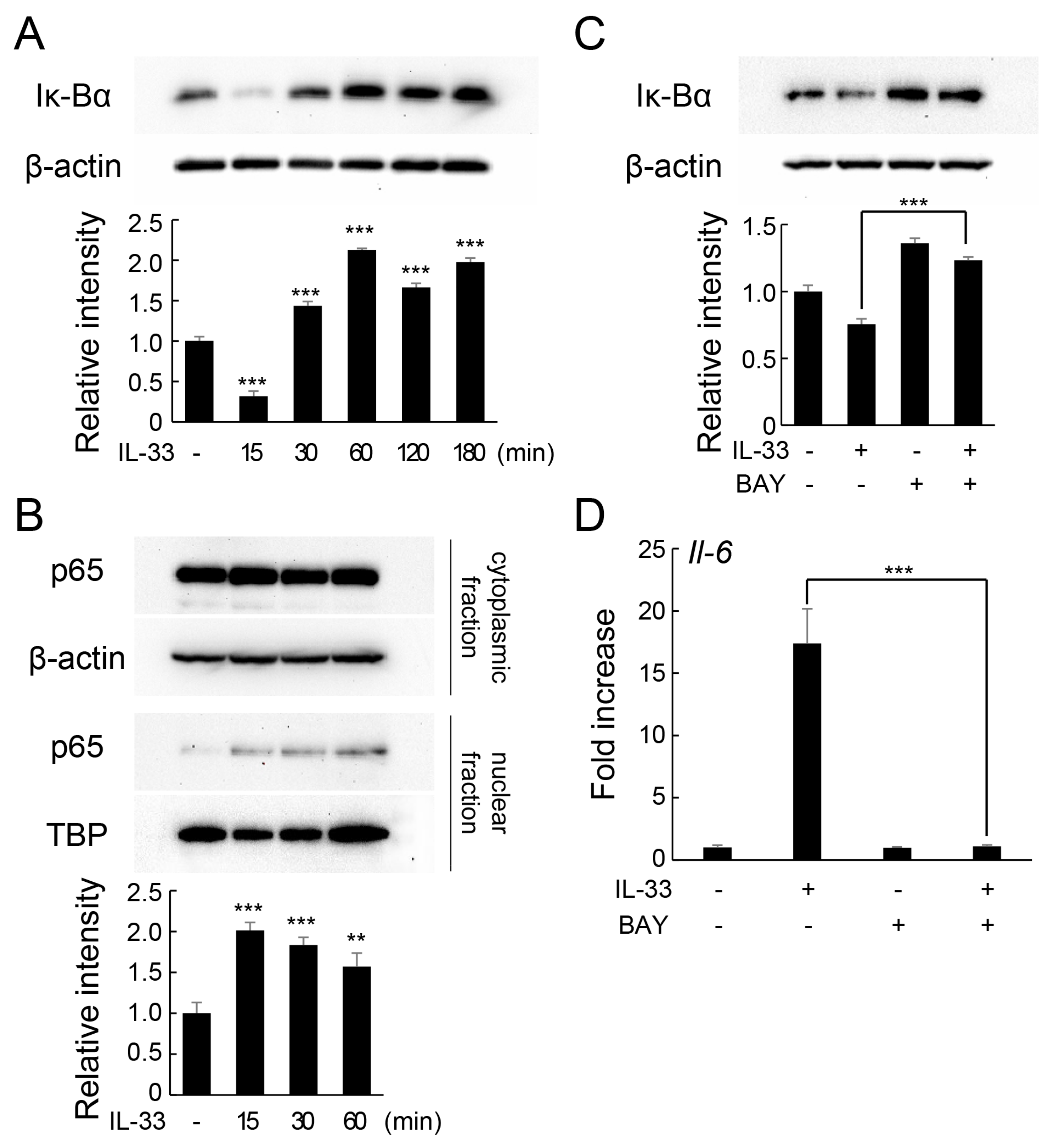
2.3. IL-33 Induces IL-6 Production via the NF-κB Signaling Pathway
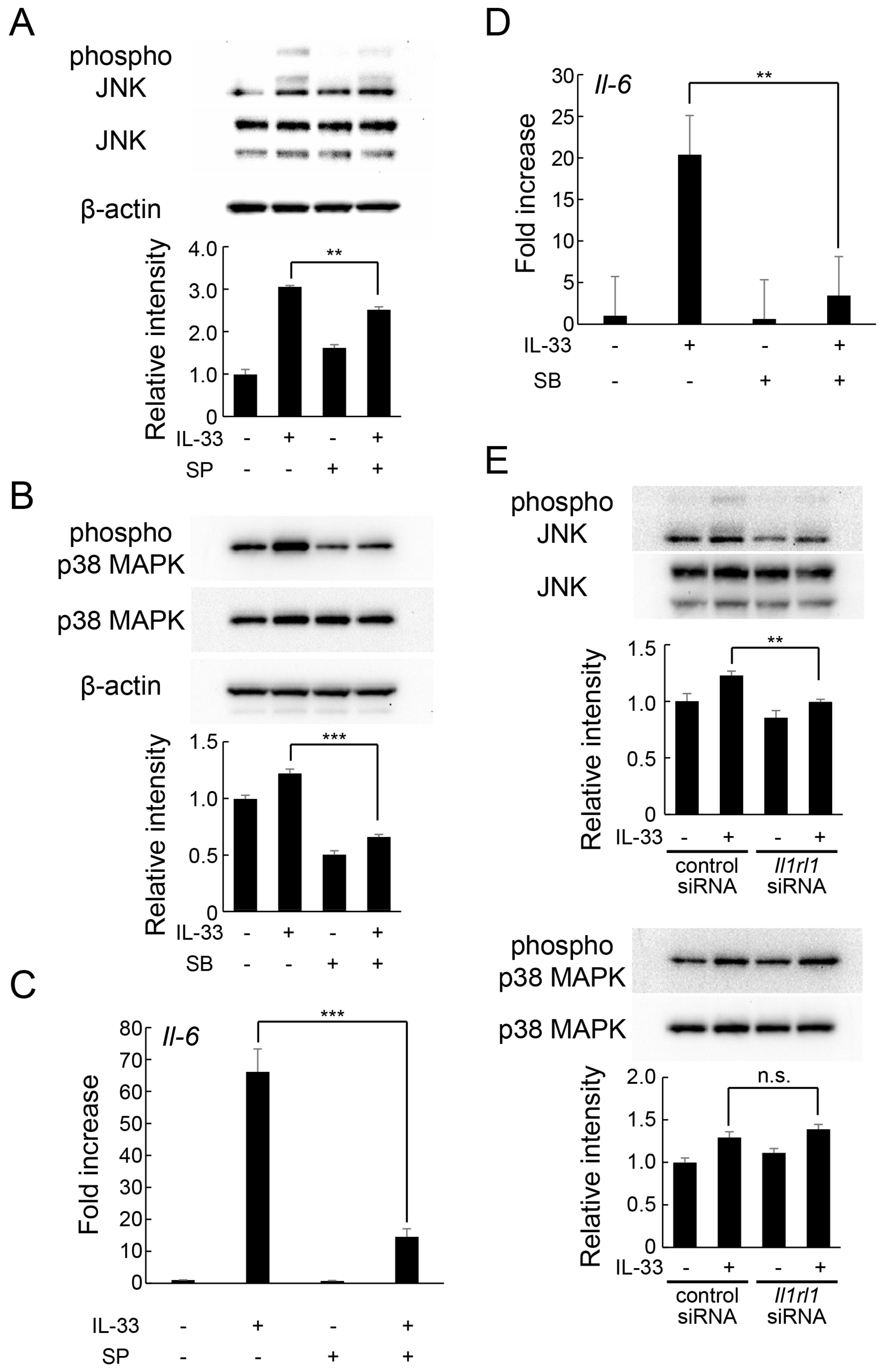
2.4. IL-33 Induces IL-6 Production via JNK and p38 MAPK-Mediated Signaling Pathways
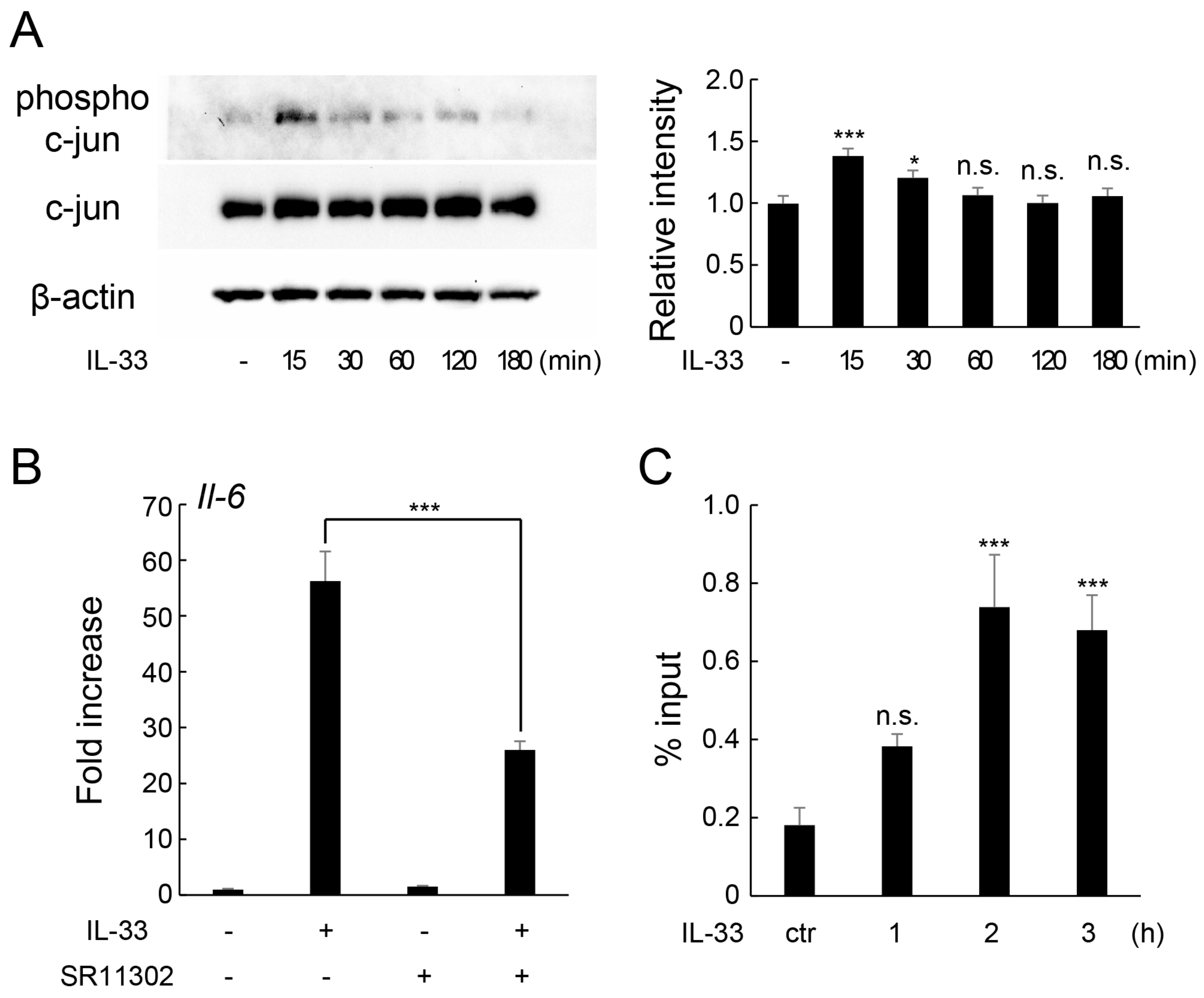
2.5. IL-33 Induces IL-6 Production via c-Jun-Mediated AP-1 Activation
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. RNA Extraction and Real-Time Quantitative PCR (Real-Time RT-qPCR)
4.4. Protein Extraction and Western Blotting
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Immunocytochemistry
4.7. Silencing of ST2L by Specific Small Interfering RNAs (siRNA)
4.8. ChIP Assay
4.9. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.; Zhan, Q.; Bao, M.; Yi, J.; Li, Y. Biomechanical and biological responses of periodontium in orthodontic tooth movement: Up-date in a new decade. Int. J. Oral Sci. 2021, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jacox, L.A.; Little, S.H.; Ko, C.C. Orthodontic tooth movement: The biology and clinical implications. Kaohsiung J. Med. Sci. 2018, 34, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Choi, Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef] [PubMed]
- Tresguerres, F.G.F.; Torres, J.; López-Quiles, J.; Hernández, G.; Vega, J.A.; Tresguerres, I.F. The osteocyte: A multifunctional cell within the bone. Ann. Anat. 2020, 227, 151422. [Google Scholar] [CrossRef]
- Palumbo, C.; Ferretti, M. The Osteocyte: From “Prisoner” to “Orchestrator”. J. Funct. Morphol. Kinesiol. 2021, 6, 28. [Google Scholar] [CrossRef]
- Padisar, P.; Hashemi, R.; Naseh, M.; Nikfarjam, B.A.; Mohammadi, M. Assessment of tumor necrosis factor alpha (TNFα) and interleukin 6 level in gingival crevicular fluid during orthodontic tooth movement: A randomized split-mouth clinical trial. Electron. Physician 2018, 10, 7146–7154. [Google Scholar] [CrossRef]
- Hashizume, M.; Hayakawa, N.; Mihara, M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology 2008, 47, 1635–1640. [Google Scholar] [CrossRef]
- Palmqvist, P.; Persson, E.; Conaway, H.H.; Lerner, U.H. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J. Immunol. 2002, 169, 3353–3362. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell. Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef]
- Kaneshiro, S.; Ebina, K.; Shi, K.; Higuchi, C.; Hirao, M.; Okamoto, M.; Koizumi, K.; Morimoto, T.; Yoshikawa, H.; Hashimoto, J. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J. Bone Miner. Metab. 2014, 32, 378–392. [Google Scholar] [CrossRef]
- Malysheva, K.; de Rooij, K.; Lowik, C.W.; Baeten, D.L.; Rose-John, S.; Stoika, R.; Korchynskyi, O. Interleukin 6/Wnt interactions in rheumatoid arthritis: Interleukin 6 inhibits Wnt signaling in synovial fibroblasts and osteoblasts. Croat. Med. J. 2016, 57, 89–98. [Google Scholar] [CrossRef]
- Ohori, F.; Kitaura, H.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Marahleh, A.; Nara, Y.; Pramusita, A.; Mizoguchi, I. IL-33 Inhibits TNF-α-Induced Osteoclastogenesis and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 1130. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Lian, J.; Yue, Y.; Zhang, Y. IL-33/ST2 as a potential target for tumor immunotherapy. Eur. J. Immunol. 2021, 51, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.L.; Macari, S.; Madeira, M.F.; Rodrigues, L.F.; Colavite, P.M.; Garlet, G.P.; Soriani, F.M.; Teixeira, M.M.; Fukada, S.Y.; Silva, T.A. Osteoprotective Effects of IL-33/ST2 Link to Osteoclast Apoptosis. Am. J. Pathol. 2015, 185, 3338–3348. [Google Scholar] [CrossRef] [PubMed]
- Saleh, H.; Eeles, D.; Hodge, J.M.; Nicholson, G.C.; Gu, R.; Pompolo, S.; Gillespie, M.T.; Quinn, J.M. Interleukin-33, a target of parathyroid hormone and oncostatin m, increases osteoblastic matrix mineral deposition and inhibits osteoclast formation in vitro. Endocrinology 2011, 152, 1911–1922. [Google Scholar] [CrossRef]
- Kiyomiya, H.; Ariyoshi, W.; Okinaga, T.; Kaneuji, T.; Mitsugi, S.; Sakurai, T.; Habu, M.; Yoshioka, I.; Tominaga, K.; Nishihara, T. IL-33 inhibits RANKL-induced osteoclast formation through the regulation of Blimp-1 and IRF-8 expression. Biochem. Biophys. Res. Commun. 2015, 460, 320–326. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, Y.; Jiang, Y.; Qin, T.; Chen, J.; Chu, X.; Yi, Q.; Gao, S.; Wang, S. Dectin-1 signaling inhibits osteoclastogenesis via IL-33-induced inhibition of NFATc1. Oncotarget 2017, 8, 53366–53374. [Google Scholar] [CrossRef]
- Kato, Y.; Windle, J.J.; Koop, B.A.; Mundy, G.R.; Bonewald, L.F. Establishment of an osteocyte-like cell line, MLO-Y4. J. Bone Miner. Res. 1997, 12, 2014–2023. [Google Scholar] [CrossRef]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef]
- Zhang, M.; Hoyle, R.G.; Ma, Z.; Sun, B.; Cai, W.; Cai, H.; Xie, N.; Zhang, Y.; Hou, J.; Liu, X.; et al. FOSL1 promotes metastasis of head and neck squamous cell carcinoma through super-enhancer-driven transcription program. Mol. Ther. 2021, 29, 2583–2600. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A critical review of its biology and the mechanisms involved in its release as a potent extracellular cytokine. Cytokine 2022, 156, 155891. [Google Scholar] [CrossRef] [PubMed]
- D’Ovidi, C. The response of immune sentinels causing inflammation in glioma and glioblastoma. Eur. J. Neurodegener. Dis. 2023, 12, 46–50. [Google Scholar]
- Besnard, A.G.; Togbe, D.; Guillou, N.; Erard, F.; Quesniaux, V.; Ryffel, B. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur. J. Immunol. 2011, 41, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Tsilioni, I. Inflammatory response mediated by cytokines. Eur. J. Neurodegener. Dis. 2023, 12, 5–10. [Google Scholar]
- Mine, Y.; Makihira, S.; Yamaguchi, Y.; Tanaka, H.; Nikawa, H. Involvement of ERK and p38 MAPK pathways on Interleukin-33-induced RANKL expression in osteoblastic cells. Cell Biol. Int. 2014, 38, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Feng, J.; Wen, J.; Bai, D.; Xu, H. Effect of interleukin-33 on cementoblast-mediated cementum repair during orthodontic tooth movement. Arch. Oral Biol. 2020, 112, 104663. [Google Scholar] [CrossRef]
- Liu, J.; Liu, L.; Su, Y.; Wang, Y.; Zhu, Y.; Sun, X.; Guo, Y.; Shan, J. IL-33 Participates in the Development of Esophageal Adenocarcinoma. Pathol. Oncol. Res. 2022, 28, 1610474. [Google Scholar] [CrossRef]
- Saidi, S.; Magne, D. Interleukin-33: A novel player in osteonecrosis of the femoral head? Jt. Bone Spine 2011, 78, 550–554. [Google Scholar] [CrossRef]
- Funakoshi-Tago, M.; Miyagawa, Y.; Ueda, F.; Mashino, T.; Moriwaki, Y.; Tago, K.; Kasahara, T.; Tamura, H. A bis-malonic acid fullerene derivative significantly suppressed IL-33-induced IL-6 expression by inhibiting NF-κB activation. Int. Immunopharmacol. 2016, 40, 254–264. [Google Scholar] [CrossRef]
- Lu, J.; Kang, J.; Zhang, C.; Zhang, X. The role of IL-33/ST2L signals in the immune cells. Immunol. Lett. 2015, 164, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.; Ran, X.; Wang, D.; Zheng, X.; Zhang, M.; Yu, B.; Sun, Y.; Wu, J. Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-κB/IL-6 signaling pathway. Cell. Mol. Life Sci. 2022, 79, 311. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Ramesh, R. Mitogen-activated protein kinases and their role in radiation response. Genes Cancer 2013, 4, 401–408. [Google Scholar] [CrossRef]
- Cui, H.; Du, X.; Liu, C.; Chen, S.; Cui, H.; Liu, H.; Wang, J.; Zheng, Z. Visfatin promotes intervertebral disc degeneration by inducing IL-6 expression through the ERK/JNK/p38 signalling pathways. Adipocyte 2021, 10, 201–215. [Google Scholar] [CrossRef]
- Huang, N.; Cui, X.; Li, W.; Zhang, C.; Liu, L.; Li, J. IL-33/ST2 promotes the malignant progression of gastric cancer via the MAPK pathway. Mol. Med. Rep. 2021, 23, 361. [Google Scholar] [CrossRef]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta. Rev. Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- Wang, J.; He, F.; Chen, L.; Li, Q.; Jin, S.; Zheng, H.; Lin, J.; Zhang, H.; Ma, S.; Mei, J.; et al. Resveratrol inhibits pulmonary fibrosis by regulating miR-21 through MAPK/AP-1 pathways. Biomed. Pharmacother. 2018, 105, 37–44. [Google Scholar] [CrossRef]
- Papavassiliou, A.G.; Musti, A.M. The Multifaceted Output of c-Jun Biological Activity: Focus at the Junction of CD8 T Cell Activation and Exhaustion. Cells 2020, 9, 2470. [Google Scholar] [CrossRef]
- Yeagley, D.; Lang, C.H. Endotoxin-Induced IL-6 Promoter Activation in Skeletal Muscle Requires an NF-κB Site. Int. J. Interferon. Cytokine Mediat. Res. 2010, 2010, 9–21. [Google Scholar]
- Yan, C.; Deng, C.; Liu, X.; Chen, Y.; Ye, J.; Cai, R.; Shen, Y.; Tang, H. TNF-α induction of IL-6 in alveolar type II epithelial cells: Contributions of JNK/c-Jun/AP-1 element, C/EBPδ/C/EBP binding site and IKK/NF-κB p65/κB site. Mol. Immunol. 2018, 101, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Umebashi, K.; Tokito, A.; Yamamoto, M.; Jougasaki, M. Interleukin-33 induces interleukin-8 expression via JNK/c-Jun/AP-1 pathway in human umbilical vein endothelial cells. PLoS ONE 2018, 13, e0191659. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Yoshida, H.; Tanaka, S. Role of interleukin-6 in bone destruction and bone repair in rheumatoid arthritis. Autoimmun. Rev. 2021, 20, 102884. [Google Scholar] [CrossRef]
- Dong, X.; Feng, J.; Li, B.; Bai, D.; Xu, H. Inhibition of osteoclastogenesis by interleukin-33 administration in the periodontal ligament under mechanical loading. J. Periodontal. Res. 2022, 57, 1003–1013. [Google Scholar] [CrossRef]
- Kanzaki, H.; Chiba, M.; Shimizu, Y.; Mitani, H. Periodontal ligament cells under mechanical stress induce osteoclastogenesis by receptor activator of nuclear factor kappaB ligand up-regulation via prostaglandin E2 synthesis. J. Bone Miner. Res. 2002, 17, 210–220. [Google Scholar] [CrossRef]
- Chen, L.; Mo, S.; Hua, Y. Compressive force-induced autophagy in periodontal ligament cells downregulates osteoclastogenesis during tooth movement. J. Periodontol. 2019, 90, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Nagai-Yoshioka, Y.; Yamasaki, R.; Kawamoto, T.; Nishihara, T.; Ariyoshi, W. Mechanisms involved in suppression of osteoclast supportive activity by transforming growth factor-β1 via the ubiquitin-proteasome system. PLoS ONE 2022, 17, e0262612. [Google Scholar] [CrossRef]
- Ariyoshi, W.; Okinaga, T.; Chaweewannakorn, W.; Akifusa, S.; Nisihara, T. Mechanisms involved in enhancement of matrix metalloproteinase-9 expression in macrophages by interleukin-33. J. Cell. Physiol. 2017, 232, 3481–3495. [Google Scholar] [CrossRef]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′ - 3′) | |
|---|---|---|
| Gapdh | forward | 5′- GAC GGC CGC ATC TTC TTG A -3′ |
| reverse | 5′- CAC ACA CCG ACC TTC ACC ATT TT -3′ | |
| Il-6 | forward | 5′- GAG GAT ACC ACT CCC AAC AGA CC -3′ |
| reverse | 5′- ATT GCT TGG GAT CCA CAC TCT CCA ACC TTT GAC -3′ | |
| Tnf-α | forward | 5′- TCA TGC ACC ACC ATC AAG GA -3′ |
| reverse | 5′- GAC ATT CGA GGC TCC AGT GAA -3′ | |
| Il-1β | forward | 5′- AAG GGC TGC TTC CAA ACC TTT GAC -3′ |
| reverse | 5′- TGG CGA GCT CAG GTA CTT CTG -3′ | |
| Tnfsf11 | forward | 5′- CTG ATG AAA GGA GGG AGC ACG -3′ |
| reverse | 5′- GGA AGG GTT GGA CAC CTG AAT G -3′ | |
| Opg | forward | 5′- GAG AGA AGC CAC GCA AAA GTG -3′ |
| reverse | 5′- TCT TGG TAG GAA CAG CAA ACC TG -3′ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noguchi, S.; Yamasaki, R.; Nagai-Yoshioka, Y.; Sato, T.; Kuroishi, K.; Gunjigake, K.; Ariyoshi, W.; Kawamoto, T. The Mechanism of Interleukin 33-Induced Stimulation of Interleukin 6 in MLO-Y4 Cells. Int. J. Mol. Sci. 2023, 24, 14842. https://doi.org/10.3390/ijms241914842
Noguchi S, Yamasaki R, Nagai-Yoshioka Y, Sato T, Kuroishi K, Gunjigake K, Ariyoshi W, Kawamoto T. The Mechanism of Interleukin 33-Induced Stimulation of Interleukin 6 in MLO-Y4 Cells. International Journal of Molecular Sciences. 2023; 24(19):14842. https://doi.org/10.3390/ijms241914842
Chicago/Turabian StyleNoguchi, Sae, Ryota Yamasaki, Yoshie Nagai-Yoshioka, Tsuyoshi Sato, Kayoko Kuroishi, Kaori Gunjigake, Wataru Ariyoshi, and Tatsuo Kawamoto. 2023. "The Mechanism of Interleukin 33-Induced Stimulation of Interleukin 6 in MLO-Y4 Cells" International Journal of Molecular Sciences 24, no. 19: 14842. https://doi.org/10.3390/ijms241914842
APA StyleNoguchi, S., Yamasaki, R., Nagai-Yoshioka, Y., Sato, T., Kuroishi, K., Gunjigake, K., Ariyoshi, W., & Kawamoto, T. (2023). The Mechanism of Interleukin 33-Induced Stimulation of Interleukin 6 in MLO-Y4 Cells. International Journal of Molecular Sciences, 24(19), 14842. https://doi.org/10.3390/ijms241914842