
Long-Term Evaluation of Biomarkers in the Czech Cohort of Gaucher Patients

 , ,
, ,
Abstract
:1. Introduction
2. Results
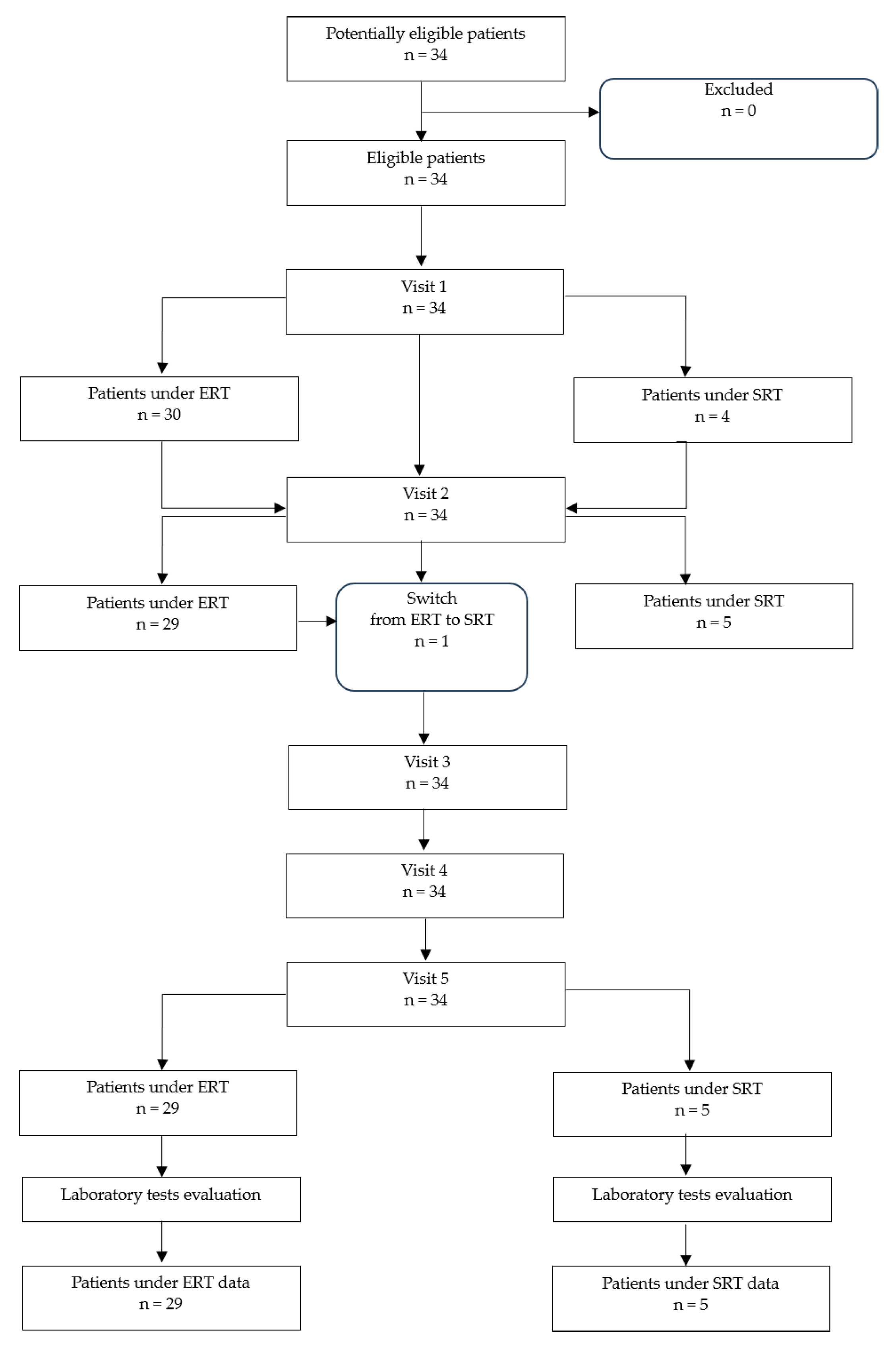
2.1. Study Population Description

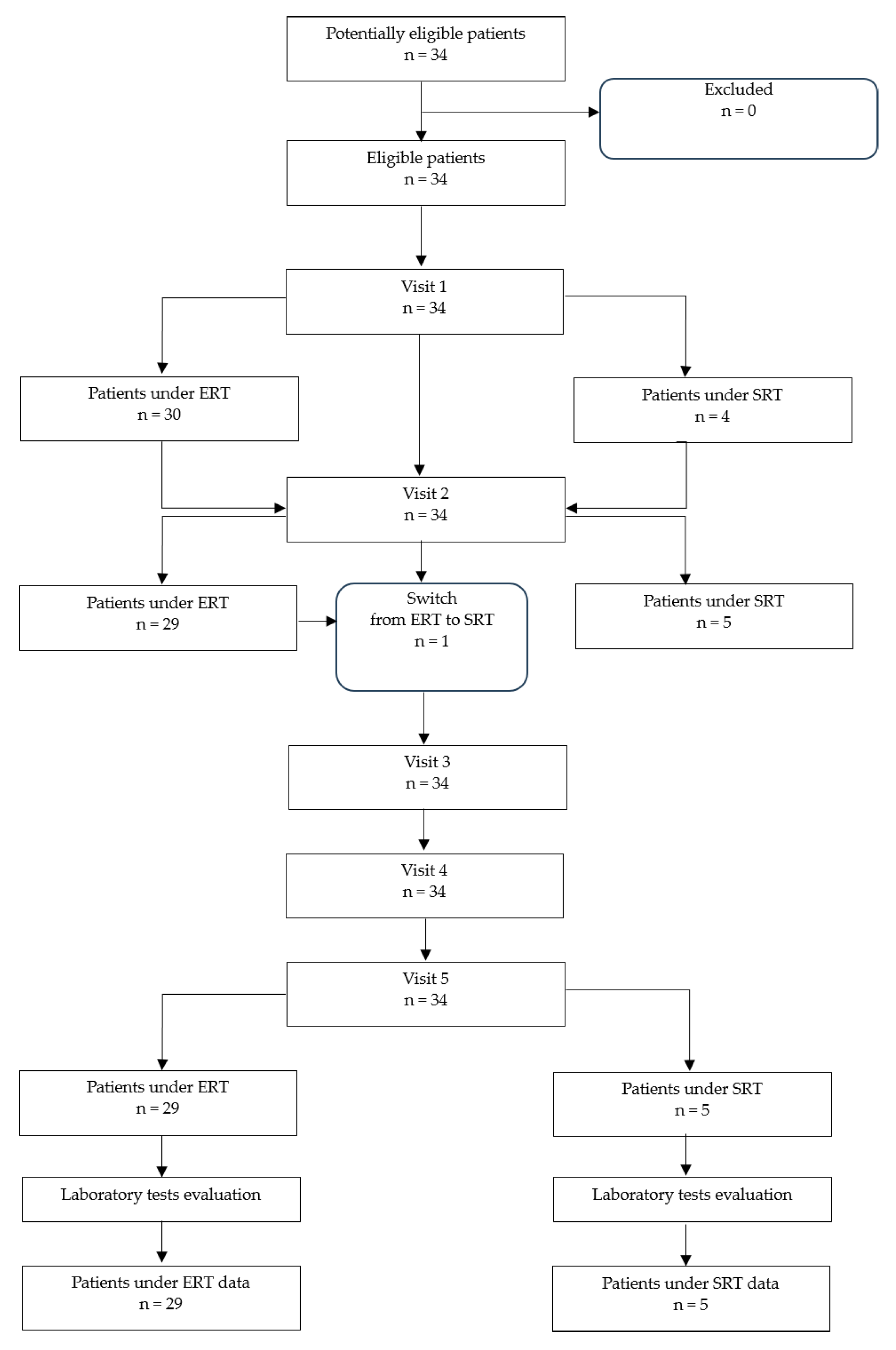{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | Males: 11 (31.4%) | Females: 23 (68.6%) | ||
| Age (mean, yrs) | 42.9 (3–69) | |||
| Genotype | Asn409Ser/Asn409Ser: 1 (2.9%) | Asn409Ser/Leu483Pro: 9 (26.5%) | Asn409Ser/Other or Leu483Pro/Leu483Pro: 24 (70.6%) | |
| Splenectomy | 3 (8.6%) | |||
| Therapy | ERT (n = 29) | SRT (n = 5) | ||
| CHITO (nmol/h/mL) | 608.3 ± 214.8 | 4846.0 ± 1638.0 | ||
| Lyso-Gb1 (ng/mL) | 81.5 ± 12.4 | 454.9 ± 168.0 | ||
| TRAP (nkat/L) | 60.9 ± 3.5 | 124.4 ± 18.1 | ||
| Platelets (109/L) | 203.9 ± 8.8 | 116.6 ± 26.7 | ||
| Patient No. | Gender | Age (Years) | GBA Genotype (NM_001005741.3; NP_001005741.1) $ | CHIT1 Genotype (NM_003465.3) | Initial Visit | Follow-Up |
|---|---|---|---|---|---|---|
| 1 | M | 37 | p.[Asn409Ser];[Leu483Pro] | p.[Gly102Ser];[=] | Cerdelga® 168 mg/D | Cerdelga® 168 mg/D |
| 2 | F | 54 | p.[Asn409Ser]; [Asp448His;Leu483Pro; Ala495Pro;Val499Val] | p.[Gly102Ser];[=] | Cerezyme® 24 U/kg/EOW | Cerezyme® 26 U/kg/EOW |
| 3 | F | 41 | p.[Asn409Ser]; c.[1265_1319del55] | c.[1049_1072dup];[=] | Cerezyme® 20 U/kg/EOW | Cerezyme® 19 U/kg/EOW |
| 4 | M | 59 | p.[Asn409Ser];[Leu483Pro] | p.[Gly102Ser];[=] | Cerdelga® 168 mg/D | Cerdelga® 168 mg/D |
| 5 | M | 10 | p.[Asn409Ser];[Asp448His] | p.[Gly102Ser];[=] | VPRIV® 50 U/kg/EOW | VPRIV® 39 U/kg/EOW |
| 6 | F | 47 | p.[Asn409Ser];[Leu483Pro] | c.1049_1072dup(;) p.Gly102Ser | Cerezyme® 19 U/kg/EOW | Cerezyme® 19 U/kg/EOW |
| 7 | M | 47 | p.[Asn409Ser]; [Asp448His;Leu483Pro; Ala495Pro;Val499Val] | WT | Cerezyme® 24 U/kg/EOW | Cerezyme® 22 U/kg/EOW |
| 8 | M | 10 | p.[His312Asp];[Asn409Ser] | c.[1049_1072dup];[=] | VPRIV® 61 U/kg/EOW | VPRIV® 42 U/kg/EOW |
| 9 | F | 3 | p.[Arg398Term];[Asn409Ser] | N/D | VPRIV® 32 U/kg/EOW | VPRIV® 24 U/kg/EOW |
| 10 | F | 25 | p.[Arg87Gln];[Asn409Ser] | p.[Gly102Ser];[=] | Cerezyme® 22 U/kg/EOW | Cerezyme® 21 U/kg/EOW |
| 11 | F | 63 | p.[Asn409Ser]; [Leu483Pro;Ala495Pro;Val499Val] | N/D | VPRIV® 31 U/kg/EOW | VPRIV® 29 U/kg/EOW |
| 12 * | F | 69 | p.[Asn409Ser];[Arg202Term] | p.[Gly102Ser]; [Gly102Ser] | Cerezyme® 41 U/kg/EOW | Cerezyme® 44 U/kg/EOW |
| 13 | F | 53 | p.[Asn409Ser];[Leu483Pro] | p.[Gly102Ser]; [Gly102Ser](;)Ala442Gly (;)Pro451Ser | Cerezyme® 25 U/kg/EOW | Cerezyme® 25 U/kg/EOW |
| 14 | F | 49 | p.[Asn409Ser];[Leu483Pro] | c.1049_1072dup(;) p.Gly102Ser(;) p.Ala442Gly | Cerezyme® 33 U/kg/EOW | Cerezyme® 29 U/kg/EOW |
| 15 | M | 29 | p.[ Asn409Ser];c.[115+1G>A] | WT | Cerezyme® 24 U/kg/EOW | Cerezyme® 22 U/kg/EOW |
| 16 | M | 33 | p.[Asn409Ser];[Leu483Pro] | WT | Cerezyme® 18 U/kg/EOW | Cerezyme® 18 U/kg/EOW |
| 17 * | F | 59 | p.[Asn409Ser];[Leu483Pro] | WT | Cerezyme® 26 U/kg/EOW | Cerezyme® 26 U/kg/EOW |
| 18 | M | 28 | p.[Asn409Ser];[Gly416Ser] | p.Gly102Ser(;) p.Ala442Gly | Cerezyme® 17 U/kg/EOW | Cerezyme® 17 U/kg/EOW |
| 19 # | M | 32 | p.[Leu483Pro];[Leu483Pro] | c.[1049_1072dup];[=] | Cerezyme® 41 U/kg/EOW | Cerezyme® 44 U/kg/EOW |
| 20 | F | 31 | p.[Asn409Ser];[Leu483Pro, Val499Val] | c.[1049_1072dup];[=] | Cerezyme® 24 U/kg/EOW | Cerezyme® 25 U/kg/EOW |
| 21 | M | 42 | p.[Asn409Ser];c.[1326dupT] | p.[Gly102Ser];[=] | VPRIV® 28 U/kg/EOW | Cerdelga® 28 U/kg/EOW |
| 22 * | F | 56 | p.[Asn409Ser]; c.[1265_1319del55] | WT | VPRIV® 23 U/kg/EOW | VPRIV® 22 U/kg/EOW |
| 23 | F | 66 | p.[ Asn409Ser];[Gly241Glu] | p.[Gly102Ser];[=] | Cerezyme® 15 U/kg/EOW | Cerezyme® 15 U/kg/EOW |
| 24 | F | 19 | c. [Gly228Term]; p.[Asn409Ser] | N/D | Cerezyme® 40 U/kg/EOW | Cerezyme® 37 U/kg/EOW |
| 25 | F | 57 | p.[Asn409Ser];[Leu483Pro; Ala495Pro;Val499Val] | c.[1049_1072dup];[=] | VPRIV® 26 U/kg/EOW | VPRIV® 25 U/kg/EOW |
| 26 | F | 33 | p.[Asn409Ser];[Ser235Pro] | p.[Gly102Ser];[=] | Cerezyme® 18 U/kg/EOW | Cerezyme® 16 U/kg/EOW |
| 27 | F | 46 | p.[Asn409Ser];[Leu483Pro; Ala495Pro;Val499Val] | p.[Gly102Ser];[=] | Zavesca® 200 mg/D | Zavesca® 200 mg/D |
| 28 | F | 59 | p.[ Asn409Ser];c.[115+1G>A] | c.[1049_1072dup];[=] | VPRIV® 28 U/kg/EOW | VPRIV® 25 U/kg/EOW |
| 29 | F | 69 | p.[Asn409Ser];[Leu483Pro; Ala495Pro;Val499Val] | c.[1049_1072dup];[=] | Cerezyme® 34 U/kg/EOW | Cerezyme® 35 U/kg/EOW |
| 30 | M | 45 | p.[Asn409Ser]; c.[1265_1319del55] | c.[1049_1072dup];[=] | VPRIV® 28 U/kg/EOW | VPRIV® 29 U/kg/EOW |
| 31 | F | 45 | p.[Asn409Ser];[Leu483Pro; Ala495Pro;Val499Val] | p.[Gly102Ser];[=] | Cerezyme® 28 U/kg/EOW | Cerezyme® 29 U/kg/EOW |
| 32 | F | 46 | p.[Arg398Gln ];[Asn409Ser] | WT | Cerezyme® 23 U/kg/EOW | Cerezyme® 28 U/kg/EOW |
| 33 | F | 39 | p.[Asn409Ser];[Leu483Pro] | p.[Gly102Ser];[=] | Cerdelga® 164 mg/D | Cerdelga® 164 mg/D |
| 34 | F | 54 | p.[Asn409Ser];[Asn409Ser] | p.[Ser308Ile];[=] | Cerezyme® 26 U/kg/EOW | Cerezyme® 24 U/kg/EOW |
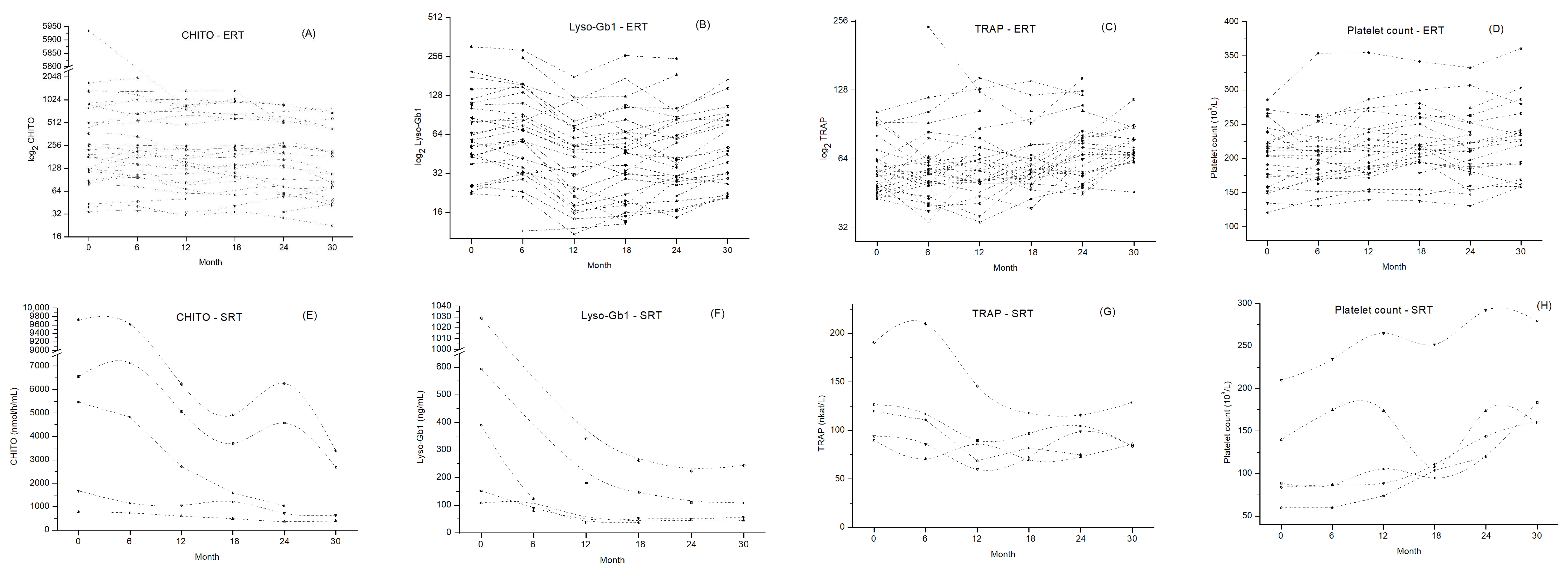
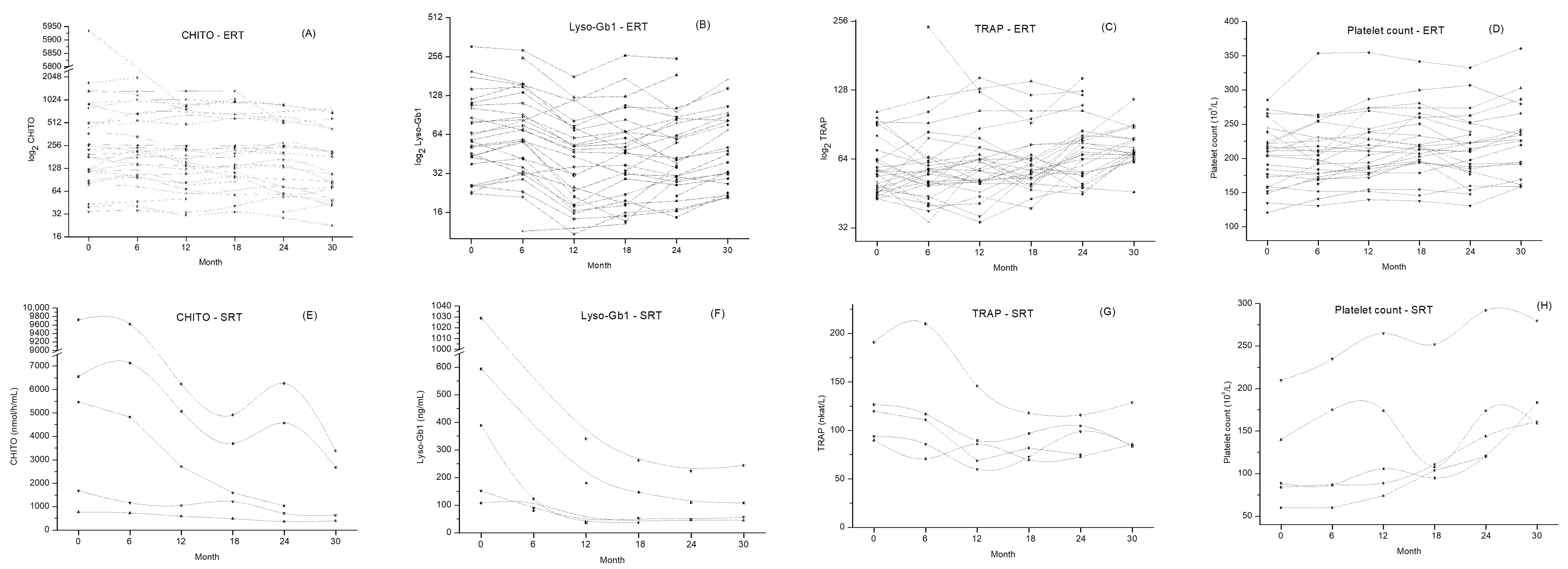
2.2. Biomarker Evolution during Study Period
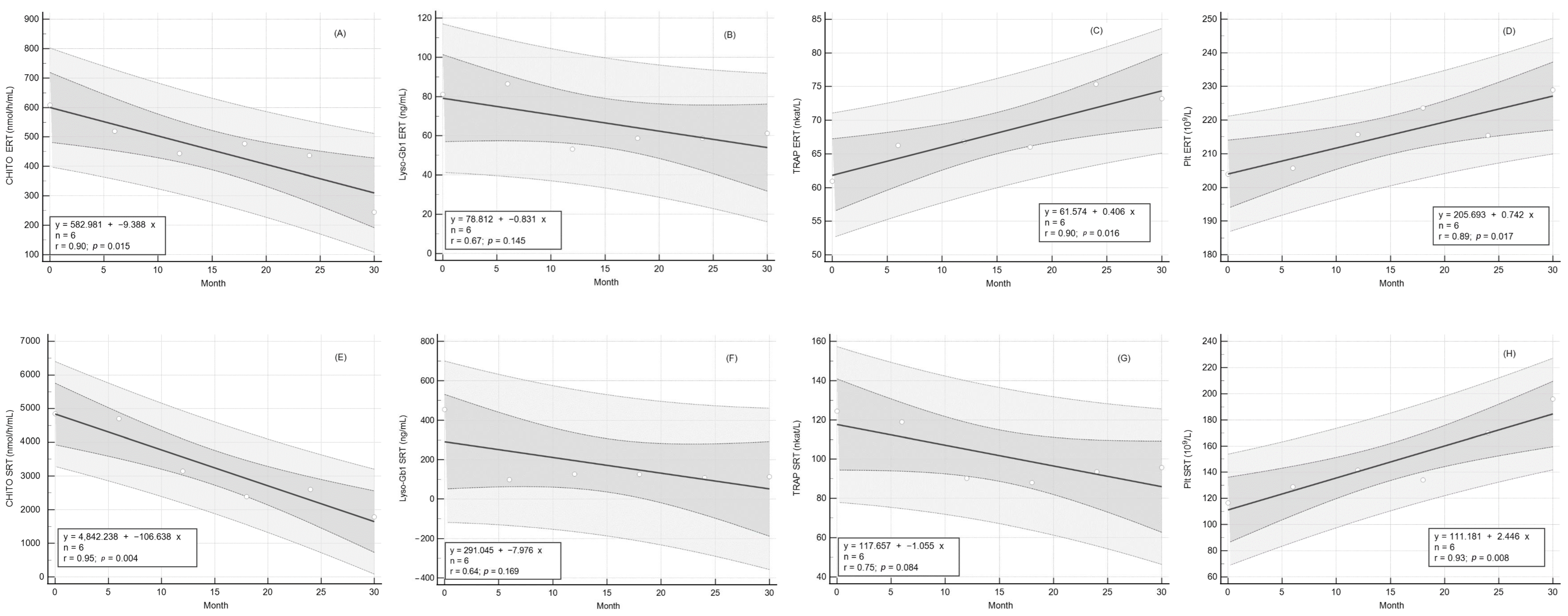
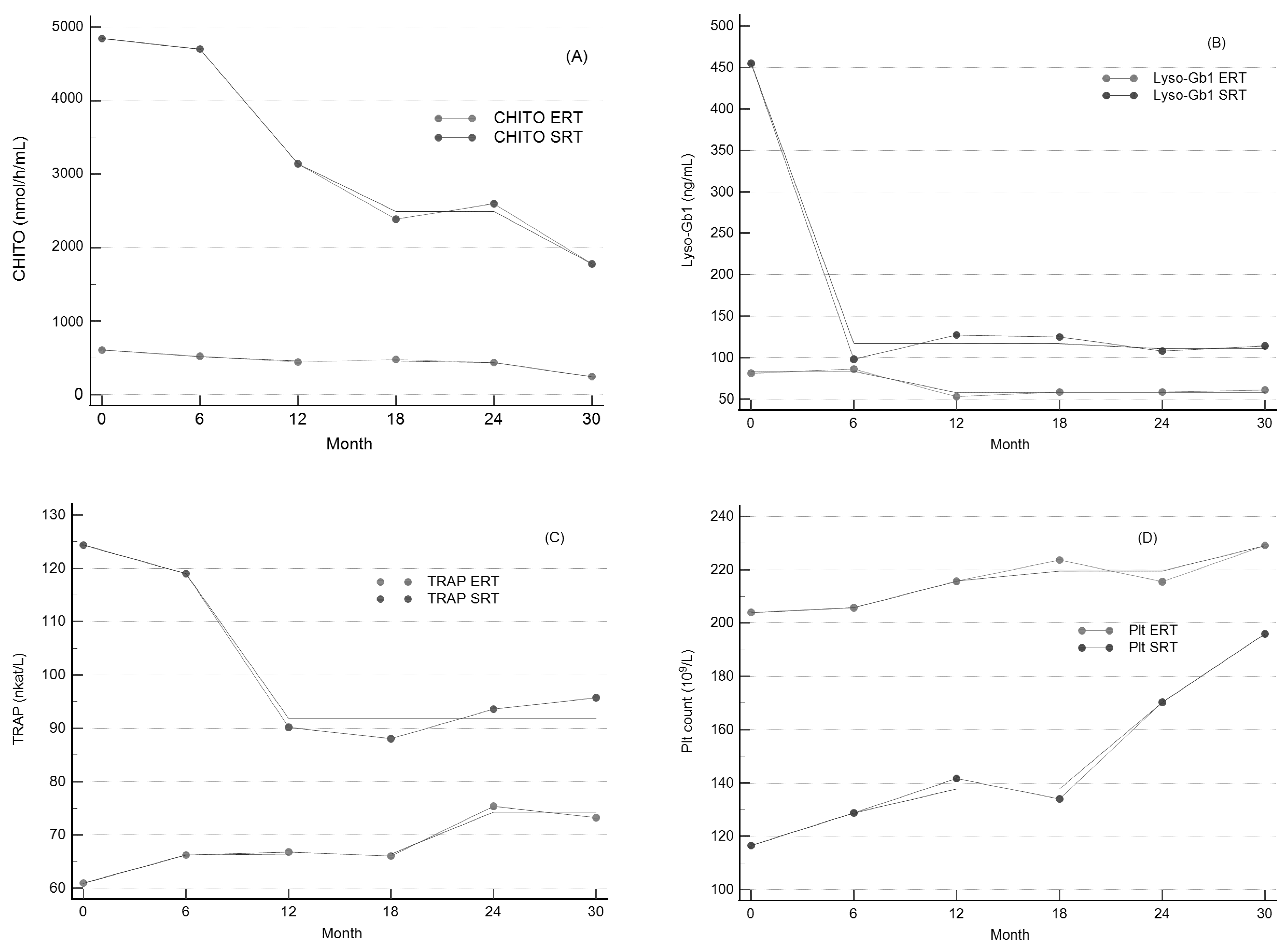
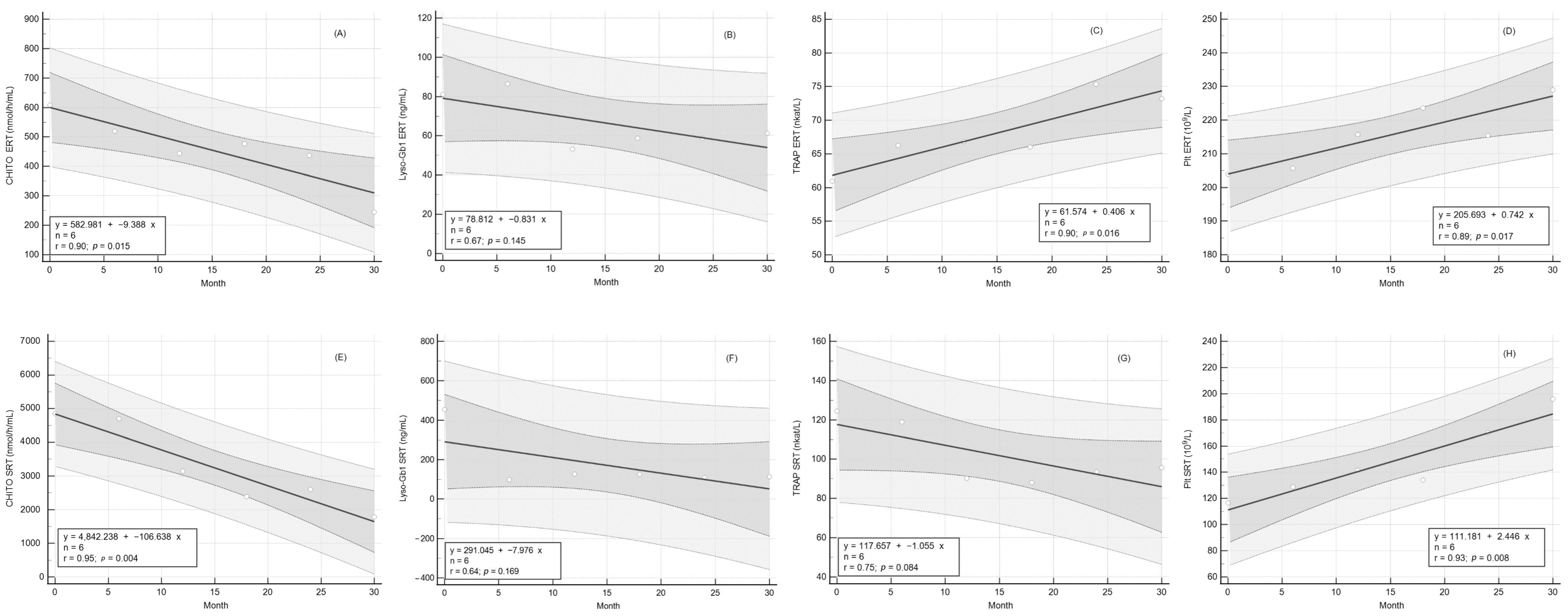
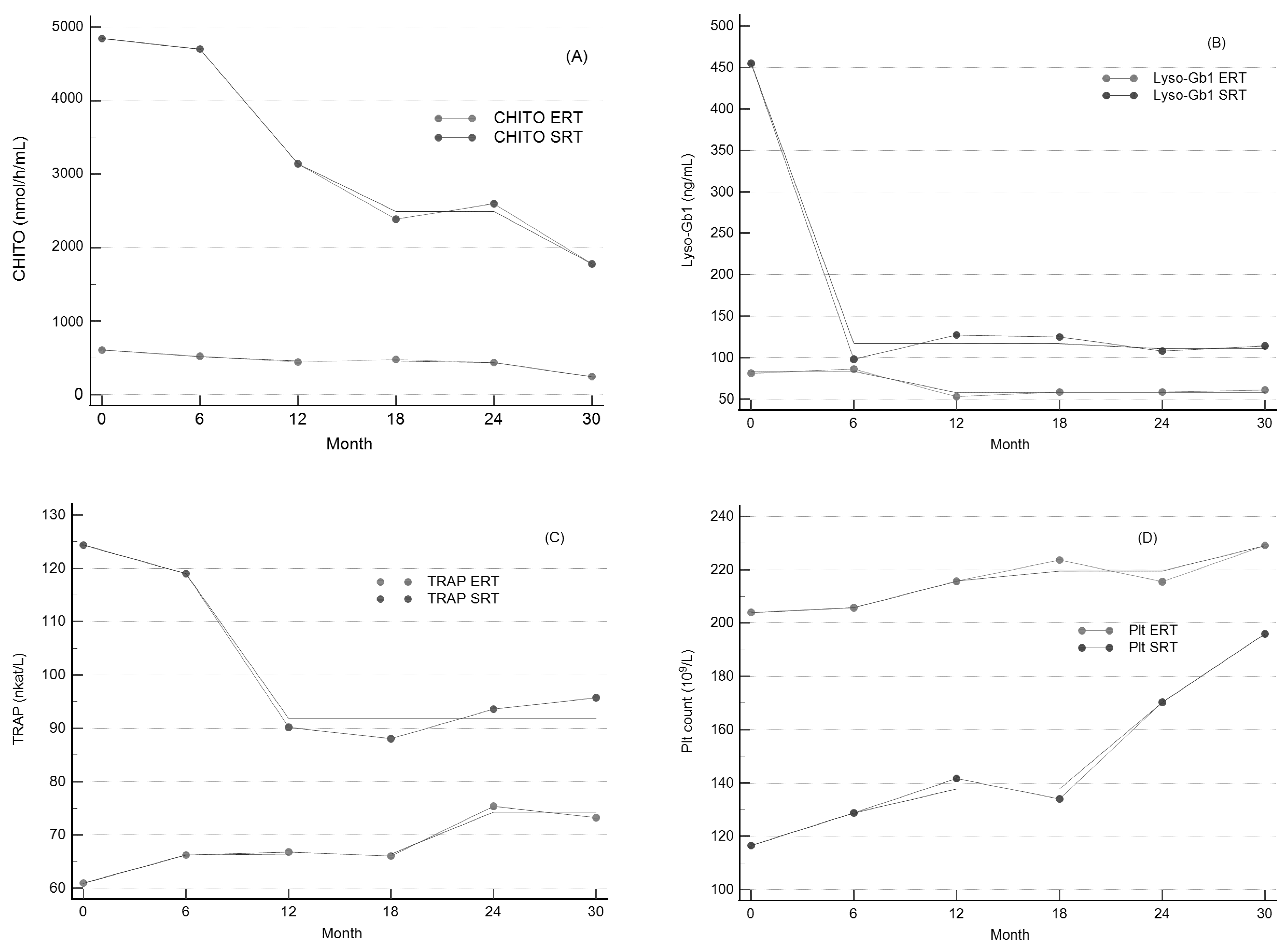
2.3. Response of CHITO Activity to Long-Term Therapy
2.4. Response of Lyso-Gb1 Levels to Long-Term Therapy
2.5. Response of TRAP Activity to Long Term Therapy
2.6. Response of Platelet Count to Long-Term Therapy
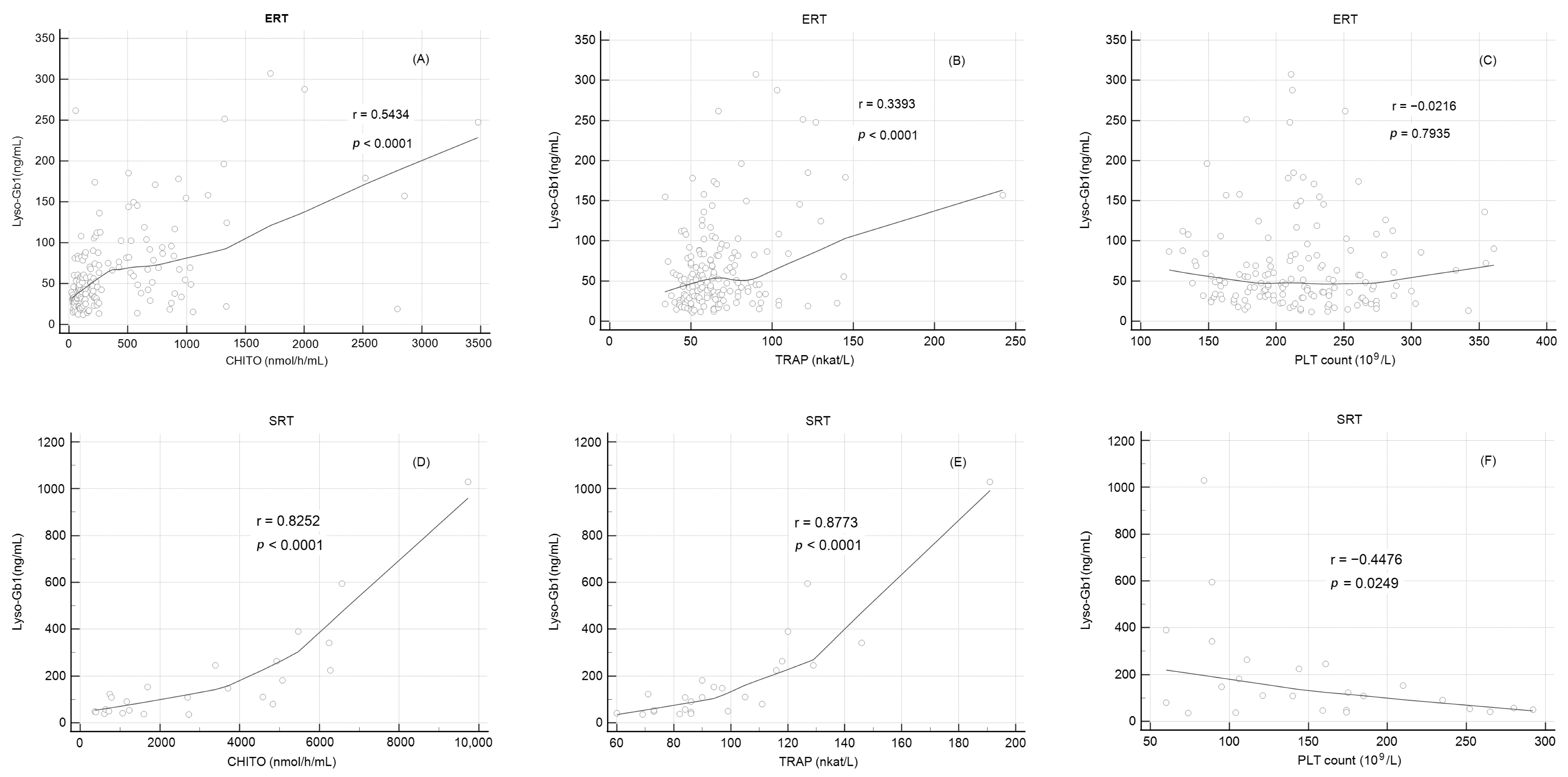
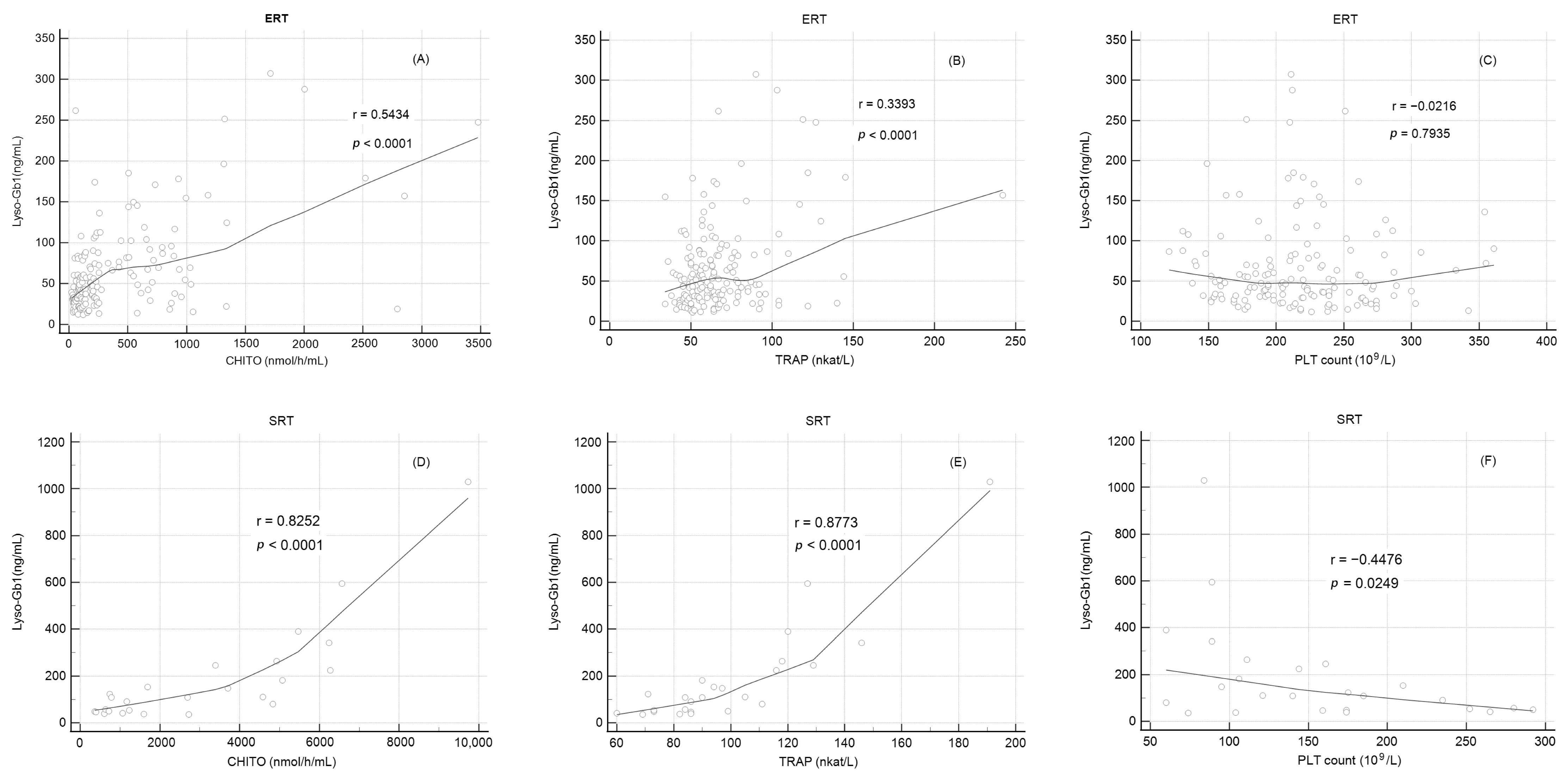
2.7. Correlation of Lyso-Gb1 Levels with Other Biomarkers
3. Discussion
4. Materials and Methods
4.1. Biochemical Plasma Markers
4.1.1. CHITO Activity
4.1.2. Chit-1 Genotyping
4.1.3. Lyso-Gb1
4.1.4. TRAP Activity
4.2. Statistical Analysis
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaplan, P.; Andersson, H.C.; Kacena, K.A.; Yee, J.D. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch. Pediatr. Adolescent Med. 2006, 160, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Thaler, A.; Gurevich, T.; Bar Shira, A.; Gana Weisz, M.; Ash, E.; Shiner, T.; Orr-Urtreger, A.; Giladi, N.; Mirelman, A. A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Park. Relat. Disord. 2017, 36, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Tayebi, N.; Woo, E.G.; Lopez, G.; Fierro, L.; Toffoli, M.; Limbachiya, N.; Hughes, D.; Pitz, V.; Patel, D.; et al. Polygenic Parkinson’s Disease Genetic Risk Score as Risk Modifier of Parkinsonism in Gaucher Disease. Mov. Disord. 2023, 38, 899–903. [Google Scholar] [CrossRef]
- Roshan Lal, T.; Sidransky, E. The Spectrum of Neurological Manifestations Associated with Gaucher Disease. Diseases 2017, 5, 10. [Google Scholar] [CrossRef]
- Poupetová, H.; Ledvinová, J.; Berná, L.; Dvoráková, L.; Kozich, V.; Elleder, M. The birth prevalence of lysosomal storage disorders in the Czech Republic: Comparison with data in different populations. J. Inherit. Metab. Dis. 2010, 33, 387–396. [Google Scholar] [CrossRef]
- Zimran, A.; Gelbart, T.; Westwood, B.; Grabowski, G.A.; Beutler, E. High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews. Am. J. Hum. Genet. 1991, 49, 855–859. [Google Scholar]
- Charrow, J.; Fraga, C.; Gu, X.; Ida, H.; Longo, N.; Lukina, E.; Nonino, A.; Gaemers, S.J.M.; Jouvin, M.H.; Li, J.; et al. Once- versus twice-daily dosing of eliglustat in adults with Gaucher disease type 1: The Phase 3, randomized, double-blind EDGE trial. Mol. Genet. Metab. 2018, 123, 347–356. [Google Scholar] [CrossRef]
- Mistry, P.K.; Belmatoug, N.; von Dahl, S.; Giugliani, R. Understanding the natural history of Gaucher disease. Am. J. Hematol. 2015, 90 (Suppl. 1), S6–S11. [Google Scholar] [CrossRef]
- Fairley, C.; Zimran, A.; Phillips, M.; Cizmarik, M.; Yee, J.; Weinreb, N.; Packman, S. Phenotypic heterogeneity of N370S homozygotes with type I Gaucher disease: An analysis of 798 patients from the ICGG Gaucher Registry. J. Inherit. Metab. Dis. 2008, 31, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, P.; López de Frutos, L.; Cebolla, J.J. Biomarker combination is necessary for the assessment of Gaucher disease? Ann. Transl. Med. 2018, 6, S81. [Google Scholar] [CrossRef] [PubMed]
- Boot, R.G.; Renkema, G.H.; Verhoek, M.; Strijland, A.; Bliek, J.; de Meulemeester, T.M.; Mannens, M.M.; Aerts, J.M. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J. Biol. Chem. 1998, 273, 25680–25685. [Google Scholar] [CrossRef]
- Stiles, A.R.; Huggins, E.; Fierro, L.; Jung, S.H.; Balwani, M.; Kishnani, P.S. The role of glucosylsphingosine as an early indicator of disease progression in early symptomatic type 1 Gaucher disease. Mol. Genet. Metab. Rep. 2021, 27, 100729. [Google Scholar] [CrossRef]
- Nilsson, O.; Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef]
- Cabrera-Salazar, M.A.; O’Rourke, E.; Henderson, N.; Wessel, H.; Barranger, J.A. Correlation of surrogate markers of Gaucher disease. Implications for long-term follow up of enzyme replacement therapy. Clin. Chim. Acta 2004, 344, 101–107. [Google Scholar] [CrossRef]
- Aerts, J.M.; Kallemeijn, W.W.; Wegdam, W.; Joao Ferraz, M.; van Breemen, M.J.; Dekker, N.; Kramer, G.; Poorthuis, B.J.; Groener, J.E.; Cox-Brinkman, J.; et al. Biomarkers in the diagnosis of lysosomal storage disorders: Proteins, lipids, and inhibodies. J. Inherit. Metab. Dis. 2011, 34, 605–619. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef] [PubMed]
- Hodaňová, K.; Hřebíček, M.; Červenková, M.; Mrázová, L.; Vepřeková, L.; Zeman, J. Analysis of the beta-glucocerebrosidase gene in Czech and Slovak Gaucher patients: Mutation profile and description of six novel mutant alleles. Blood Cells, Mol. Dis. 1999, 25, 287–298. [Google Scholar] [CrossRef]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Böttcher, T.; Lukas, J.; Hübner, R.; Gölnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef]
- Dao, J.; Goker-Alpan, O.; Limgala, R. Evaluation of disease burden and therapy modifications using glucosylsphingosine (lyso-GL1) in Gaucher disease. Mol. Genet. Metab. 2019, 126, S45. [Google Scholar] [CrossRef]
- Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef]
- Peterschmitt, J.M.; Foster, M.; Zhang, K.; Ji, A.; Cox, G. Correlations between glucosylsphingosine (lyso-GL-1) and baseline disease severity as well as response to treatment in two clinical trials of eliglustat in treatment-naïve adults with type 1 Gaucher disease. Mol. Genet. Metab. 2019, 126, S117. [Google Scholar] [CrossRef]
- Hollak, C.E.; Maas, M.; Aerts, J.M. Clinically relevant therapeutic endpoints in type I Gaucher disease. J. Inherit. Metab. Dis. 2001, 24, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B.; Moran, M.T.; McFarlane, I.; Schofield, J.P.; Boot, R.G.; Aerts, J.M.; Cox, T.M. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cell Mol. Dis. 2005, 35, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.; Lachmann, R.; Hollak, C.; Aerts, J.; van Weely, S.; Hrebicek, M.; Platt, F.; Butters, T.; Dwek, R.; Moyses, C.; et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000, 355, 1481–1485. [Google Scholar] [CrossRef]
- Zimran, A.; Altarescu, G.; Philips, M.; Attias, D.; Jmoudiak, M.; Deeb, M.; Wang, N.; Bhirangi, K.; Cohn, G.M.; Elstein, D. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood 2010, 115, 4651–4656. [Google Scholar] [CrossRef]
- van Dussen, L.; Hendriks, E.J.; Groener, J.E.; Boot, R.G.; Hollak, C.E.; Aerts, J.M. Value of plasma chitotriosidase to assess non-neuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J. Inherit. Metab. Dis. 2014, 37, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Raskovalova, T.; Deegan, P.B.; Yang, R.; Pavlova, E.; Stirnemann, J.; Labarere, J.; Zimran, A.; Mistry, P.K.; Berger, M. Plasma chitotriosidase activity versus CCL18 level for assessing type I Gaucher disease severity: Protocol for a systematic review with meta-analysis of individual participant data. Syst. Rev. 2017, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Vigan, M.; Stirnemann, J.; Caillaud, C.; Froissart, R.; Boutten, A.; Fantin, B.; Belmatoug, N.; Mentré, F. Modeling changes in biomarkers in Gaucher disease patients receiving enzyme replacement therapy using a pathophysiological model. Orphanet J. Rare Dis. 2014, 9, 95. [Google Scholar] [CrossRef]
- Ben Turkia, H.; Riahi, I.; Azzouz, H.; Ladab, S.; Cherif, W.; Ben Chehida, A.; Abdelmoula, M.S.; Caillaud, C.; Chemli, J.; Abdelhak, S.; et al. Phénotype clinique et spectre mutationnel de la maladie de Gaucher pédiatrique en Tunisie. Tunis. Med. 2010, 88, 158–162. [Google Scholar] [PubMed]
- Malaguarnera, L.; Simpore, J.; Prodi, D.A.; Angius, A.; Sassu, A.; Persico, I.; Barone, R.; Musumeci, S. A 24-bp duplication in exon 10 of human chitotriosidase gene from the sub-Saharan to the Mediterranean area: Role of parasitic diseases and environmental conditions. Genes. Immun. 2003, 4, 570–574. [Google Scholar] [CrossRef]
- Grace, M.E.; Balwani, M.; Nazarenko, I.; Prakash-Cheng, A.; Desnick, R.J. Type 1 Gaucher disease: Null and hypomorphic novel chitotriosidase mutations—Implications for diagnosis and therapeutic monitoring. Hum. Mutat. 2007, 28, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Waalen, J.; Crain, K.; Smargon, A.; Beutler, E. Human chitotriosidase polymorphisms G354R and A442V associated with reduced enzyme activity. Blood Cells Mol. Dis. 2007, 39, 353–360. [Google Scholar] [CrossRef]
- Bussink, A.P.; Verhoek, M.; Vreede, J.; Ghauharali-van der Vlugt, K.; Donker-Koopman, W.E.; Sprenger, R.R.; Hollak, C.E.; Aerts, J.M.; Boot, R.G. Common G102S polymorphism in chitotriosidase differentially affects activity towards 4-methylumbelliferyl substrates. FEBS J. 2009, 276, 5678–5688. [Google Scholar] [CrossRef] [PubMed]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker Cohen, M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef]
- Elstein, D.; Mellgard, B.; Dinh, Q.; Lan, L.; Qiu, Y.; Cozma, C.; Eichler, S.; Bottcher, T.; Zimran, A. Reductions in glucosylsphingosine (lyso-Gb1) in treatment-naive and previously treated patients receiving velaglucerase alfa for type 1 Gaucher disease: Data from phase 3 clinical trials. Mol. Genet. Metab. 2017, 122, 113–120. [Google Scholar] [CrossRef]
- Lukina, E.; Watman, N.; Dragosky, M.; Lau, H.; Avila Arreguin, E.; Rosenbaum, H.; Zimran, A.; Foster, M.C.; Gaemers, S.J.M.; Peterschmitt, M.J. Outcomes after 8 years of eliglustat therapy for Gaucher disease type 1: Final results from the Phase 2 trial. Am. J. Hematol. 2019, 94, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, J.; Pinto, R.; Lemos, M.; Sa Miranda, M.C.; Poenaru, L. Age dependency of serum acid phosphatase in controls and Gaucher patients. Enzyme 1984, 32, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Sims, K.B.; Pastores, G.M.; Weinreb, N.J.; Barranger, B.; Rosenbllom, B.E.; Packman, S.; Kaplan, P.; Mankin, H.; Xavier, R.; Angell, J.; et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: Results of a 48-month longitudinal cohort study. Clin. Genet. 2008, 73, 430–440. [Google Scholar] [CrossRef]
- Alfonso, P.; Pampin, S.; Estrada, J.; Rodriguez-Rey, J.C.; Giraldo, P.; Sancho, J.; Pocovi, M. Miglustat (NB-DNJ) works as a chaperone for mutated acid b-glucosidase in cells transfected with several Gaucher disease mutations. Blood Cells Mol. Dis. 2005, 35, 268–276. [Google Scholar] [CrossRef]
- Wraith, J.E.; Imrie, J. New therapies in the management of Niemann-Pick type C disease: Clinical utility of miglustat. Ther. Clin. Risk Manag. 2009, 5, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Radin, N.S.; Shayman, J.A.; Wotring, L.L.; Zipkin, R.E.; Sivakumar, R.; Ruggieri, J.M.; Carson, K.G.; Ganem, B. Structural and stereochemical studies of potent inhibitors of glucosylceramide synthase and tumor cell growth. J. Lipid Res. 1995, 36, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Leng, H.; Zhang, H.; Li, L.; Zhang, S.; Wang, Y.; Ersek, A.; Morris, E.; Sezgin, E.; Lee, Y.H.; Li, Y.; et al. The glycosphingolipid inhibitor eliglustat inhibits autophagy in osteoclasts to increase bone mass and reduce myeloma bone disease. bioRxiv 2021. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Vincent, C.; Fain, O.; Fantin, B.; Mentré, F. Bone events and evolution of biologic markers in Gaucher disease before and during treatment. Arthritis Res. Ther. 2010, 12, R156. [Google Scholar] [CrossRef]
- van Dussen, L.; Lips, P.; Everts, V.E.; Bravenboer, N.; Jansen, I.D.C.; Groener, J.E.M.; Maas, M.; Blokland, J.A.K.; Aerts, M.F.G.; Hollak, C.E. Markers of bone turnover in Gaucher disease: Modeling the evolution of bone disease. J. Clin. Endocrinol. Metab. 2011, 96, 2194–2205. [Google Scholar] [CrossRef]
- Giraldo, P.; Latre, P.; Alfonso, P.; Acedo, A.; Alonso, D.; Barez, A.; Corrales, A.; Franco, R.; Roldan, V.; Serrano, S.; et al. Short-term effect of miglustat in every day clinical use in treatment-naïve or previously treated patients with type 1 Gaucher’s disease. Haematologica 2006, 91, 703–706. [Google Scholar]
- Mistry, P.K.; Davies, S.; Corfield, A.; Dixon, A.K.; Cox, T.M. Successful treatment of bone marrow failure in Gaucher’s disease with low-dose modified glucocerebrosidase. Q. J. Med. 1992, 83, 541–546. [Google Scholar]
- Elstein, D.; Altarescu, G.; Maayan, H.; Phillips, M.; Abrahamov, A.; Hadas-Halpern, I.; Tiomkin, M.; Zimran, A. Booster-effect with velaglucerase alfa in patients with Gaucher disease switched from long-term imiglucerase therapy: Early Access Program results from Jerusalem. Blood Cells Mol. Dis. 2012, 48, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.I.; DeArmey, S.; Cope, H.; Rairikar, M.; Kishnani, P.S. Treatment of profound thrombocytopenia in a patient with Gaucher disease type 1: Is there a role for substrate reduction therapy. Mol. Genet. Metab. Rep. 2017, 12, 82–84. [Google Scholar] [CrossRef]
- Fuller, M.; Szer, J.; Stark, S.; Fletcher, J.M. Rapid, single-phase extraction of glucosylsphingosine from plasma: A universal screening and monitoring tool. Clin. Chim. Acta 2015, 450, 6–10. [Google Scholar] [CrossRef]
- Franco, M.; Reihani, N.; Marin, M.; De Person, M.; Billette de Villemeur, T.; Rose, C.; Colin, Y.; Moussa, F.; Belmatoug, N.; Le Van Kim, C. Effect of velaglucerase alfa enzyme replacement therapy on red blood cell properties in Gaucher disease. Am. J. Hematol. 2017, 92, E561–E563. [Google Scholar] [CrossRef] [PubMed]
- Tylki-Szymańska, A.; Szymańska-Rożek, P.; Hasiński, P.; Ługowska, A. Plasma chitotriosidase activity versus plasma glucosylsphingosine in wide spectrum of Gaucher disease phenotypes—A statistical insight. Mol. Genet. Metab. 2018, 123, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Cohen, P.; West, S.G.; Aiken, L.S. Applied Multiple Regression/Correlation Analysis for the Behavioural Sciences, 3rd ed.; Lawrence Earlbaum Associates: Mahwah, NJ, USA, 2003. [Google Scholar]
| ERT (n = 29) | SRT (n = 5) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Duration of Therapy before Entering the Study |
15.6 Years
(Range 2.0 to 25.0 Years) |
2.8 Years
(Range 0.0 to 11 Years) | ||||||||
| Month 0 | Month 30 | p-Value | Trend | Month 0 | Month 30 | p-Value | Trend | |||
| CHITO (nmol/h/mL) | 608.3 ± 214.8 | 244.7 ± 52.6 | 0.123 | ↓ | 4846.0 ± 1638.0 | 1780.0 ± 743.0 | 0.045 | ↓ | ||
| Lyso-Gb1 (ng/mL) | 81.5 ± 12.4 | 60.9 ± 8.6 | 0.048 | ↓ | 454.9 ± 168.0 | 114.4 ± 45.7 | 0.002 | ↓ | ||
| TRAP (nkat/L) | 60.9 ± 3.5 | 73.5 ± 3.1 | 0.001 | ↑ | 124.4 ± 18.1 | 95.8 ± 11.1 | 0.041 | ↓ | ||
| Platelet (109/L) | 203.9 ± 8.8 | 229.0 ± 12.4 | 0.006 | ↑ | 116.6 ± 26.7 | 196.0 ± 28.6 | 0.028 | ↑ | ||
| Lyso-Gb1 Correlation with CHITO, TRAP, and Platelets Count | ||||||||||
| ERT (n = 30) | SRT (n = 5) | |||||||||
| Lyso-Gb1 | Lyso-Gb1 | |||||||||
| R | p-value | R | p-value | |||||||
| CHITO | 0.54 | <0.0001 | 0.83 | <0.0001 | ||||||
| TRAP | 0.34 | <0.0001 | 0.88 | <0.0001 | ||||||
| Platelets | −0.01 | 0.92 | −0.45 | 0.0249 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malinová, V.; Poupětová, H.; Řeboun, M.; Dvořáková, L.; Reichmannová, S.; Švandová, I.; Murgašová, L.; Kasper, D.C.; Magner, M. Long-Term Evaluation of Biomarkers in the Czech Cohort of Gaucher Patients. Int. J. Mol. Sci. 2023, 24, 14440. https://doi.org/10.3390/ijms241914440
Malinová V, Poupětová H, Řeboun M, Dvořáková L, Reichmannová S, Švandová I, Murgašová L, Kasper DC, Magner M. Long-Term Evaluation of Biomarkers in the Czech Cohort of Gaucher Patients. International Journal of Molecular Sciences. 2023; 24(19):14440. https://doi.org/10.3390/ijms241914440
Chicago/Turabian StyleMalinová, Věra, Helena Poupětová, Martin Řeboun, Lenka Dvořáková, Stella Reichmannová, Ivana Švandová, Lenka Murgašová, David C. Kasper, and Martin Magner. 2023. "Long-Term Evaluation of Biomarkers in the Czech Cohort of Gaucher Patients" International Journal of Molecular Sciences 24, no. 19: 14440. https://doi.org/10.3390/ijms241914440
APA StyleMalinová, V., Poupětová, H., Řeboun, M., Dvořáková, L., Reichmannová, S., Švandová, I., Murgašová, L., Kasper, D. C., & Magner, M. (2023). Long-Term Evaluation of Biomarkers in the Czech Cohort of Gaucher Patients. International Journal of Molecular Sciences, 24(19), 14440. https://doi.org/10.3390/ijms241914440