
Exome Sequencing and Optical Genome Mapping in Molecularly Unsolved Cases of Duchenne Muscular Dystrophy: Identification of a Causative X-Chromosomal Inversion Disrupting the DMD Gene

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Overview of DMD Patients in Our Center
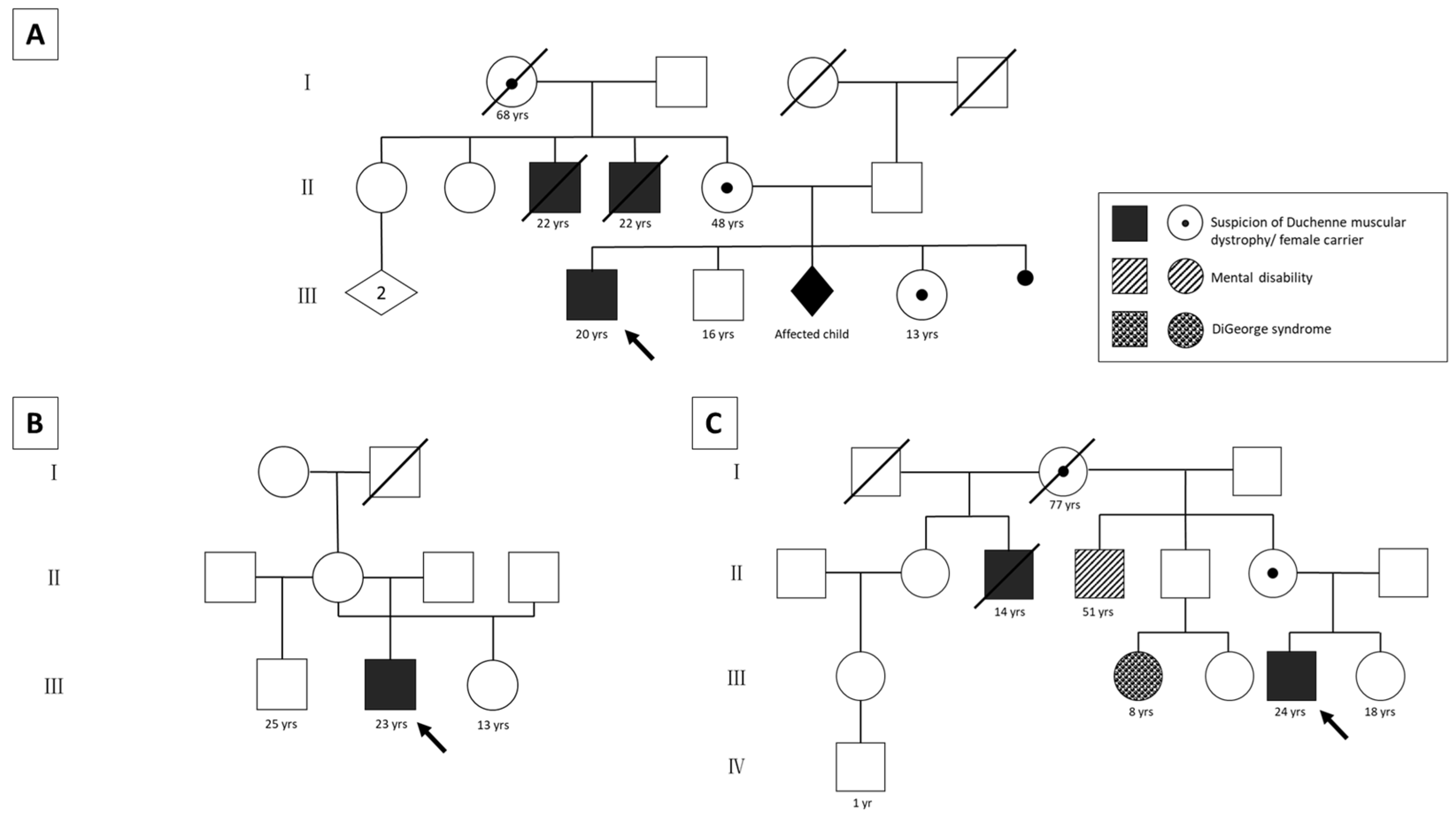
2.2. Clinical Characterization of the DMD Patients without Molecular Diagnosis
2.2.1. Patient 1
2.2.2. Patient 2
2.2.3. Patient 3
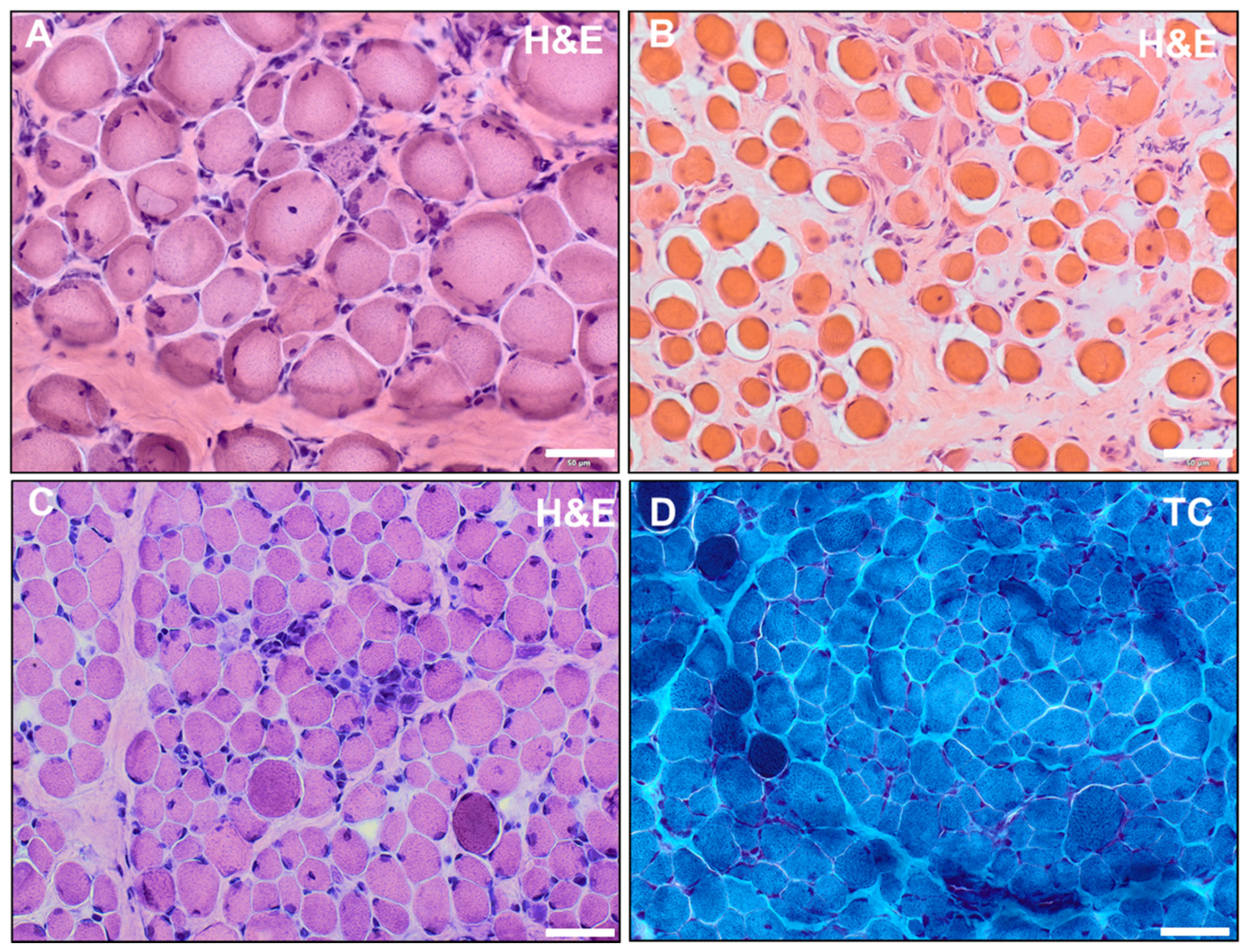
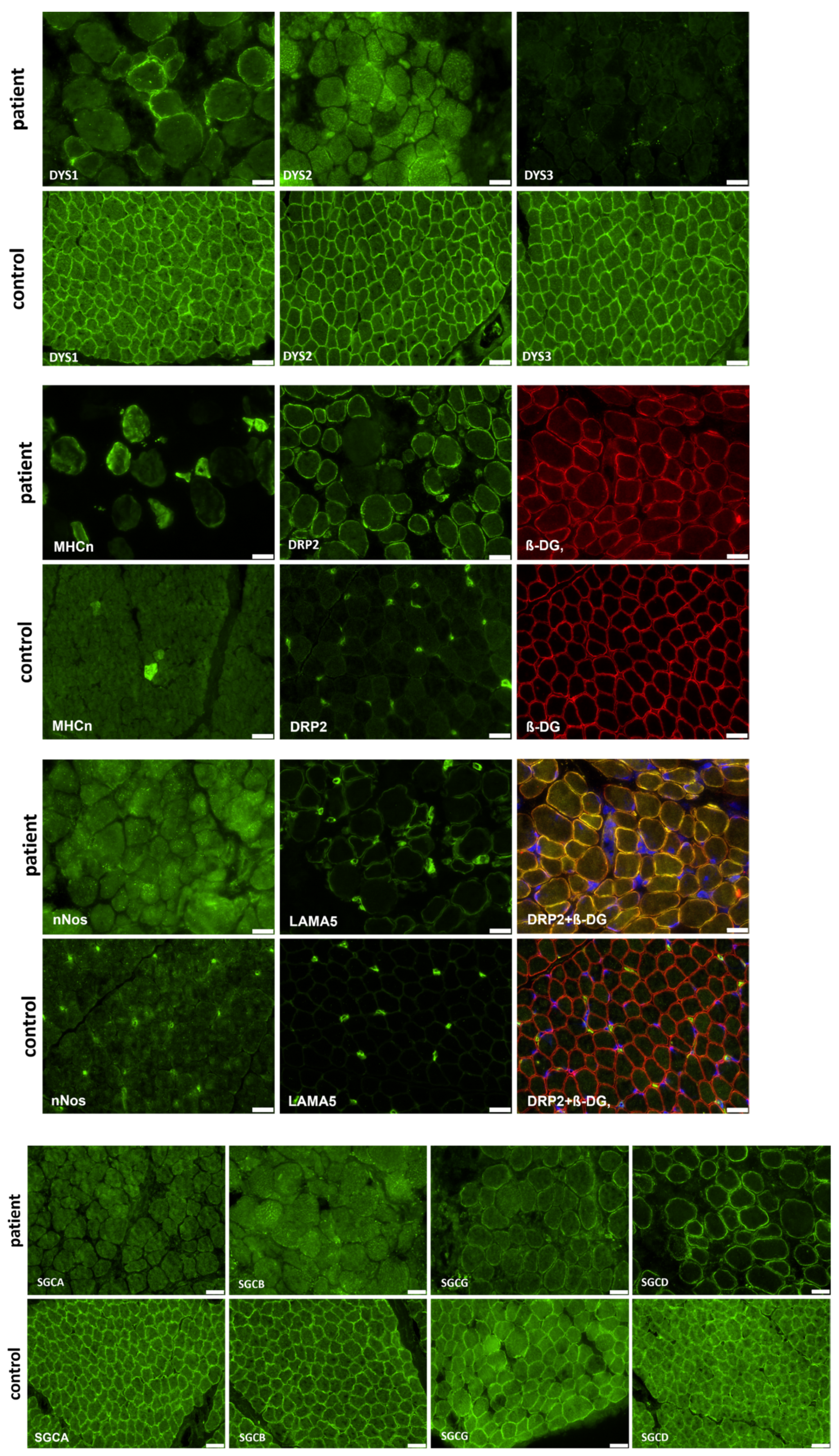
2.3. Muscle Biopsy
2.4. Results of Whole-Exome Sequencing
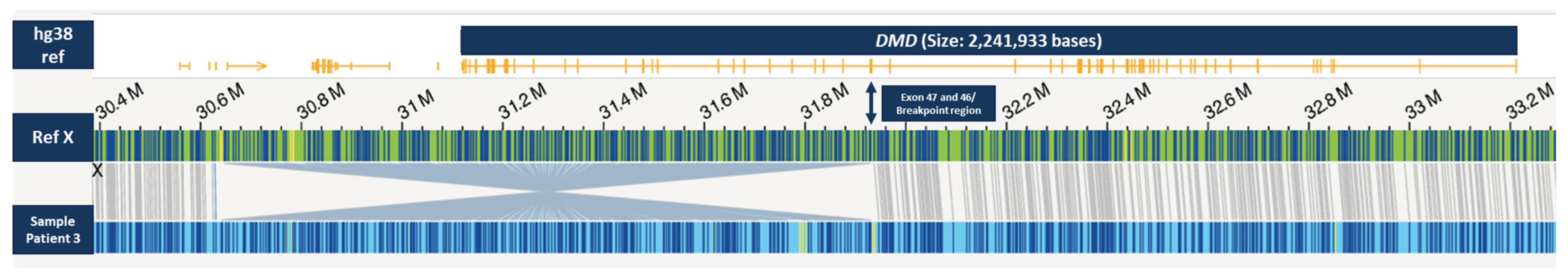
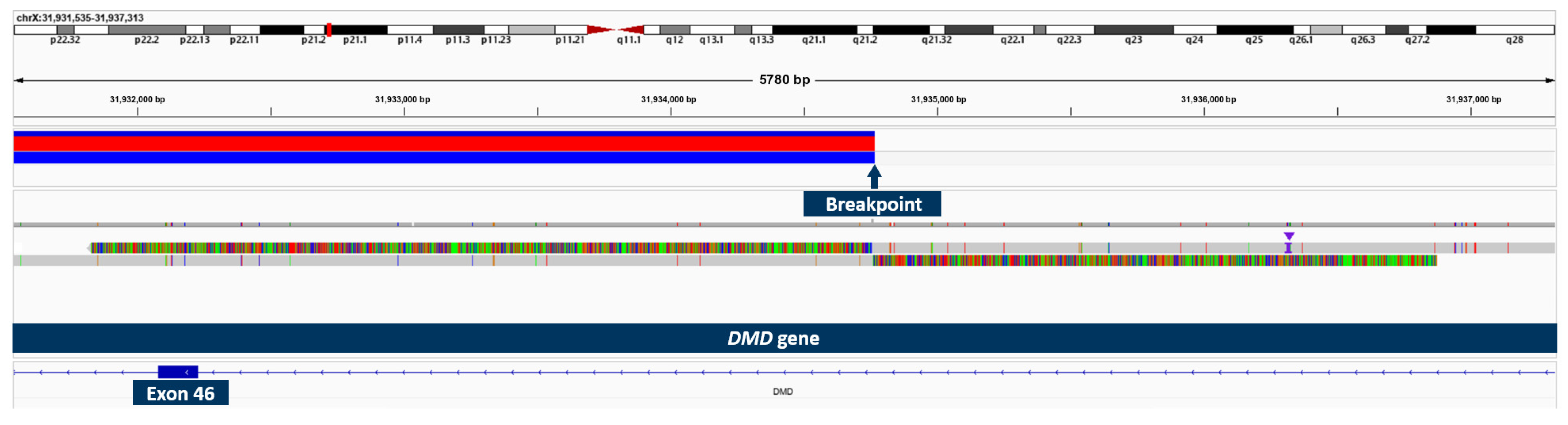
2.5. Results of OGM and Long-Read Sequencing in Patient 3
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Muscle Biopsy
4.3. Whole-Exome Sequencing and Segregation Analyses
4.4. Optical Genome Mapping
4.5. Long-Read Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child Health 2015, 51, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Landfeldt, E.; Thompson, R.; Sejersen, T.; McMillan, H.J.; Kirschner, J.; Lochmüller, H. Life expectancy at birth in Duchenne muscular dystrophy: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 643–653. [Google Scholar] [CrossRef]
- Waldrop, M.A.; Flanigan, K.M. Update in Duchenne and Becker muscular dystrophy. Curr. Opin. Neurol. 2019, 32, 722–727. [Google Scholar] [CrossRef]
- Viggiano, E.; Picillo, E.; Passamano, L.; Onore, M.E.; Piluso, G.; Scutifero, M.; Torella, A.; Nigro, V.; Politano, L. Spectrum of Genetic Variants in the Dystrophin Gene: A Single Centre Retrospective Analysis of 750 Duchenne and Becker Patients from Southern Italy. Genes 2023, 14, 214. [Google Scholar] [CrossRef]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; van Deutekom, J.C.T.; Fokkema, I.F.; van Ommen, G.-J.B.; den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Okubo, M.; Goto, K.; Komaki, H.; Nakamura, H.; Mori-Yoshimura, M.; Hayashi, Y.K.; Mitsuhashi, S.; Noguchi, S.; Kimura, E.; Nishino, I. Comprehensive analysis for genetic diagnosis of Dystrophinopathies in Japan. Orphanet J. Rare Dis. 2017, 12, 149. [Google Scholar] [CrossRef]
- Dent, K.M.; Dunn, D.M.; von Niederhausern, A.C.; Aoyagi, A.T.; Kerr, L.; Bromberg, M.B.; Hart, K.J.; Tuohy, T.; White, S.; den Dunnen, J.T.; et al. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am. J. Med. Genet. A 2005, 134, 295–298. [Google Scholar] [CrossRef]
- Waddell, L.B.; Bryen, S.J.; Cummings, B.B.; Bournazos, A.; Evesson, F.J.; Joshi, H.; Marshall, J.L.; Tukiainen, T.; Valkanas, E.; Weisburd, B.; et al. WGS and RNA Studies Diagnose Noncoding DMD Variants in Males with High Creatine Kinase. Neurol. Genet. 2021, 7, e554. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Sun, C.; Liu, Y.; Yu, M.; Zheng, Y.; Meng, L.; Wang, G.; Cornejo-Sanchez, D.M.; Bharadwaj, T.; Yan, J.; et al. Practical approach to the genetic diagnosis of unsolved dystrophinopathies: A stepwise strategy in the genomic era. J. Med. Genet. 2021, 58, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Dobrescu, M.A.; Chelu, G.; Tache, D.E.; Purcaru, S.O.; Petrescu, I.O. Differential Diagnosis between Duchenne Muscular Dystrophy and Limb Girdle Muscular Dystrophy 2a. Curr. Health Sci. J. 2015, 41, 385–389. [Google Scholar] [CrossRef]
- Schwartz, M.; Hertz, J.M.; Sveen, M.L.; Vissing, J. LGMD2I presenting with a characteristic Duchenne or Becker muscular dystrophy phenotype. Neurology 2005, 64, 1635–1637. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Clemens, P.R.; Hoffman, E.P. Exon-Skipping in Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, S343–S358. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. [Google Scholar] [CrossRef]
- Nilius-Eliliwi, V.; Gerding, W.M.; Schroers, R.; Nguyen, H.P.; Vangala, D.B. Optical Genome Mapping for Cytogenetic Diagnostics in AML. Cancers 2023, 15, 1684. [Google Scholar] [CrossRef]
- Godfrey, C.; Clement, E.; Mein, R.; Brockington, M.; Smith, J.; Talim, B.; Straub, V.; Robb, S.; Quinlivan, R.; Feng, L.; et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007, 130, 2725–2735. [Google Scholar] [CrossRef]
- Vuillaumier-Barrot, S.; Quijano-Roy, S.; Bouchet-Seraphin, C.; Maugenre, S.; Peudenier, S.; van den Bergh, P.; Marcorelles, P.; Avila-Smirnow, D.; Chelbi, M.; Romero, N.B.; et al. Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromuscul. Disord. 2009, 19, 182–188. [Google Scholar] [CrossRef]
- Mercuri, E.; Messina, S.; Bruno, C.; Mora, M.; Pegoraro, E.; Comi, G.P.; D’Amico, A.; Aiello, C.; Biancheri, R.; Berardinelli, A.; et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: A population study. Neurology 2009, 72, 1802–1809. [Google Scholar] [CrossRef]
- Chung Liang, L.; Sulaiman, N.; Yazid, M.D. A Decade of Progress in Gene Targeted Therapeutic Strategies in Duchenne Muscular Dystrophy: A Systematic Review. Front. Bioeng. Biotechnol. 2022, 10, 833833. [Google Scholar] [CrossRef] [PubMed]
- Saito, K. GeneReviews®: Fukuyama Congenital Muscular Dystrophy; National Institutes of Health: Seattle, WA, USA, 1993. [Google Scholar]
- Puckett, R.L.; Moore, S.A.; Winder, T.L.; Willer, T.; Romansky, S.G.; Covault, K.K.; Campbell, K.P.; Abdenur, J.E. Further evidence of Fukutin mutations as a cause of childhood onset limb-girdle muscular dystrophy without mental retardation. Neuromuscul. Disord. 2009, 19, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Zhang, C.; Li, P.; Tong, Y.; Zhu, B.; He, J.; Zhao, Y.; Yao, F.; Cui, L.-Y.; Liang, F.; et al. Identification and characterization of two DMD pedigrees with large inversion mutations based on a long-read sequencing pipeline. Eur. J. Hum. Genet. 2023, 31, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Zaum, A.-K.; Nanda, I.; Kress, W.; Rost, S. Detection of pericentric inversion with breakpoint in DMD by whole genome sequencing. Mol. Genet. Genom. Med. 2022, 10, e2028. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Grilli, A.; Zia, S.; Fang, M.; Rossi, R.; Gualandi, F.; Rimessi, P.; El Dani, R.; Fabris, M.; Lu, Z.; et al. RNA-seq in DMD urinary stem cells recognized muscle-related transcription signatures and addressed the identification of atypical mutations by whole-genome sequencing. Hum. Genet. Genom. Adv. 2022, 3, 100054. [Google Scholar] [CrossRef]
- Barseghyan, H.; Tang, W.; Wang, R.T.; Almalvez, M.; Segura, E.; Bramble, M.S.; Lipson, A.; Douine, E.D.; Lee, H.; Délot, E.C.; et al. Next-generation mapping: A novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med. 2017, 9, 90. [Google Scholar] [CrossRef]
- Bionano Genomics. Bionano Solve Theory of Operation: Structural Variant Calling. Available online: https://bionanogenomics.com/wp-content/uploads/2018/04/30110-Bionano-Solve-Theory-of-Operation-Structural-Variant-Calling.pdf (accessed on 1 June 2023).
- Shaffer, L.G.; Bejjani, B.A. A cytogeneticist’s perspective on genomic microarrays. Hum. Reprod. Update 2004, 10, 221–226. [Google Scholar] [CrossRef]
- Gerding, W.M.; Tembrink, M.; Nilius-Eliliwi, V.; Mika, T.; Dimopoulos, F.; Ladigan-Badura, S.; Eckhardt, M.; Pohl, M.; Wünnenberg, M.; Farshi, P.; et al. Optical genome mapping reveals additional prognostic information compared to conventional cytogenetics in AML/MDS patients. Int. J. Cancer 2022, 150, 1998–2011. [Google Scholar] [CrossRef]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef]
- Schnause, A.C.; Komlosi, K.; Herr, B.; Neesen, J.; Dremsek, P.; Schwarz, T.; Tzschach, A.; Jägle, S.; Lausch, E.; Fischer, J.; et al. Marfan Syndrome Caused by Disruption of the FBN1 Gene due to A Reciprocal Chromosome Translocation. Genes 2021, 12, 1836. [Google Scholar] [CrossRef]
- Cope, H.; Barseghyan, H.; Bhattacharya, S.; Fu, Y.; Hoppman, N.; Marcou, C.; Walley, N.; Rehder, C.; Deak, K.; Alkelai, A.; et al. Detection of a mosaic CDKL5 deletion and inversion by optical genome mapping ends an exhaustive diagnostic odyssey. Mol. Genet. Genomic Med. 2021, 9, e1665. [Google Scholar] [CrossRef] [PubMed]
- Alesi, V.; Lepri, F.R.; Dentici, M.L.; Genovese, S.; Sallicandro, E.; Bejo, K.; Dallapiccola, B.; Capolino, R.; Novelli, A.; Digilio, M.C. Intragenic inversions in NF1 gene as pathogenic mechanism in neurofibromatosis type 1. Eur. J. Hum. Genet. 2022, 30, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical genome mapping as a next-generation cytogenomic tool for detection of structural and copy number variations for prenatal genomic analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics-Its Advantages and Limitations. Genes 2021, 12, 1958. [Google Scholar] [CrossRef]
- Ohno, K.; Tsujino, A.; Brengman, J.M.; Harper, C.M.; Bajzer, Z.; Udd, B.; Beyring, R.; Robb, S.; Kirkham, F.J.; Engel, A.G. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc. Natl. Acad. Sci. USA 2001, 98, 2017–2022. [Google Scholar] [CrossRef]
- O’Grady, G.L.; Verschuuren, C.; Yuen, M.; Webster, R.; Menezes, M.; Fock, J.M.; Pride, N.; Best, H.A.; Benavides Damm, T.; Turner, C.; et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology 2016, 87, 1442–1448. [Google Scholar] [CrossRef]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef]
- Cotton, S.; Voudouris, N.J.; Greenwood, K.M. Intelligence and Duchenne muscular dystrophy: Full-scale, verbal, and performance intelligence quotients. Dev. Med. Child Neurol. 2001, 43, 497–501. [Google Scholar] [CrossRef]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2020, 57, 1748–1767. [Google Scholar] [CrossRef]
- Iskandar, K.; Triono, A.; Sunartini; Dwianingsih, E.K.; Indraswari, B.W.; Kirana, I.R.; Ivana, G.; Sutomo, R.; Patria, S.Y.; Herini, E.S.; et al. Dp71 and intellectual disability in Indonesian patients with Duchenne muscular dystrophy. PLoS ONE 2022, 17, e0276640. [Google Scholar] [CrossRef]
- Roberts, R.G.; Freeman, T.C.; Kendall, E.; Vetrie, D.L.; Dixon, A.K.; Shaw-Smith, C.; Bone, Q.; Bobrow, M. Characterization of DRP2, a novel human dystrophin homologue. Nat. Genet. 1996, 13, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Krag, T.O.; Gyrd-Hansen, M.; Khurana, T.S. Harnessing the potential of dystrophin-related proteins for ameliorating Duchenne’s muscular dystrophy. Acta Physiol. Scand. 2001, 171, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.T.; Wolfe, D. Tissue processing and hematoxylin and eosin staining. Methods Mol. Biol. 2014, 1180, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Variants in the DMD Gene | Count (%) |
|---|---|
| Deletion of one or more exons | 40 (~66%) |
| Duplication of one or more exons | 3 (~5%) |
| Point mutation Nonsense mutation | 14 (~23%) 6 (~10%) |
| No mutation detected | 3 (~5%) |
| Total | 60 |
| Chromosome | Size and Type | Start Labeling Position (hg38) | End Labeling Position (hg38) | Muscle Genes | Parents |
|---|---|---|---|---|---|
| X | ~1.28 Mb Inversion | 30646419 | 31930149 | DMD | Mother as heterozygous carrier |
| 10 | ~4.3 Mb Duplication | 45702207 | 50040591 | CHAT, SLC18A3 | Maternally inherited |
| 14 | ~3 kb Insertion | 63964082 | 63991908 | SYNE2 | Paternally inherited |
| Antibody | Dilution | Abbreviation | Supplier |
|---|---|---|---|
| DYS1, mmc | 1:3 | NCL-DYS1, Lot: 6066097 | Novocastra |
| DYS2, mmc | 1:10 | NCL-DYS2, Lot: 6065996 | Novocastra |
| DYS3, mmc | 1:10 | NCL-DYS3, Lot: 6066797 | Novocastra |
| Alpha-sarcoglycan, mmc | 1:50 | NCL-L-a-SARC, | Novocastra |
| Beta-sarcoglycan, mmc | 1:50 | NCL-L-b-SARC, Lot: 6083740 | Novocastra |
| Gamma-sarcoglycan, mmc | 1:25 | NCL-g-SARC, Lot: 6084768 | Novocastra |
| Delta-sarcoglycan, mmc | 1:10 | NCL-d-SARC, Lot: 6069438 | Novocastra |
| LAMA5, mmc | 1:500 | MAB1924 Lot: 21031281 | Chemicon, Merck |
| DRP2, mmc | 1:5 | NCL-DRP2, Lot: 6035452 | Novocastra |
| MHCn, mmc | 1:5 | NCL-MHCn, Lot: 6091144 | Novocastra |
| Beta-dystroglycan, mmc | 1:10 | NCL-b-DG | Novocastra |
| nNos, rpc | 1:200 | 06-528, Lot: #18537 | Upstate biotechnology |
| Spectrin, mmc | 1:100 | NCL-SPEC1, Lot: 6084548 | Novocastra |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erbe, L.S.; Hoffjan, S.; Janßen, S.; Kneifel, M.; Krause, K.; Gerding, W.M.; Döring, K.; Güttsches, A.-K.; Roos, A.; Buena Atienza, E.; et al. Exome Sequencing and Optical Genome Mapping in Molecularly Unsolved Cases of Duchenne Muscular Dystrophy: Identification of a Causative X-Chromosomal Inversion Disrupting the DMD Gene. Int. J. Mol. Sci. 2023, 24, 14716. https://doi.org/10.3390/ijms241914716
Erbe LS, Hoffjan S, Janßen S, Kneifel M, Krause K, Gerding WM, Döring K, Güttsches A-K, Roos A, Buena Atienza E, et al. Exome Sequencing and Optical Genome Mapping in Molecularly Unsolved Cases of Duchenne Muscular Dystrophy: Identification of a Causative X-Chromosomal Inversion Disrupting the DMD Gene. International Journal of Molecular Sciences. 2023; 24(19):14716. https://doi.org/10.3390/ijms241914716
Chicago/Turabian StyleErbe, Leoni S., Sabine Hoffjan, Sören Janßen, Moritz Kneifel, Karsten Krause, Wanda M. Gerding, Kristina Döring, Anne-Katrin Güttsches, Andreas Roos, Elena Buena Atienza, and et al. 2023. "Exome Sequencing and Optical Genome Mapping in Molecularly Unsolved Cases of Duchenne Muscular Dystrophy: Identification of a Causative X-Chromosomal Inversion Disrupting the DMD Gene" International Journal of Molecular Sciences 24, no. 19: 14716. https://doi.org/10.3390/ijms241914716
APA StyleErbe, L. S., Hoffjan, S., Janßen, S., Kneifel, M., Krause, K., Gerding, W. M., Döring, K., Güttsches, A.-K., Roos, A., Buena Atienza, E., Gross, C., Lücke, T., Nguyen, H. H. P., Vorgerd, M., & Köhler, C. (2023). Exome Sequencing and Optical Genome Mapping in Molecularly Unsolved Cases of Duchenne Muscular Dystrophy: Identification of a Causative X-Chromosomal Inversion Disrupting the DMD Gene. International Journal of Molecular Sciences, 24(19), 14716. https://doi.org/10.3390/ijms241914716