

Clinical, Genetic, and Histological Characterization of Patients with Rare Neuromuscular and Mitochondrial Diseases Presenting with Different Cardiomyopathy Phenotypes
 , ,
, ,  ,
,  ,
,  , ,
, , 
 , and
, and 
Abstract
1. Introduction
2. Results
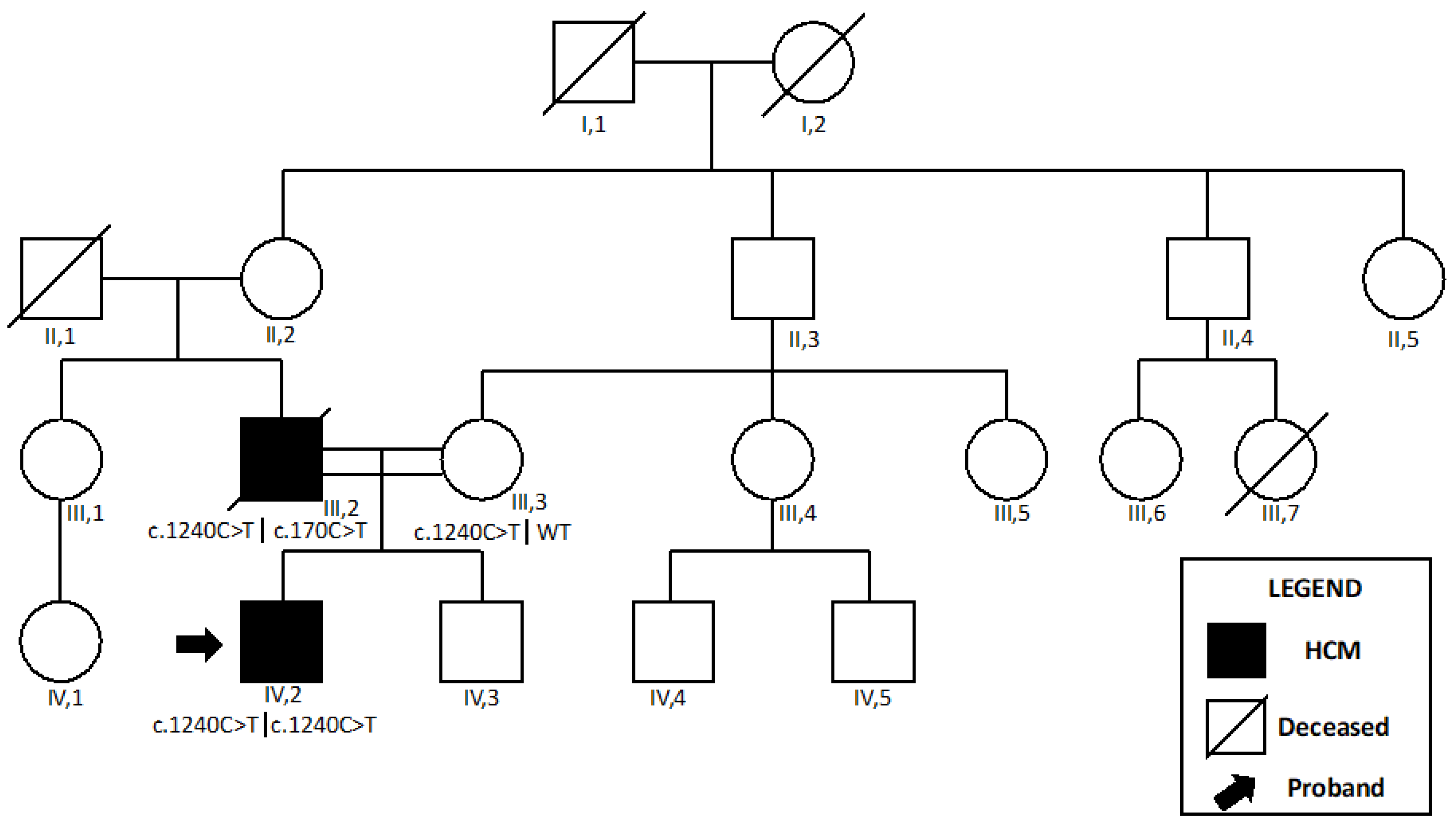
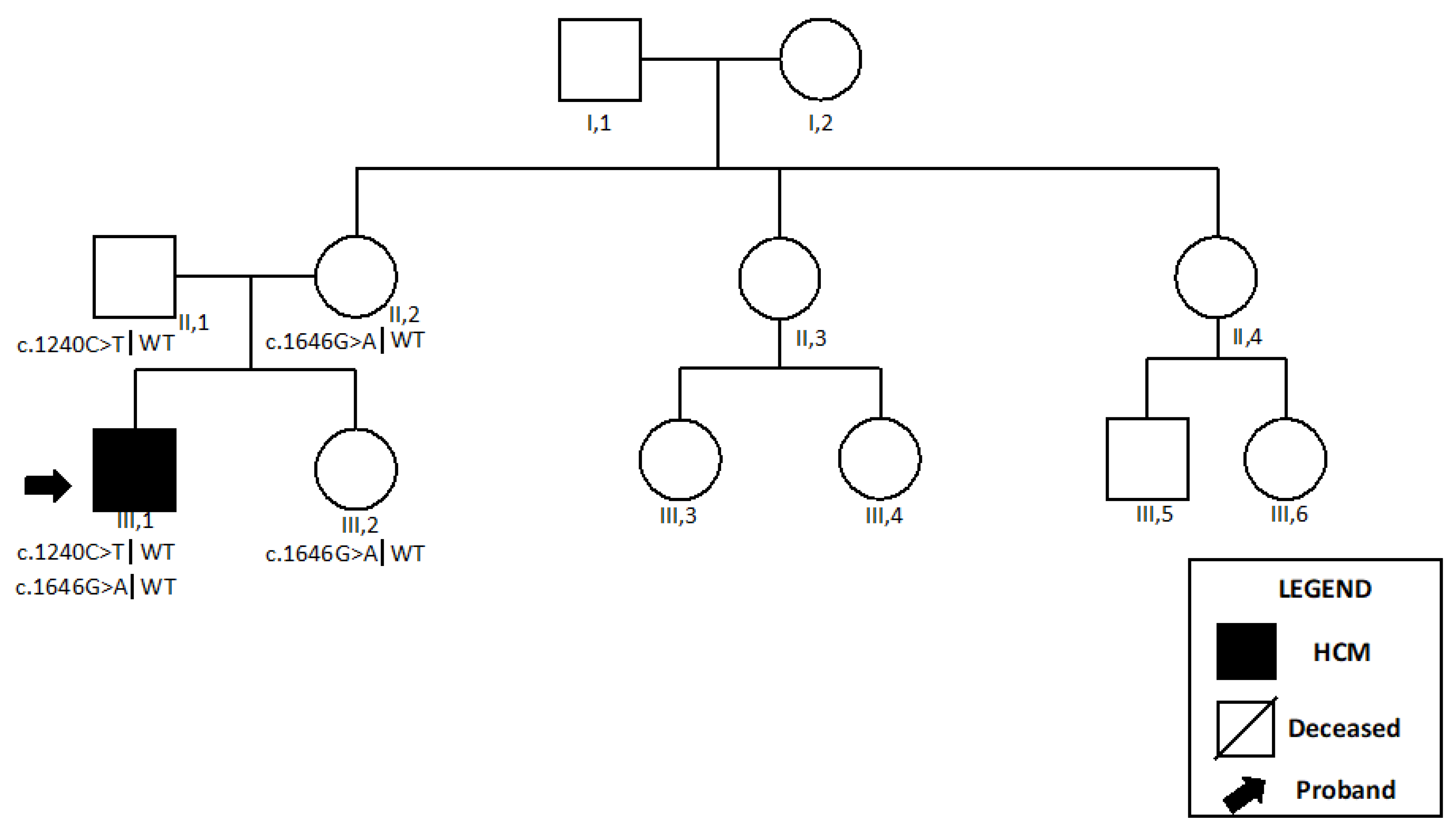
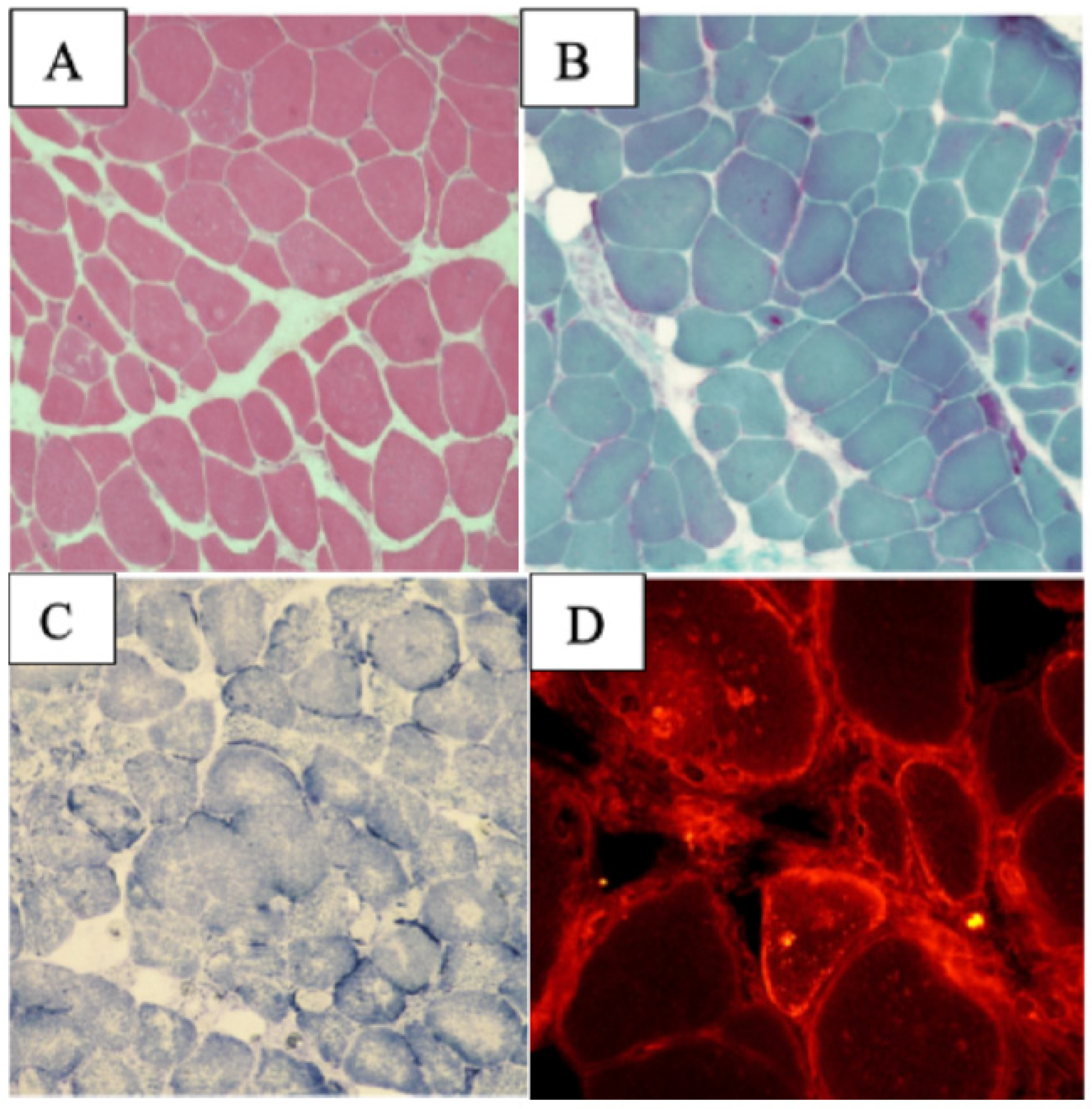
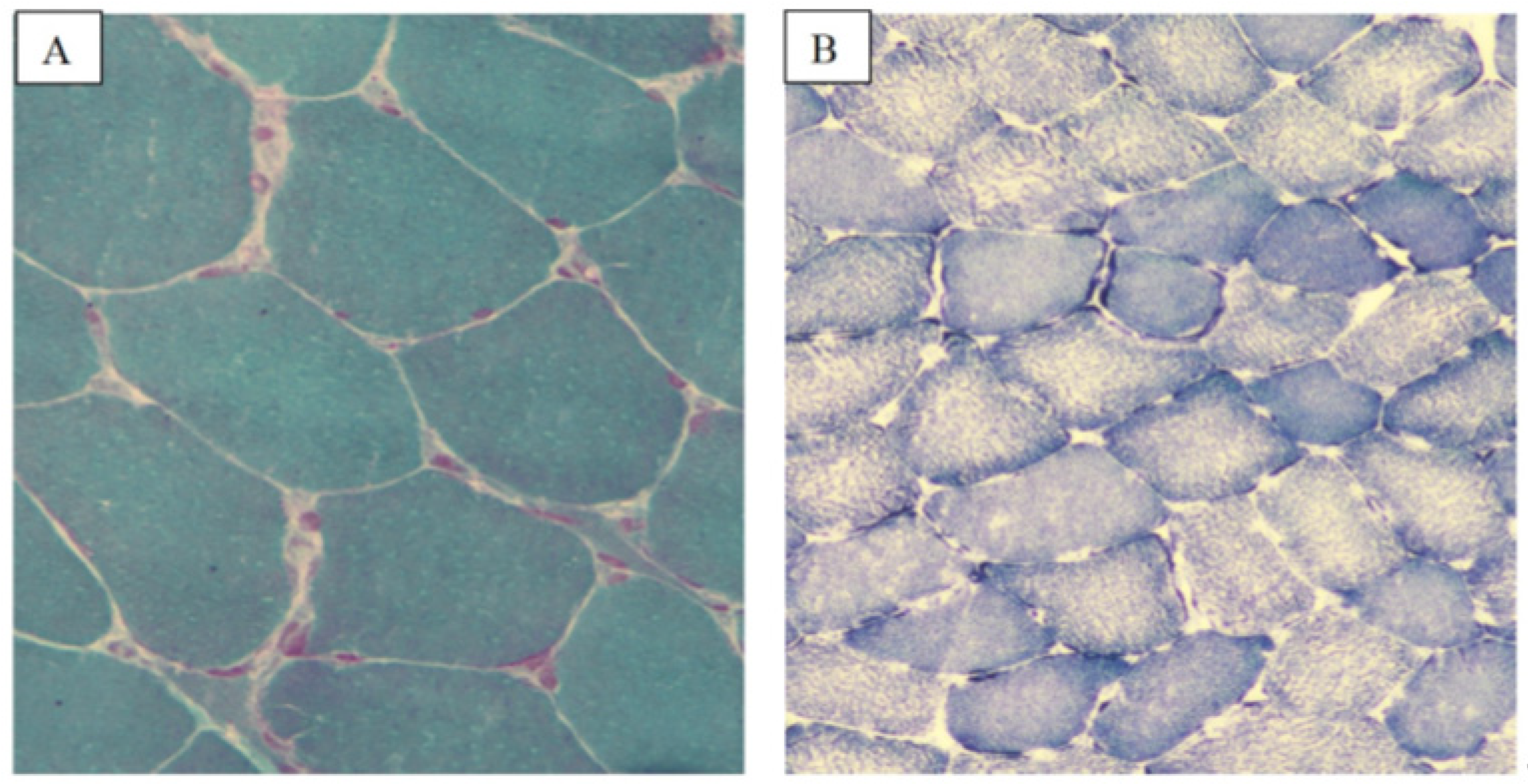
2.1. ACAD9
2.1.1. Patient 1
2.1.2. Patient 2
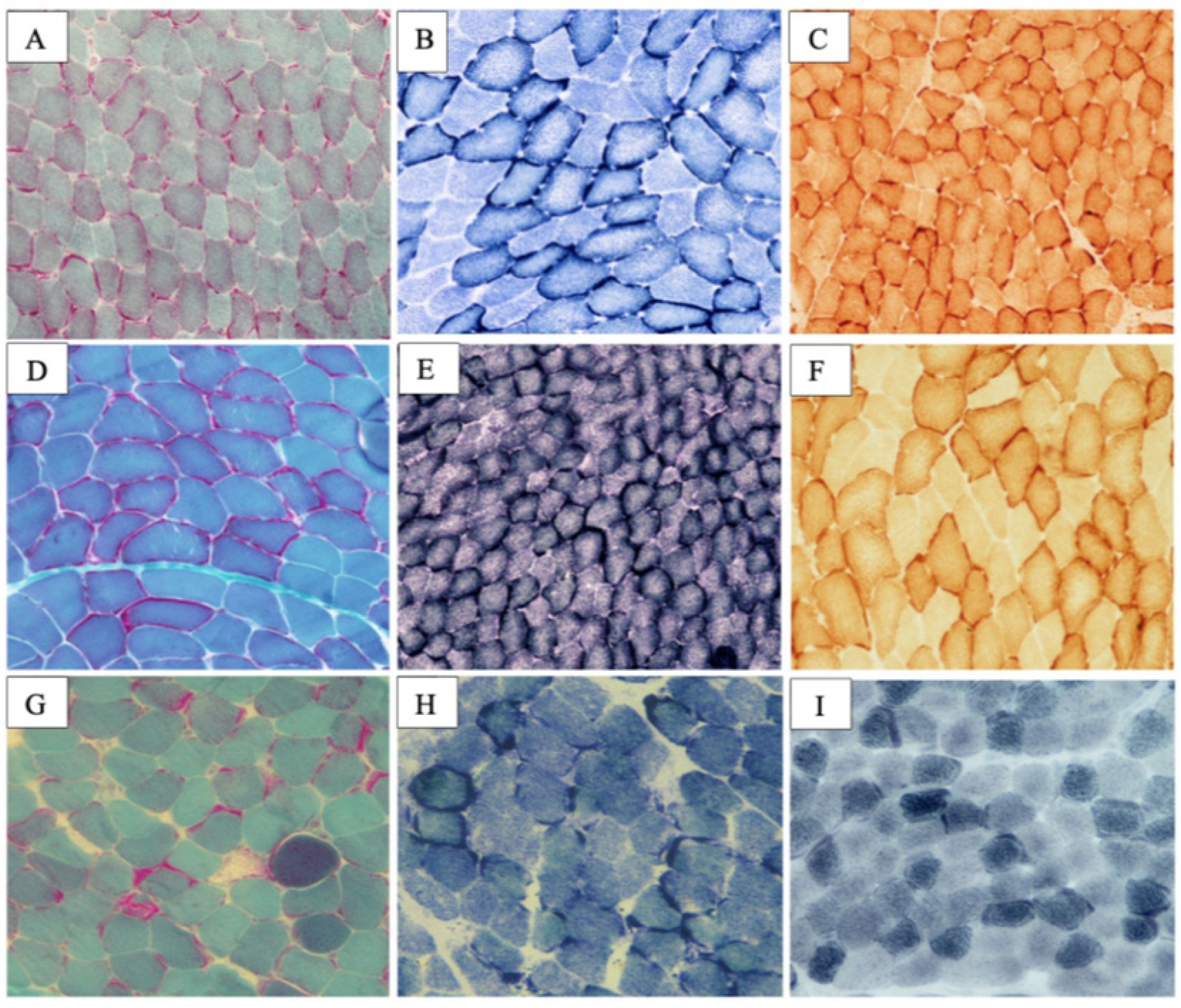
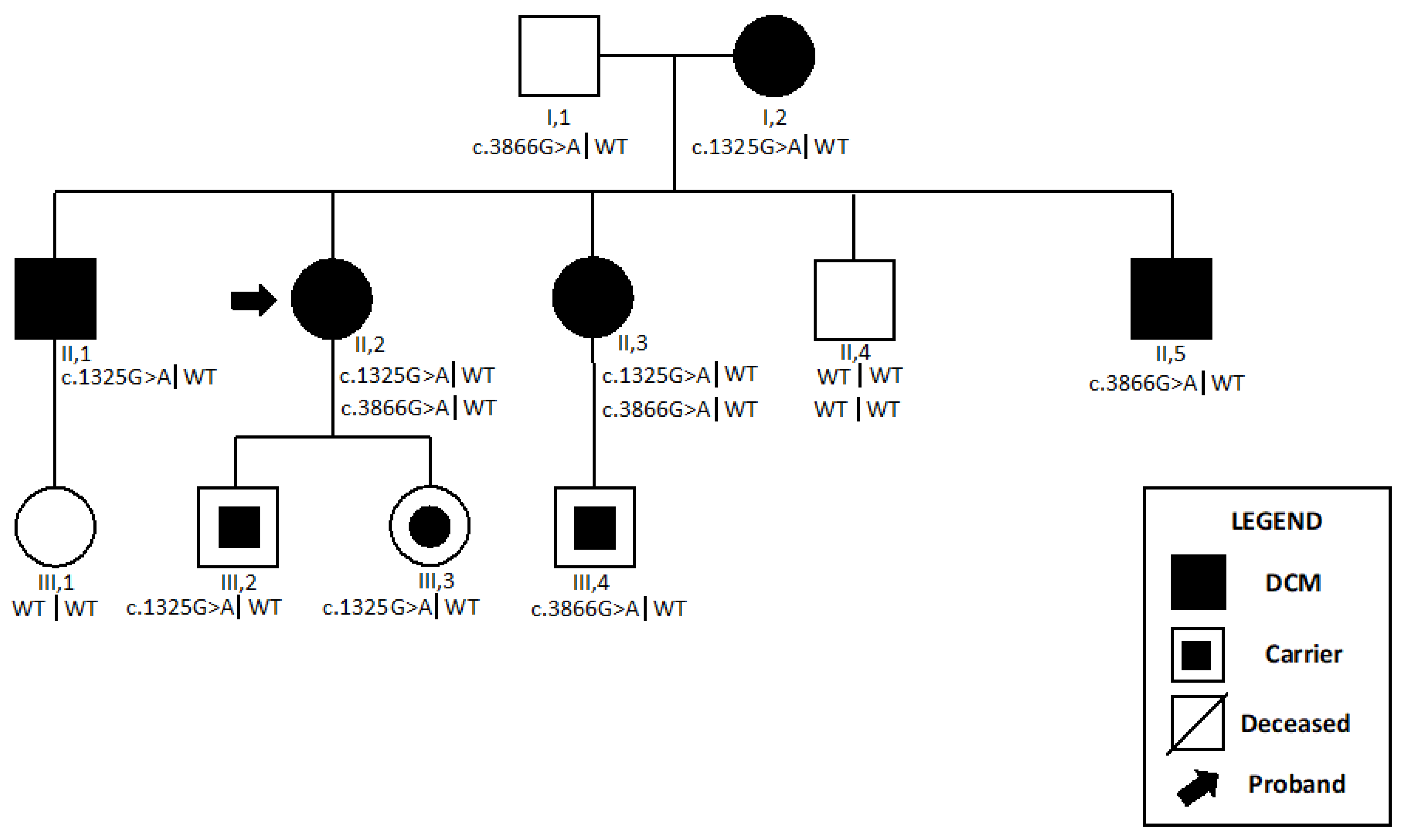
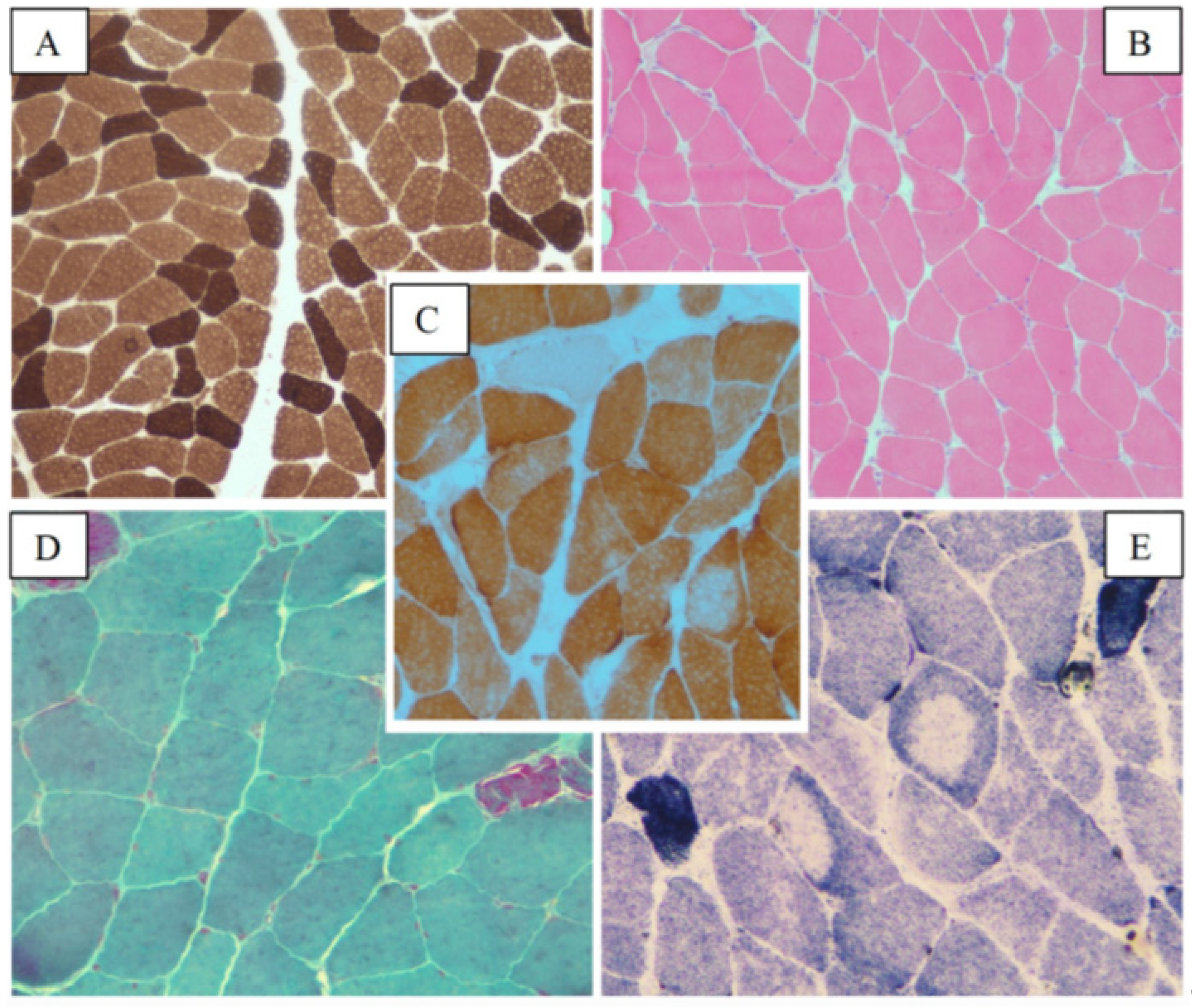
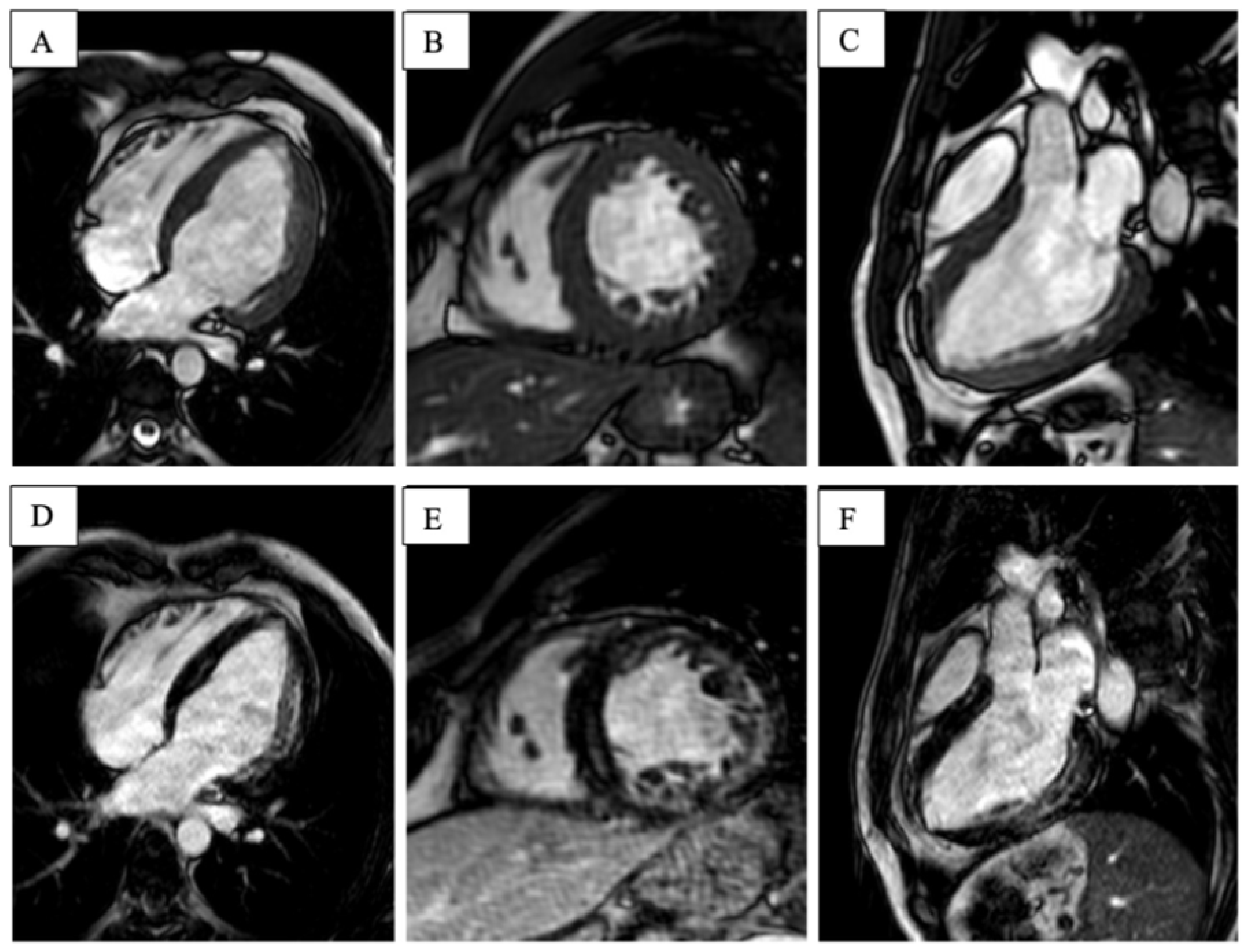
2.2. MYH7-Related Myopathy
2.2.1. Patient 3
2.2.2. Patient 4
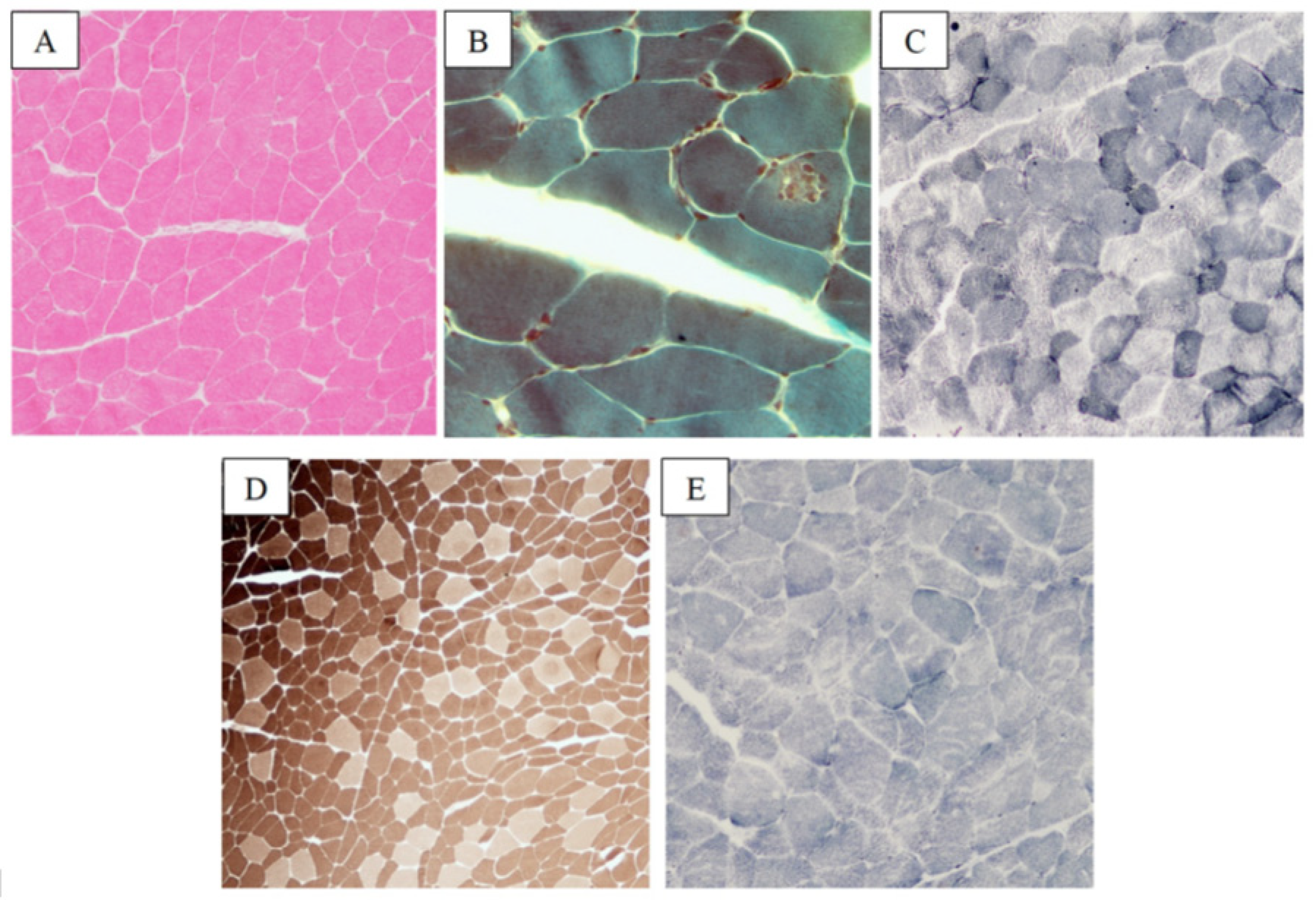
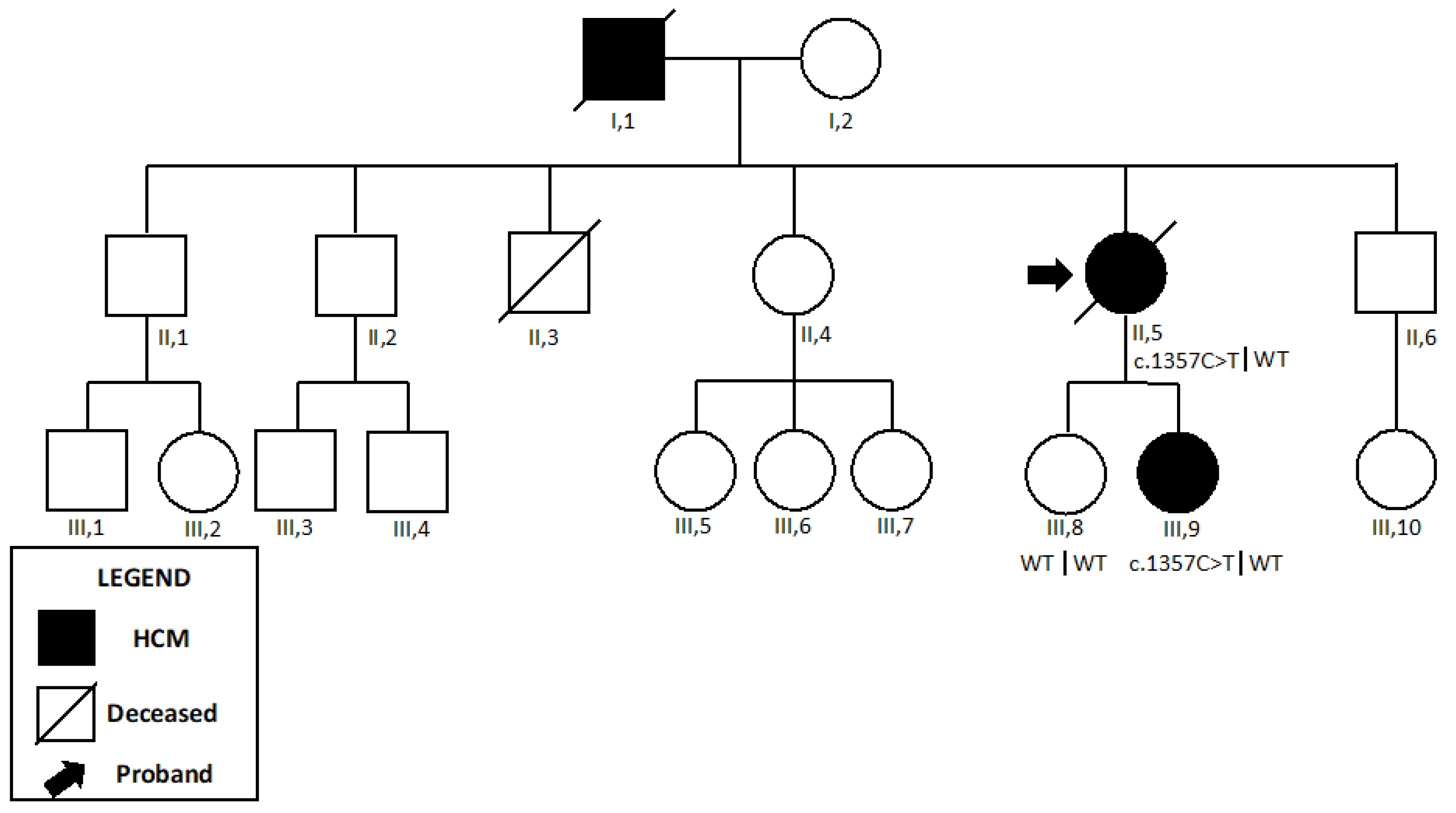
2.3. Desminopathy
2.4. Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes (MELAS) Syndrome
2.5. Mitochondrial tRna Translation Optimization (MTO1)
3. Discussion
4. Materials and Methods
4.1. Study Design
- The enrolment phase occurred between 2010 and 2012 at the Inherited and Rare Cardiovascular Disease Unit of the University of Campania “Luigi Vanvitelli”, Naples, Italy. During the first phase, 425 consecutive patients with cardiomyopathy were identified;
- Between 2012 and 2013, 24 cardiomyopathy patients showing a persistent increase in serum creatine kinase enzyme were selected for neuromuscular evaluation;
- Between 2012 and 2022, 11 patients with high suspicion for NMDs underwent a comprehensive neurological, histological, and molecular evaluation, according to the study protocol, and seven patients had a definitive diagnosis of NMD or MD. Preliminary data and the diagnostic work-up is described elsewhere [22].
4.2. Study Protocol
4.3. Cardiovascular Protocol
4.4. Neuromuscular Protocol
4.5. Genetic Evaluation
5. Study Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the Cardiomyopathies: A Position Statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2007, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Narula, N.; Dec, G.W.; Reddy, K.S.; Greenberg, B.; Kushwaha, S.; Marwick, T.; Pinney, S.; Bellazzi, R.; Favalli, V.; et al. The MOGE(S) Classification for a Phenotype–Genotype Nomenclature of Cardiomyopathy. J. Am. Coll. Cardiol. 2013, 62, 2046–2072. [Google Scholar] [CrossRef] [PubMed]
- Charron, P.; Arad, M.; Arbustini, E.; Basso, C.; Bilinska, Z.; Elliott, P.; Helio, T.; Keren, A.; McKenna, W.J.; Monserrat, L.; et al. Genetic Counselling and Testing in Cardiomyopathies: A Position Statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2010, 31, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef]
- Task Force members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Vasilescu, C.; Ojala, T.H.; Brilhante, V.; Ojanen, S.; Hinterding, H.M.; Palin, E.; Alastalo, T.-P.; Koskenvuo, J.; Hiippala, A.; Jokinen, E.; et al. Genetic Basis of Severe Childhood-Onset Cardiomyopathies. J. Am. Coll. Cardiol. 2018, 72, 2324–2338. [Google Scholar] [CrossRef]
- Arbustini, E.; Di Toro, A.; Giuliani, L.; Favalli, V.; Narula, N.; Grasso, M. Cardiac Phenotypes in Hereditary Muscle Disorders. J. Am. Coll. Cardiol. 2018, 72, 2485–2506. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement From the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019, 140, 31–41. [Google Scholar] [CrossRef]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchía, J.; García-Pinilla, J.M.; Pascual-Figal, D.A.; Nuñez, J.; Guzzo-Merello, G.; et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J. Am. Coll. Cardiol. 2018, 71, 2293–2302. [Google Scholar] [CrossRef]
- Monda, E.; Limongelli, G. Is There a Role for Genetic Testing in Patients With Myocarditis? Circ. Genom. Precis. Med. 2022, 15, e003824. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.P.; Charron, P.; Gimeno-Blanes, J.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic Work-up in Cardiomyopathies: Bridging the Gap between Clinical Phenotypes and Final Diagnosis. A Position Statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Mirra, B.; Barretta, F.; Caiazza, M.; Lombardo, B.; Scudiero, O.; Tinto, N.; Limongelli, G.; Frisso, G. Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes. Int. J. Mol. Sci. 2021, 22, 5742. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for Mitochondrial Disorders. Cochrane Database Syst. Rev. 2012, CD004426. [Google Scholar] [CrossRef]
- Lioncino, M.; Monda, E.; Caiazza, M.; Fusco, A.; Cirillo, A.; Dongiglio, F.; Simonelli, V.; Sampaolo, S.; Ruggiero, L.; Scarano, G.; et al. Cardiovascular Involvement in MtDNA Disease. Heart Fail. Clin. 2022, 18, 51–60. [Google Scholar] [CrossRef]
- Lund, M.; Diaz, L.J.; Ranthe, M.F.; Petri, H.; Duno, M.; Juncker, I.; Eiberg, H.; Vissing, J.; Bundgaard, H.; Wohlfahrt, J.; et al. Cardiac Involvement in Myotonic Dystrophy: A Nationwide Cohort Study. Eur. Heart J. 2014, 35, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- van Rijsingen, I.A.W.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk Factors for Malignant Ventricular Arrhythmias in Lamin A/C Mutation Carriers. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
- Bates, M.G.D.; Bourke, J.P.; Giordano, C.; d’Amati, G.; Turnbull, D.M.; Taylor, R.W. Cardiac Involvement in Mitochondrial DNA Disease: Clinical Spectrum, Diagnosis, and Management. Eur. Heart J. 2012, 33, 3023–3033. [Google Scholar] [CrossRef]
- Limongelli, G.; Adorisio, R.; Baggio, C.; Bauce, B.; Biagini, E.; Castelletti, S.; Favilli, S.; Imazio, M.; Lioncino, M.; Merlo, M.; et al. Diagnosis and Management of Rare Cardiomyopathies in Adult and Paediatric Patients. A Position Paper of the Italian Society of Cardiology (SIC) and Italian Society of Paediatric Cardiology (SICP). Int. J. Cardiol. 2022, 357, 55–71. [Google Scholar] [CrossRef]
- Constantinides, V.C.; Papahatzaki, M.M.; Papadimas, G.K.; Karandreas, N.; Zambelis, T.; Kokotis, P.; Manda, P. Diagnostic Accuracy of Muscle Biopsy and Electromyography in 123 Patients with Neuromuscular Disorders. In Vivo 2018, 32, 1647–1652. [Google Scholar] [CrossRef]
- Feingold, B.; Mahle, W.T.; Auerbach, S.; Clemens, P.; Domenighetti, A.A.; Jefferies, J.L.; Judge, D.P.; Lal, A.K.; Markham, L.W.; Parks, W.J.; et al. Management of Cardiac Involvement Associated With Neuromuscular Diseases: A Scientific Statement From the American Heart Association. Circulation 2017, 136, e200–e231. [Google Scholar] [CrossRef] [PubMed]
- Lioncino, M.; Monda, E.; Caiazza, M.; Simonelli, V.; Nesti, C.; Mauriello, A.; Budillon, A.; Di Santo, A.; Bruno, G.; Varone, A.; et al. A Combined Clinical, Molecular and Muscle Biopsy Approach to Unveil Prevalence and Clinical Features of Rare Neuromuscular and Mitochondrial Diseases in Patients with Cardiomyopathies. Circ. Genom. Precis. Med. 2023, e004122. [Google Scholar] [CrossRef]
- Fiorillo, C.; Astrea, G.; Savarese, M.; Cassandrini, D.; Brisca, G.; Trucco, F.; Pedemonte, M.; Trovato, R.; Ruggiero, L.; Vercelli, L.; et al. MYH7-Related Myopathies: Clinical, Histopathological and Imaging Findings in a Cohort of Italian Patients. Orphanet J. Rare Dis. 2016, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure: Developed by the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure of the European Society of Cardiology (ESC). With the Special Contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131. [Google Scholar] [CrossRef]
- Arbustini, E.; Pasotti, M.; Pilotto, A.; Pellegrini, C.; Grasso, M.; Previtali, S.; Repetto, A.; Bellini, O.; Azan, G.; Scaffino, M.; et al. Desmin Accumulation Restrictive Cardiomyopathy and Atrioventricular Block Associated with Desmin Gene Defects. Eur. J. Heart Fail. 2006, 8, 477–483. [Google Scholar] [CrossRef]
- Limongelli, G.; D’Alessandro, R.; Maddaloni, V.; Rea, A.; Sarkozy, A.; McKenna, W.J. Skeletal Muscle Involvement in Cardiomyopathies. J. Cardiovasc. Med. 2013, 14, 837–861. [Google Scholar] [CrossRef] [PubMed]
- Esposito, T.; Sampaolo, S.; Limongelli, G.; Varone, A.; Formicola, D.; Diodato, D.; Farina, O.; Napolitano, F.; Pacileo, G.; Gianfrancesco, F.; et al. Digenic Mutational Inheritance of the Integrin Alpha 7 and the Myosin Heavy Chain 7B Genes Causes Congenital Myopathy with Left Ventricular Non-Compact Cardiomyopathy. Orphanet J. Rare Dis. 2013, 8, 91. [Google Scholar] [CrossRef]
- Schram, G.; Fournier, A.; Leduc, H.; Dahdah, N.; Therien, J.; Vanasse, M.; Khairy, P. All-Cause Mortality and Cardiovascular Outcomes With Prophylactic Steroid Therapy in Duchenne Muscular Dystrophy. J. Am. Coll. Cardiol. 2013, 61, 948–954. [Google Scholar] [CrossRef]
- Monda, E.; Lioncino, M.; Rubino, M.; Passantino, S.; Verrillo, F.; Caiazza, M.; Cirillo, A.; Fusco, A.; Di Fraia, F.; Fimiani, F.; et al. Diagnosis and Management of Cardiovascular Involvement in Friedreich Ataxia. Heart Fail. Clin. 2022, 18, 31–37. [Google Scholar] [CrossRef]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and Clinical Significance of Red Flags in Patients with Hypertrophic Cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Lombardi, R.; Mirra, B.; Barretta, F.; Esposito, M.V.; Uomo, F.; Caiazza, M.; Monda, E.; Losi, M.A.; Limongelli, G.; et al. Next-Generation Sequencing Gene Panels in Inheritable Cardiomyopathies and Channelopathies: Prevalence of Pathogenic Variants and Variants of Unknown Significance in Uncommon Genes. Biomolecules 2022, 12, 1417. [Google Scholar] [CrossRef] [PubMed]
- Maruotti, G.M.; Frisso, G.; Calcagno, G.; Fortunato, G.; Castaldo, G.; Martinelli, P.; Sacchetti, L.; Salvatore, F. Prenatal Diagnosis of Inherited Diseases: 20 Years’ Experience of an Italian Regional Reference Centre. Clin. Chem. Lab. Med. (CCLM) 2013, 51, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, Biochemical and Genetic Spectrum of 70 Patients with ACAD9 Deficiency: Is Riboflavin Supplementation Effective? Orphanet J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Zarrouk-Mahjoub, S. Mitochondrial Multiorgan Disorder Syndrome (MIMODS) Due to a Compound Heterozygous Mutation in the ACAD9 Gene. Mol. Genet. Metab. Rep. 2017, 13, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Kley, R.A.; Leber, Y.; Schrank, B.; Zhuge, H.; Orfanos, Z.; Kostan, J.; Onipe, A.; Sellung, D.; Güttsches, A.K.; Eggers, B.; et al. FLNC-Associated Myofibrillar Myopathy: New Clinical, Functional, and Proteomic Data. Neurol. Genet. 2021, 7, e590. [Google Scholar] [CrossRef]
- Monda, E.; Caiazza, M.; Limongelli, G. The Expanding Spectrum of FLNC Cardiomyopathy. Cardiogenetics 2022, 12, 276–277. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular Insights into Cardiomyopathies Associated with Desmin (DES) Mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Šaric, T.; Cakar, H.; et al. The Novel Desmin Mutant p.A120D Impairs Filament Formation, Prevents Intercalated Disk Localization, and Causes Sudden Cardiac Death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef]
- Bourke, J.P.; Ng, Y.S.; Tynan, M.; Bates, M.G.D.; Mohiddin, S.; Turnbull, D.; Gorman, G.S. Arrhythmia Prevalence and Sudden Death Risk in Adults with the m.3243A>G Mitochondrial Disorder. Open Heart 2022, 9, e001819. [Google Scholar] [CrossRef]
- Ng, Y.S.; Grady, J.P.; Lax, N.Z.; Bourke, J.P.; Alston, C.L.; Hardy, S.A.; Falkous, G.; Schaefer, A.G.; Radunovic, A.; Mohiddin, S.A.; et al. Sudden Adult Death Syndrome in m.3243A>G-Related Mitochondrial Disease: An Unrecognized Clinical Entity in Young, Asymptomatic Adults. Eur. Heart J. 2016, 37, 2552–2559. [Google Scholar] [CrossRef]
- Savvatis, K.; Vissing, C.R.; Klouvi, L.; Florian, A.; Rahman, M.; Béhin, A.; Fayssoil, A.; Masingue, M.; Stojkovic, T.; Bécane, H.M.; et al. Cardiac Outcomes in Adults with Mitochondrial Diseases. J. Am. Coll. Cardiol. 2022, 80, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Amato, A. Electrodiagnostic Evaluation of Myopathies. Phys. Med. Rehabil. Clin. N. Am. 2013, 24, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Compston, A. Aids to the Investigation of Peripheral Nerve Injuries. Medical Research Council: Nerve Injuries Research Committee. His Majesty’s Stationery Office: 1942; pp. 48 (Iii) and 74 Figures and 7 Diagrams; with Aids to the Examination of the Peripheral Nervous System. By Michael O’Brien for the Guarantors of Brain. Saunders Elsevier: 2010; pp. [8] 64 and 94 Figures. Brain 2010, 133, 2838–2844. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Frisso, G.; Precone, V.; Boccia, A.; Fienga, A.; Pacileo, G.; Limongelli, G.; Paolella, G.; Calabrò, R.; Salvatore, F. DNA Sequence Capture and Next-Generation Sequencing for the Molecular Diagnosis of Genetic Cardiomyopathies. J. Mol. Diagn. 2014, 16, 32–44. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gene | DNA Position | Protein Position | Frequency GnomAD (Exomes) | ACMG Evaluation | ACMG Criteria |
|---|---|---|---|---|---|---|
| Patient 1 | ACAD9 | c.1240C>T | p.Arg414Cys | 0.0000119 | P | PP5, PM5, PP3, PM1, PM2, PP1 |
| ACAD9 | c.1240C>T | p.Arg414Cys | 0.0000119 | P | PP5, PM5, PP3, PM1, PM2, PP1 | |
| Patient 2 | ACAD9 | c.1240C>T | p.Arg414Cys | 0.0000119 | P | PP5, PM5, PP3, PM1, PM2, PP1 |
| ACAD9 | c.1646G>A | p.Arg549Gln | 0.00000802 | VUS | PP3, PM2, BP1 | |
| Patient 3 | MYH7 | c.1325G>A | p.Arg442His | 0.0000119 | P | PS3, PM1, PM5, PP3, PP5, PM2, PP1 |
| MYH7 | c.3866G>A | p.Arg1289Gln | 0.00000795 | VUS | PP3, PM2, PP1, PP2 | |
| Patient 4 | MYH7 | c.1357C>T | p.Arg453Cys | NR | P | PP5, PM5, PP3, PM1, PM2 |
| Patient 5 | DES | c.46C>T | p.Arg16Cys | NR | LP | PP3, PM1, PP5, PM2 |
| Patient 6 | MT-TL1 | m.3243A>G | N/A | NR | P | PM2, PP3, PS4, PP5, PM5 |
| Patient 7 | MTO1 | c.253G>A | p.Gly85Arg | 0.00000795 | VUS | PP3, PM2, BP1 |
| MTO1 | c.1055C>T | p.Thr352Met | 0.00000398 | VUS | PP3, PM2, BP1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monda, E.; Lioncino, M.; Caiazza, M.; Simonelli, V.; Nesti, C.; Rubino, M.; Perna, A.; Mauriello, A.; Budillon, A.; Pota, V.; et al. Clinical, Genetic, and Histological Characterization of Patients with Rare Neuromuscular and Mitochondrial Diseases Presenting with Different Cardiomyopathy Phenotypes. Int. J. Mol. Sci. 2023, 24, 9108. https://doi.org/10.3390/ijms24109108
Monda E, Lioncino M, Caiazza M, Simonelli V, Nesti C, Rubino M, Perna A, Mauriello A, Budillon A, Pota V, et al. Clinical, Genetic, and Histological Characterization of Patients with Rare Neuromuscular and Mitochondrial Diseases Presenting with Different Cardiomyopathy Phenotypes. International Journal of Molecular Sciences. 2023; 24(10):9108. https://doi.org/10.3390/ijms24109108
Chicago/Turabian StyleMonda, Emanuele, Michele Lioncino, Martina Caiazza, Vincenzo Simonelli, Claudia Nesti, Marta Rubino, Alessia Perna, Alfredo Mauriello, Alberta Budillon, Vincenzo Pota, and et al. 2023. "Clinical, Genetic, and Histological Characterization of Patients with Rare Neuromuscular and Mitochondrial Diseases Presenting with Different Cardiomyopathy Phenotypes" International Journal of Molecular Sciences 24, no. 10: 9108. https://doi.org/10.3390/ijms24109108
APA StyleMonda, E., Lioncino, M., Caiazza, M., Simonelli, V., Nesti, C., Rubino, M., Perna, A., Mauriello, A., Budillon, A., Pota, V., Bruno, G., Varone, A., Nigro, V., Santorelli, F. M., Pacileo, G., Russo, M. G., Frisso, G., Sampaolo, S., & Limongelli, G. (2023). Clinical, Genetic, and Histological Characterization of Patients with Rare Neuromuscular and Mitochondrial Diseases Presenting with Different Cardiomyopathy Phenotypes. International Journal of Molecular Sciences, 24(10), 9108. https://doi.org/10.3390/ijms24109108