
A Side-by-Side Comparison of Wildtype and Variant Melanocortin 1 Receptor Signaling with Emphasis on Protection against Oxidative Damage to DNA

 , , , , , , , and
, , , , , , , and 
Abstract
:1. Introduction
2. Results
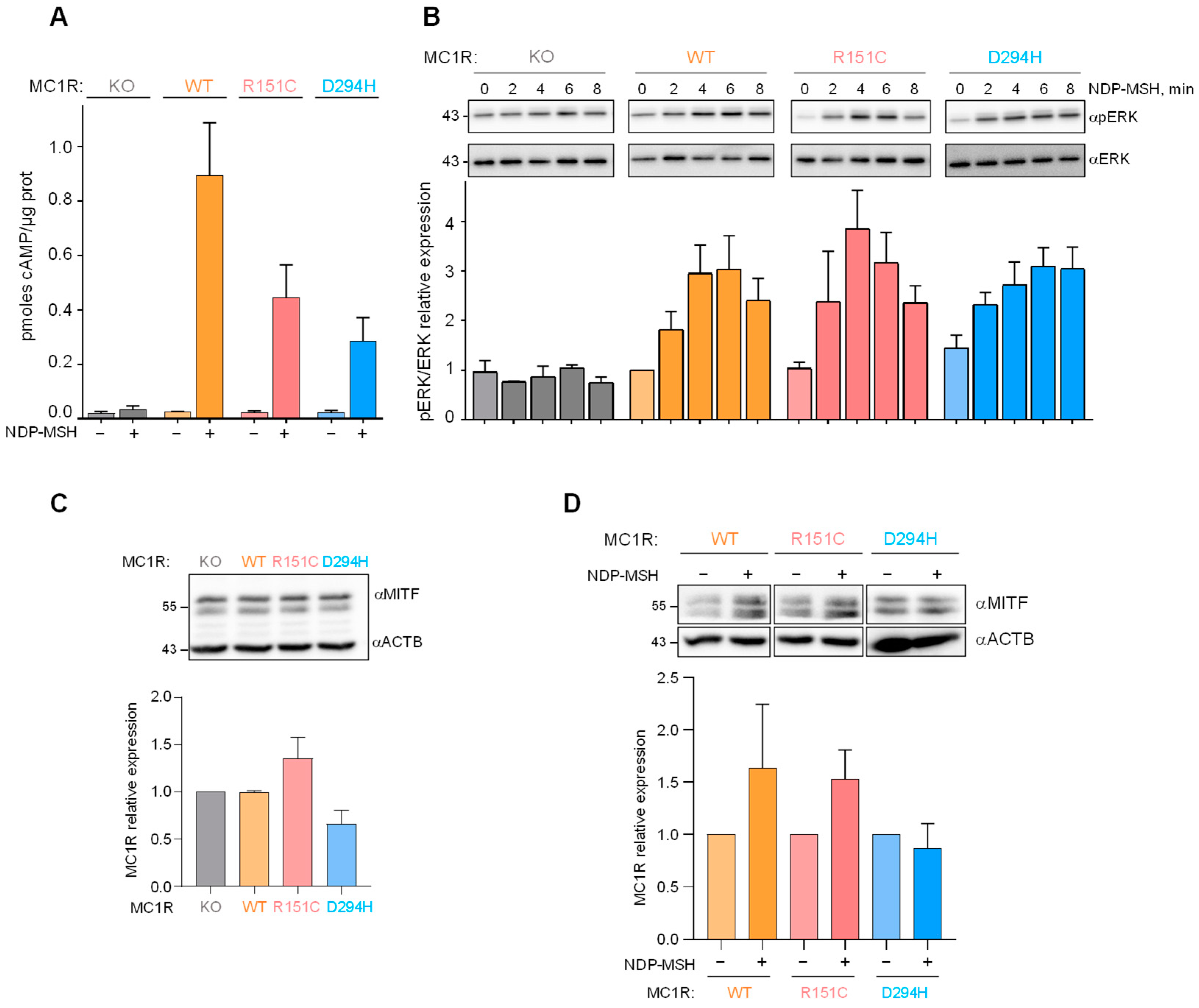
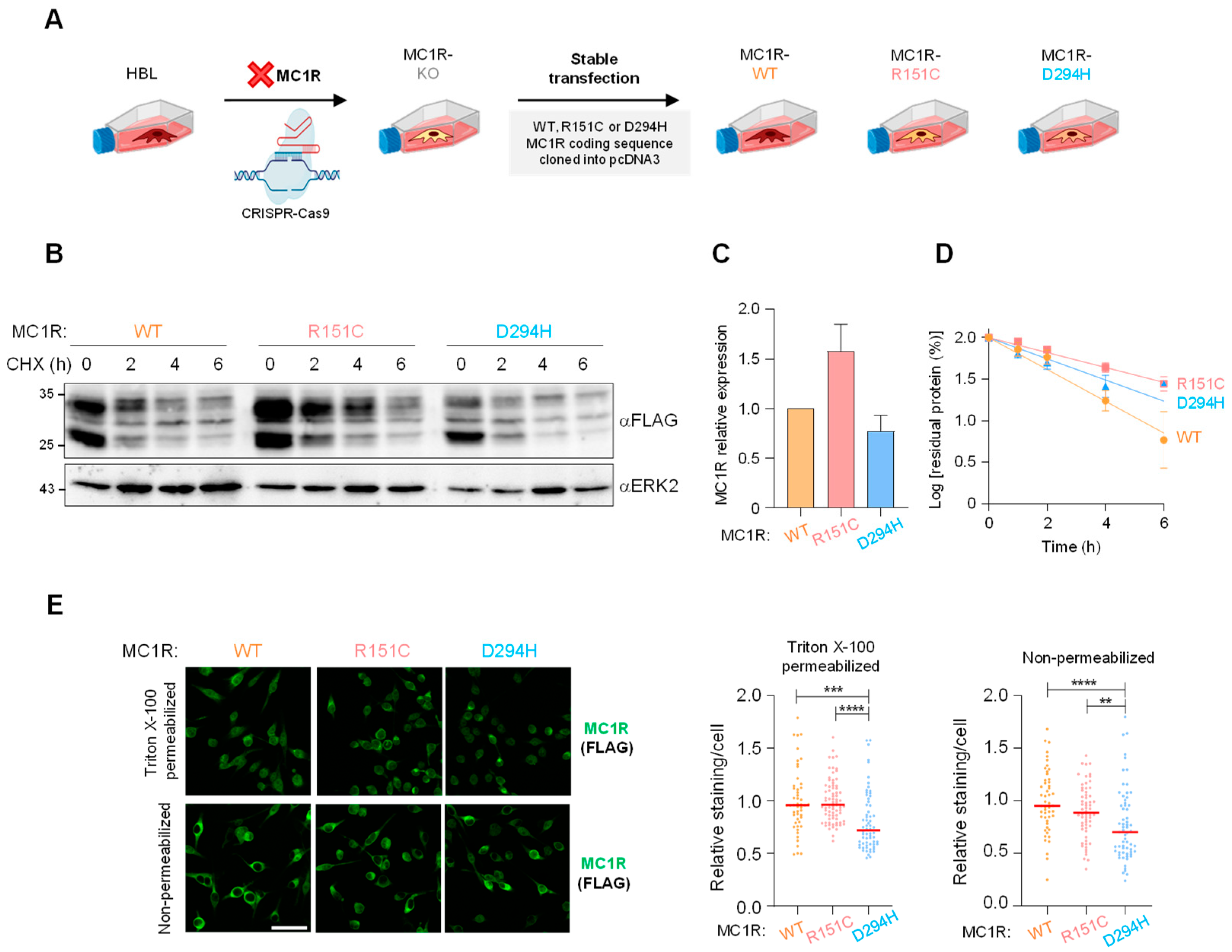
2.1. Generation of MC1R Knockout Cells (MC1R-KO) and Reconstitution with Defined MC1R Variants

2.2. Signaling Downstream of WT and Variant MC1R
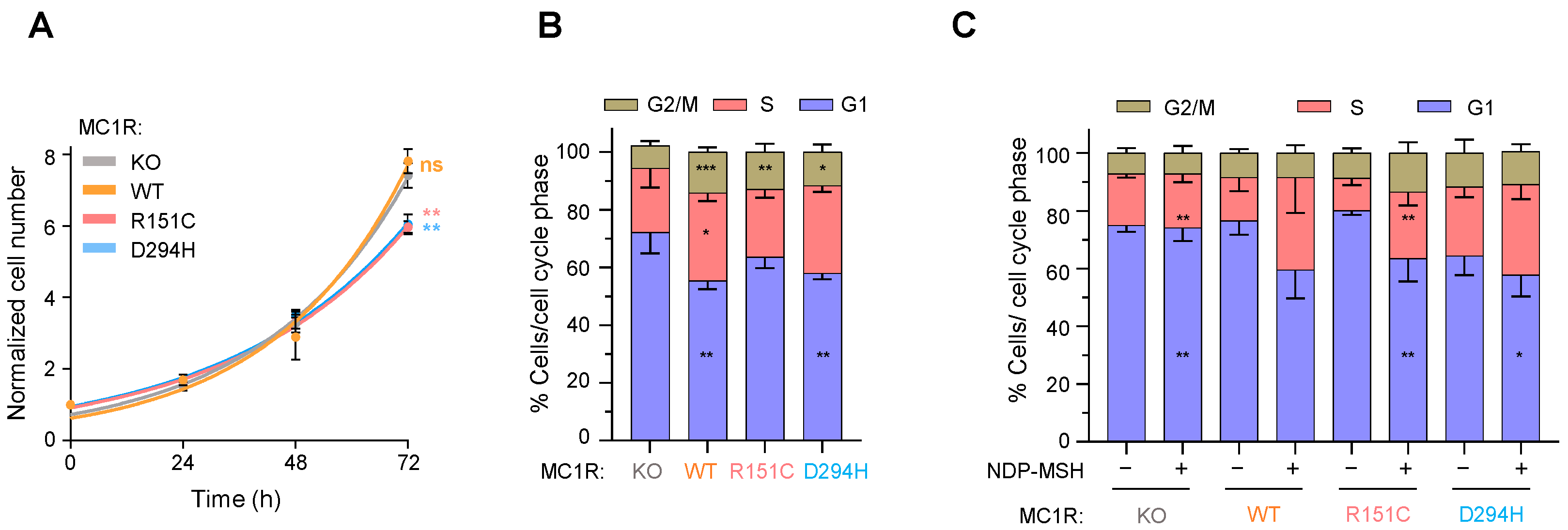
2.3. Effects of MC1R Genotype on Melanoma Cell Proliferation

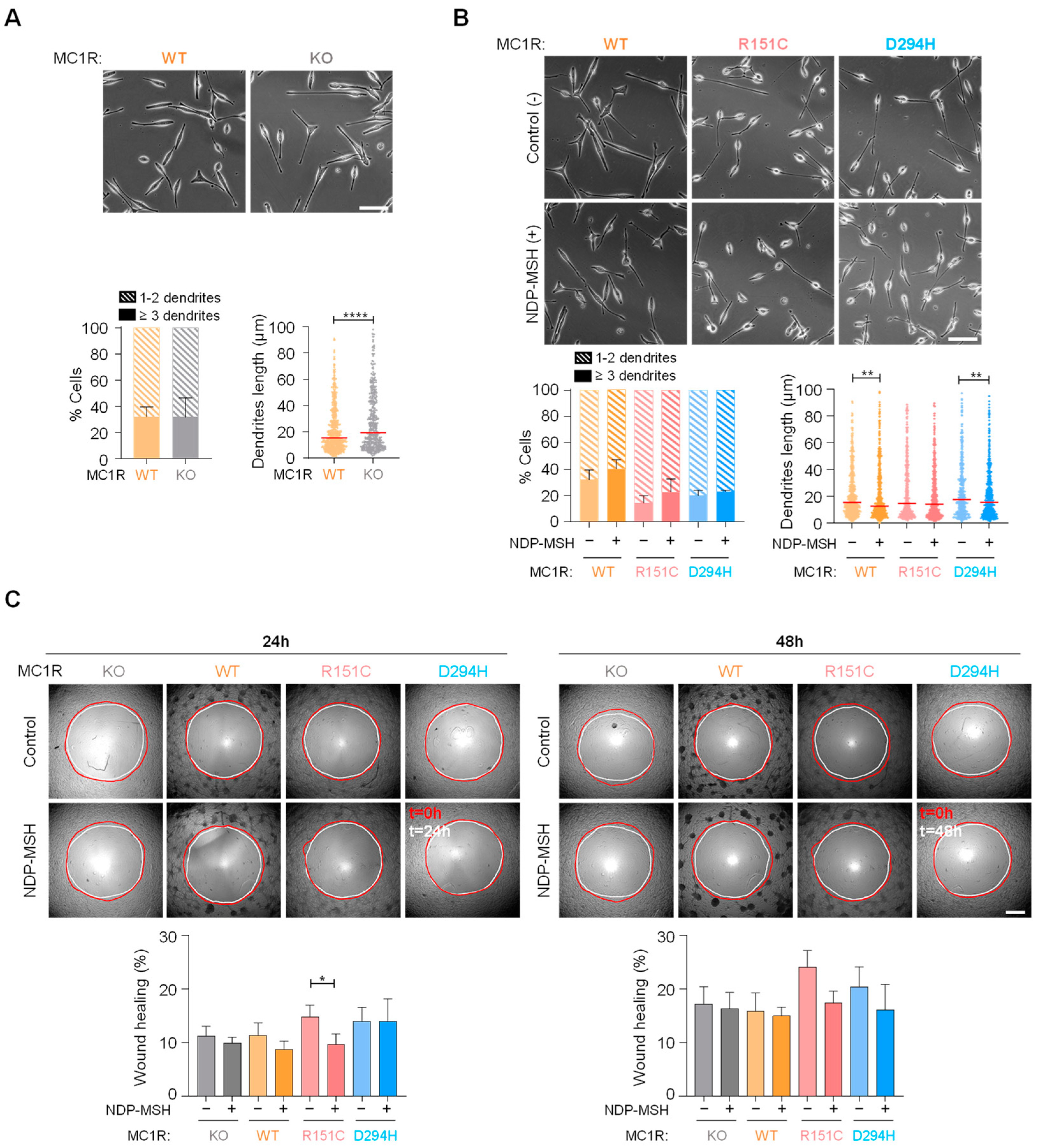
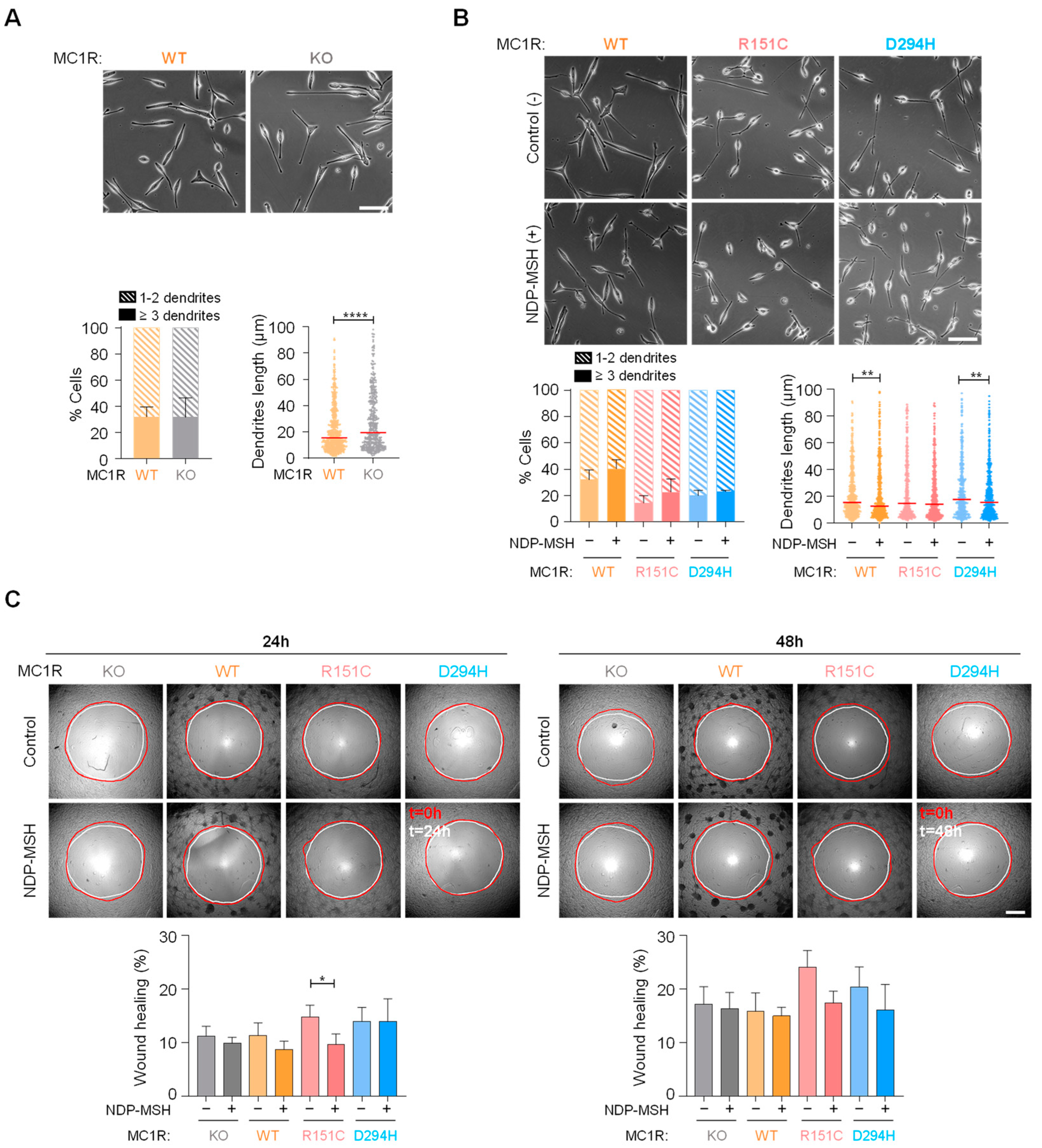
2.4. Effects of MC1R Genotype on Melanoma Cell Shape and Motility
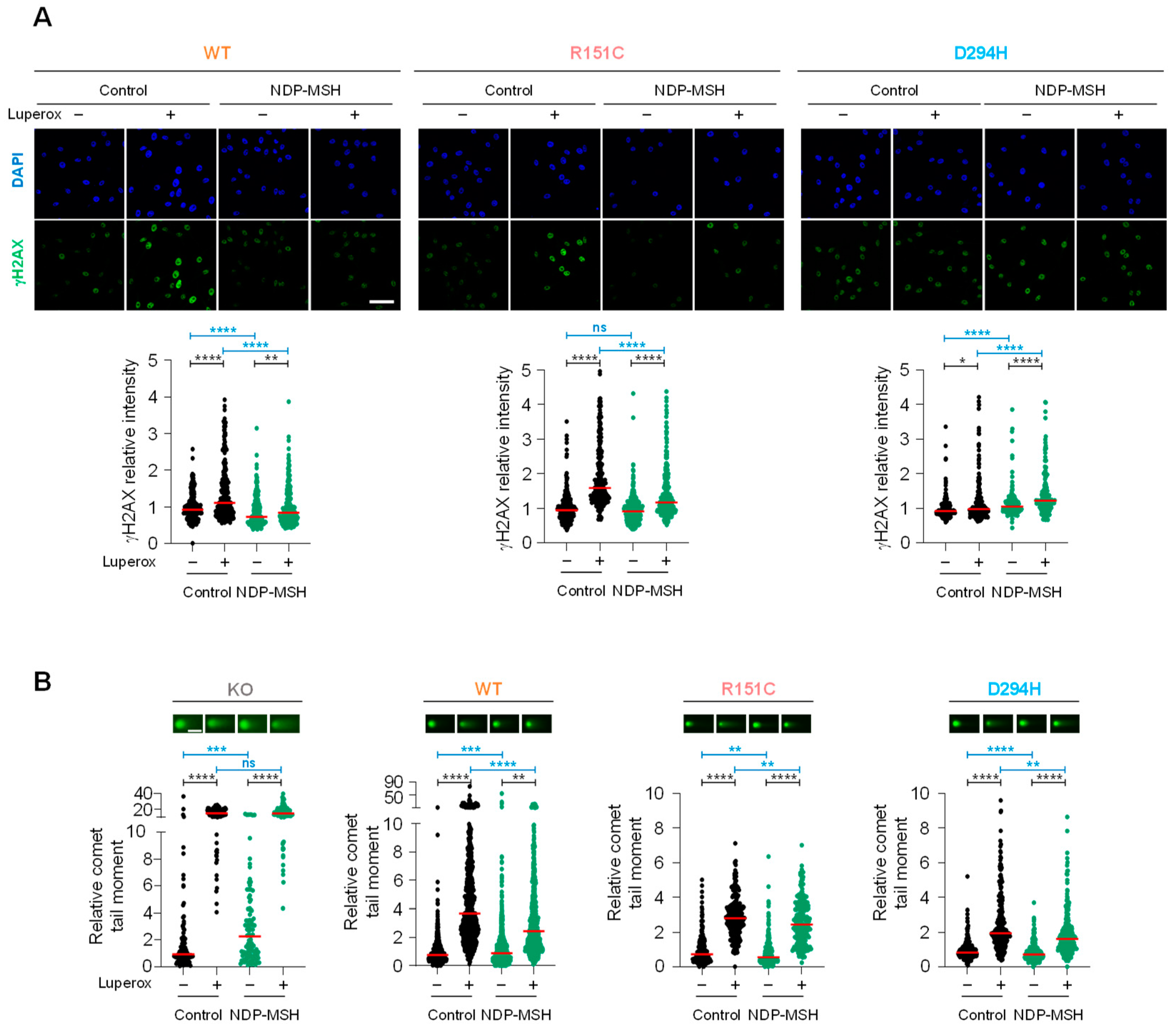
2.5. Protection of DNA from Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Generation of CRISPR/Cas9-Based MC1R-KO Cells and Reconstitution with Defined MC1R Variants
4.3. Cell Culture and Analysis of Proliferation and Cell Cycle Progression
4.4. Analysis of Cell Morphology and Migration
4.5. Analysis of DNA Integrity and Detection of DSBs
4.6. Immunoblotting and Immunofluorescence
4.7. Functional Assays
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| DSB | Double-strand break |
| ERK | Extracellular signal-regulated protein kinase |
| FCS | Fetal calf serum |
| GPCR | G protein-coupled receptor |
| MC1R | Melanocortin 1 receptor |
| αMSH | α-melanocyte-stimulating hormone |
| MITF | Microphthalmia-associated transcription factor |
| NDP-MSH | [Nle4, dphe7]-α-melanocyte-stimulating hormone |
| NER | Nucleotide excision repair |
| PPARγ | Peroxisome proliferator-activated receptor |
| γH2AX | Phosphorylated histone h2ax |
| ROS | Reactive oxygen species |
| SSB | Single-strand break |
| UVR | Ultraviolet solar radiation |
| WT | Wildtype |
References
- Rees, J.L. Genetics of Hair and Skin Color. Annu. Rev. Genet. 2003, 37, 67–90. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Borron, J.C.; Abdel-Malek, Z.; Jimenez-Cervantes, C. MC1R, the CAMP Pathway, and the Response to Solar UV: Extending the Horizon beyond Pigmentation. Pigment Cell Melanoma Res. 2014, 27, 699–720. [Google Scholar] [CrossRef] [PubMed]
- Wolf Horrell, E.M.; Boulanger, M.C.; D’Orazio, J.A. Melanocortin 1 Receptor: Structure, Function, and Regulation. Front. Genet. 2016, 7, 95. [Google Scholar] [CrossRef]
- Manganelli, M.; Guida, S.; Ferretta, A.; Pellacani, G.; Porcelli, L.; Azzariti, A.; Guida, G. Behind the Scene: Exploiting MC1R in Skin Cancer Risk and Prevention. Genes 2021, 12, 1093. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, E.; Gandini, S.; Bellocco, R.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; Lazovich, D.; Kanetsky, P.A.; Ghiorzo, P.; Gruis, N.A.; et al. MC1R Variants as Melanoma Risk Factors Independent of At-Risk Phenotypic Characteristics: A Pooled Analysis from the M-SKIP Project. Cancer Manag. Res. 2018, 10, 1143–1154. [Google Scholar] [CrossRef]
- Wilson, B.D.; Ollmann, M.M.; Kang, L.; Stoffel, M.; Bell, G.I.; Barsh, G.S. Structure and Function of ASP, the Human Homolog of the Mouse Agouti Gene. Hum. Mol. Genet. 1995, 4, 223–230. [Google Scholar] [CrossRef]
- Miller, M.W.; Duhl, D.M.; Vrieling, H.; Cordes, S.P.; Ollmann, M.M.; Winkes, B.M.; Barsh, G.S. Cloning of the Mouse Agouti Gene Predicts a Secreted Protein Ubiquitously Expressed in Mice Carrying the Lethal Yellow Mutation. Genes Dev. 1993, 7, 454–467. [Google Scholar] [CrossRef]
- Candille, S.I.; Kaelin, C.B.; Cattanach, B.M.; Yu, B.; Thompson, D.A.; Nix, M.A.; Kerns, J.A.; Schmutz, S.M.; Millhauser, G.L.; Barsh, G.S. A -Defensin Mutation Causes Black Coat Color in Domestic Dogs. Science 2007, 318, 1418–1423. [Google Scholar] [CrossRef]
- Zhou, D.; Ota, K.; Nardin, C.; Feldman, M.; Widman, A.; Wind, O.; Simon, A.; Reilly, M.; Levin, L.R.; Buck, J.; et al. Mammalian Pigmentation Is Regulated by a Distinct CAMP-Dependent Mechanism That Controls Melanosome PH. Sci. Signal. 2018, 11, eaau7987. [Google Scholar] [CrossRef]
- Yusupova, M.; Zhou, D.; You, J.; Gonzalez-Guzman, J.; Ghanta, M.B.; Pu, H.; Abdel-Malek, Z.; Chen, Q.; Gross, S.S.; D’Orazio, J.; et al. Distinct CAMP Signaling Microdomains Differentially Regulate Melanosomal PH and Pigmentation. J. Investig. Dermatol. 2023; in press. [Google Scholar] [CrossRef]
- Sánchez-Más, J.; Hahmann, C.; Gerritsen, I.; García-Borrón, J.C.; Jiménez-Cervantes, C.; Sanchez-Más, J.; Hahmann, C.; Gerritsen, I.; Garcia-Borrón, J.C.; Jiménez-Cervantes, C. Agonist-Independent, High Constitutive Activity of the Human Melanocortin 1 Receptor. Pigment Cell Res. 2004, 17, 386–395. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome Sequencing Identifies Recurrent Somatic RAC1 Mutations in Melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Noonan, F.P.; Zaidi, M.R.; Wolnicka-Glubisz, A.; Anver, M.R.; Bahn, J.; Wielgus, A.; Cadet, J.; Douki, T.; Mouret, S.; Tucker, M.A.; et al. Melanoma Induction by Ultraviolet A but Not Ultraviolet B Radiation Requires Melanin Pigment. Nat. Commun. 2012, 3, 884. [Google Scholar] [CrossRef] [PubMed]
- Robles-Espinoza, C.D.; Roberts, N.D.; Chen, S.; Leacy, F.P.; Alexandrov, L.B.; Pornputtapong, N.; Halaban, R.; Krauthammer, M.; Cui, R.; Timothy Bishop, D.; et al. Germline MC1R Status Influences Somatic Mutation Burden in Melanoma. Nat. Commun. 2016, 7, 12064. [Google Scholar] [CrossRef]
- Kim, E.; Panzella, L.; Napolitano, A.; Payne, G.F. Redox Activities of Melanins Investigated by Electrochemical Reverse Engineering: Implications for Their Roles in Oxidative Stress. J. Investig. Dermatol. 2020, 140, 537–543. [Google Scholar] [CrossRef]
- Napolitano, A.; Panzella, L.; Monfrecola, G.; d’Ischia, M. Pheomelanin-Induced Oxidative Stress: Bright and Dark Chemistry Bridging Red Hair Phenotype and Melanoma. Pigment Cell Melanoma Res. 2014, 27, 721–733. [Google Scholar] [CrossRef]
- Panzella, L.; Leone, L.; Greco, G.; Vitiello, G.; D’Errico, G.; Napolitano, A.; d’Ischia, M. Red Human Hair Pheomelanin Is a Potent Pro-Oxidant Mediating UV-Independent Contributory Mechanisms of Melanomagenesis. Pigment Cell Melanoma Res. 2014, 27, 244–252. [Google Scholar] [CrossRef]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An Ultraviolet-Radiation-Independent Pathway to Melanoma Carcinogenesis in the Red Hair/Fair Skin Background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef]
- Kadekaro, A.L.; Chen, J.; Yang, J.; Chen, S.; Jameson, J.; Swope, V.B.; Cheng, T.; Kadakia, M.; Abdel-Malek, Z. Alpha-Melanocyte-Stimulating Hormone Suppresses Oxidative Stress through a P53-Mediated Signaling Pathway in Human Melanocytes. Mol. Cancer Res. 2012, 10, 778–786. [Google Scholar] [CrossRef]
- Kadekaro, A.L.; Kavanagh, R.J.; Kanto, H.; Terzieva, S.; Hauser, J.E.; Kobayashi, N.; Schwemberger, S.; Cornelius, J.; Babcock, G.; Shertzer, H.G.; et al. Alpha-Melanocortin and Endothelin-1 Activate Antiapoptotic Pathways and Reduce DNA Damage in Human Melanocytes. Cancer Res. 2005, 65, 4292–4299. [Google Scholar] [CrossRef]
- Mosca, S.; Cardinali, G.; Flori, E.; Briganti, S.; Bottillo, I.; Mileo, A.M.; Maresca, V. The PI3K Pathway Induced by AMSH Exerts a Negative Feedback on Melanogenesis and Contributes to the Release of Pigment. Pigment Cell Melanoma Res. 2021, 34, 72–88. [Google Scholar] [CrossRef] [PubMed]
- Flori, E.; Rosati, E.; Cardinali, G.; Kovacs, D.; Bellei, B.; Picardo, M.; Maresca, V. The α-Melanocyte Stimulating Hormone/Peroxisome Proliferator Activated Receptor-γ Pathway down-Regulates Proliferation in Melanoma Cell Lines. J. Exp. Clin. Cancer Res. 2017, 36, 142. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, G.; Kovacs, D.; Mosca, S.; Bellei, B.; Flori, E.; Morrone, A.; Mileo, A.M.; Maresca, V. The AMSH-Dependent PI3K Pathway Supports Energy Metabolism, via Glucose Uptake, in Melanoma Cells. Cells 2023, 12, 1099. [Google Scholar] [CrossRef] [PubMed]
- Castejón-Griñán, M.; Herraiz, C.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. CAMP-Independent Non-Pigmentary Actions of Variant Melanocortin 1 Receptor: AKT-Mediated Activation of Protective Responses to Oxidative DNA Damage. Oncogene 2018, 37, 3631–3646. [Google Scholar] [CrossRef]
- Herraiz, C.; Jiménez-Cervantes, C.; Zanna, P.; García-Borrón, J.C. Melanocortin 1 Receptor Mutations Impact Differentially on Signalling to the CAMP and the ERK Mitogen-Activated Protein Kinase Pathways. FEBS Lett. 2009, 583, 3269–3274. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Overcoming Resistance to BRAF Inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef]
- Hacker, E.; Boyce, Z.; Kimlin, M.G.; Wockner, L.; Pollak, T.; Vaartjes, S.A.; Hayward, N.K.; Whiteman, D.C. The Effect of MC1R Variants and Sunscreen on the Response of Human Melanocytes in Vivo to Ultraviolet Radiation and Implications for Melanoma. Pigment Cell Melanoma Res. 2013, 26, 835–844. [Google Scholar] [CrossRef]
- Robinson, S.J.; Healy, E. Human Melanocortin 1 Receptor (MC1R) Gene Variants Alter Melanoma Cell Growth and Adhesion to Extracellular Matrix. Oncogene 2002, 21, 8037–8046. [Google Scholar] [CrossRef]
- Halaban, R. The Regulation of Normal Melanocyte Proliferation. Pigment Cell Res. 2000, 13, 4–14. [Google Scholar] [CrossRef]
- Suzuki, I.; Cone, R.D.; Im, S.; Nordlund, J.; Abdel-Malek, Z.A. Binding of Melanotropic Hormones to the Melanocortin Receptor MC1R on Human Melanocytes Stimulates Proliferation and Melanogenesis. Endocrinology 1996, 137, 1627–1633. [Google Scholar] [CrossRef]
- Lyons, J.; Bastian, B.C.; McCormick, F. MC1R and CAMP Signaling Inhibit Cdc25B Activity and Delay Cell Cycle Progression in Melanoma Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13845–13850. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Laorden, B.L.; Sanchez-Mas, J.; Martinez-Alonso, E.; Martinez-Menarguez, J.A.; Garcia-Borron, J.C.; Jimenez-Cervantes, C. Dimerization of the Human Melanocortin 1 Receptor: Functional Consequences and Dominant-Negative Effects. J. Investig. Dermatol. 2006, 126, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, C.; Sánchez-Laorden, B.L.; Jiménez-Cervantes, C.; García-Borrón, J.C. N-Glycosylation of the Human Melanocortin 1 Receptor: Occupancy of Glycosylation Sequons and Functional Role. Pigment Cell Melanoma Res. 2011, 24, 479–489. [Google Scholar] [CrossRef]
- Buntenbroich, I.; Simões, T.; Escobar-Henriques, M. Analysis of Protein Stability by Synthesis Shutoff. Bio-Protocol 2021, 11, e4225. [Google Scholar] [CrossRef]
- Sánchez-Laorden, B.L.; Jiménez-Cervantes, C.; García-Borrón, J.C. Regulation of Human Melanocortin 1 Receptor Signaling and Trafficking by Thr-308 and Ser-316 and Its Alteration in Variant Alleles Associated with Red Hair and Skin Cancer. J. Biol. Chem. 2007, 282, 3241–3251. [Google Scholar] [CrossRef]
- Hsiao, J.J.; Fisher, D.E. The Roles of Microphthalmia-Associated Transcription Factor and Pigmentation in Melanoma. Arch. Biochem. Biophys. 2014, 563, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Goding, C.R.; Arnheiter, H. Mitf—The First 25 Years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef]
- Guida, S.; Guida, G.; Goding, C.R. MC1R Functions, Expression, and Implications for Targeted Therapy. J. Investig. Dermatol. 2022, 142, 293–302.e1. [Google Scholar] [CrossRef]
- Herraiz, C.; Martínez-Vicente, I.; Maresca, V. The α-Melanocyte-Stimulating Hormone/Melanocortin-1 Receptor Interaction: A Driver of Pleiotropic Effects beyond Pigmentation. Pigment Cell Melanoma Res. 2021, 34, 748–761. [Google Scholar] [CrossRef]
- Agarwal, A.; Barbăroșie, C.; Ambar, R.; Finelli, R. The Impact of Single- and Double-Strand DNA Breaks in Human Spermatozoa on Assisted Reproduction. Int. J. Mol. Sci. 2020, 21, 3882. [Google Scholar] [CrossRef]
- Alnajjar, K.S.; Sweasy, J.B. A New Perspective on Oxidation of DNA Repair Proteins and Cancer. DNA Repair 2019, 76, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.F.; Olsen, C.M.; Hayward, N.K.; Whiteman, D.C. Melanocortin 1 Receptor and Risk of Cutaneous Melanoma: A Meta-Analysis and Estimates of Population Burden. Int. J. Cancer 2011, 129, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Urban, K.; Mehrmal, S.; Uppal, P.; Giesey, R.L.; Delost, G.R. The Global Burden of Skin Cancer: A Longitudinal Analysis from the Global Burden of Disease Study, 1990–2017. JAAD Int. 2021, 2, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, K.A.; Shekar, S.L.; Newton, R.A.; James, M.R.; Stow, J.L.; Duffy, D.L.; Sturm, R.A. Receptor Function, Dominant Negative Activity and Phenotype Correlations for MC1R Variant Alleles. Hum. Mol. Genet. 2007, 16, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Hayward, R.; Martin, J.; Ogilvie, L.; Hedley, D.; Curtin, J.A.; Bastian, B.C.; Springer, C.; Marais, R. In Melanoma, RAS Mutations Are Accompanied by Switching Signaling from BRAF to CRAF and Disrupted Cyclic AMP Signaling. Cancer Res. 2006, 66, 9483–9491. [Google Scholar] [CrossRef]
- Beaumont, K.A.; Shekar, S.N.; Cook, A.L.; Duffy, D.L.; Sturm, R.A. Red Hair Is the Null Phenotype of MC1R. Hum. Mutat. 2008, 29, E88–E94. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA Damage Response and Cancer Therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Holcomb, N.C.; Bautista, R.-M.; Jarrett, S.G.; Carter, K.M.; Gober, M.K.; D’Orazio, J.A. CAMP-Mediated Regulation of Melanocyte Genomic Instability: A Melanoma-Preventive Strategy. Adv. Protein Chem. Struct. Biol. 2019, 115, 247–295. [Google Scholar]
- Schuch, A.P.; Moreno, N.C.; Schuch, N.J.; Menck, C.F.M.; Garcia, C.C.M. Sunlight Damage to Cellular DNA: Focus on Oxidatively Generated Lesions. Free Radic. Biol. Med. 2017, 107, 110–124. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Oliveros, J.C.; Franch, M.; Tabas-Madrid, D.; San-León, D.; Montoliu, L.; Cubas, P.; Pazos, F. Breaking-Cas-Interactive Design of Guide RNAs for CRISPR-Cas Experiments for ENSEMBL Genomes. Nucleic Acids Res. 2016, 44, W267–W271. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Culture Conditions | Doubling Time (h) 1 | |||
|---|---|---|---|---|
| MC1R-KO | WT | R151C | D294H | |
| 10% FCS | 21.5 ± 0.7 | 19.8 ± 1.5 | 26.5 ± 1.4 | 26.7 ± 0.6 |
| 1% FCS | 64.6 ± 1.6 * | 45.3 ± 2.8 * | 56.0 ± 3.8 * | 42.7 ± 2.4 * |
| 1% FCS + 100 nM NDP-MSH | 55.8 ± 6.4 * | >100 | 59.1 ± 8.3 | 41.3 ± 3.0 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerdido, S.; Sánchez-Beltrán, J.; Lambertos, A.; Abrisqueta, M.; Padilla, L.; Herraiz, C.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. A Side-by-Side Comparison of Wildtype and Variant Melanocortin 1 Receptor Signaling with Emphasis on Protection against Oxidative Damage to DNA. Int. J. Mol. Sci. 2023, 24, 14381. https://doi.org/10.3390/ijms241814381
Cerdido S, Sánchez-Beltrán J, Lambertos A, Abrisqueta M, Padilla L, Herraiz C, Olivares C, Jiménez-Cervantes C, García-Borrón JC. A Side-by-Side Comparison of Wildtype and Variant Melanocortin 1 Receptor Signaling with Emphasis on Protection against Oxidative Damage to DNA. International Journal of Molecular Sciences. 2023; 24(18):14381. https://doi.org/10.3390/ijms241814381
Chicago/Turabian StyleCerdido, Sonia, José Sánchez-Beltrán, Ana Lambertos, Marta Abrisqueta, Lidia Padilla, Cecilia Herraiz, Conchi Olivares, Celia Jiménez-Cervantes, and José C. García-Borrón. 2023. "A Side-by-Side Comparison of Wildtype and Variant Melanocortin 1 Receptor Signaling with Emphasis on Protection against Oxidative Damage to DNA" International Journal of Molecular Sciences 24, no. 18: 14381. https://doi.org/10.3390/ijms241814381
APA StyleCerdido, S., Sánchez-Beltrán, J., Lambertos, A., Abrisqueta, M., Padilla, L., Herraiz, C., Olivares, C., Jiménez-Cervantes, C., & García-Borrón, J. C. (2023). A Side-by-Side Comparison of Wildtype and Variant Melanocortin 1 Receptor Signaling with Emphasis on Protection against Oxidative Damage to DNA. International Journal of Molecular Sciences, 24(18), 14381. https://doi.org/10.3390/ijms241814381