
Role of Rho/MRTF in Aggressive Vemurafenib-Resistant Murine Melanomas and Immune Checkpoint Upregulation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
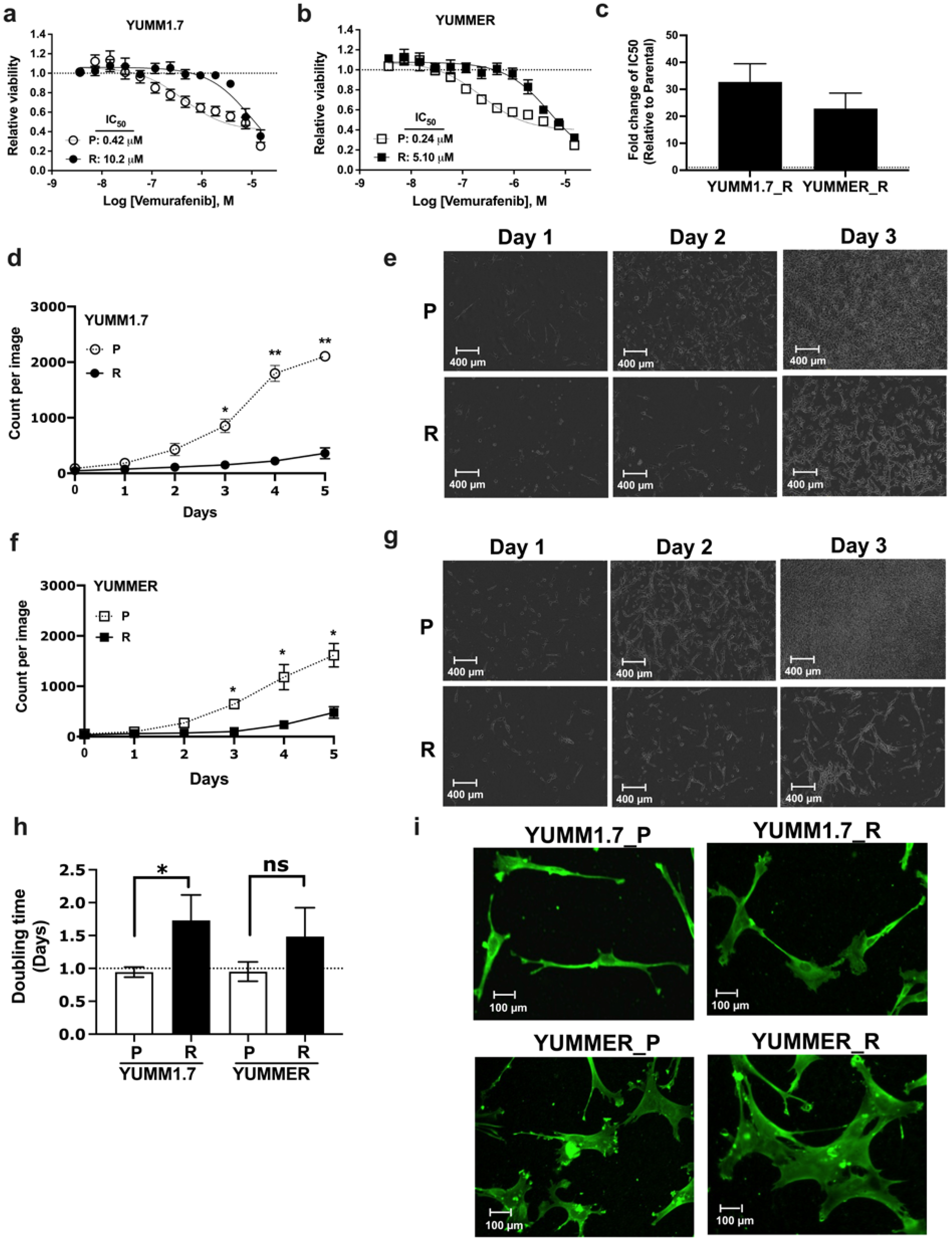
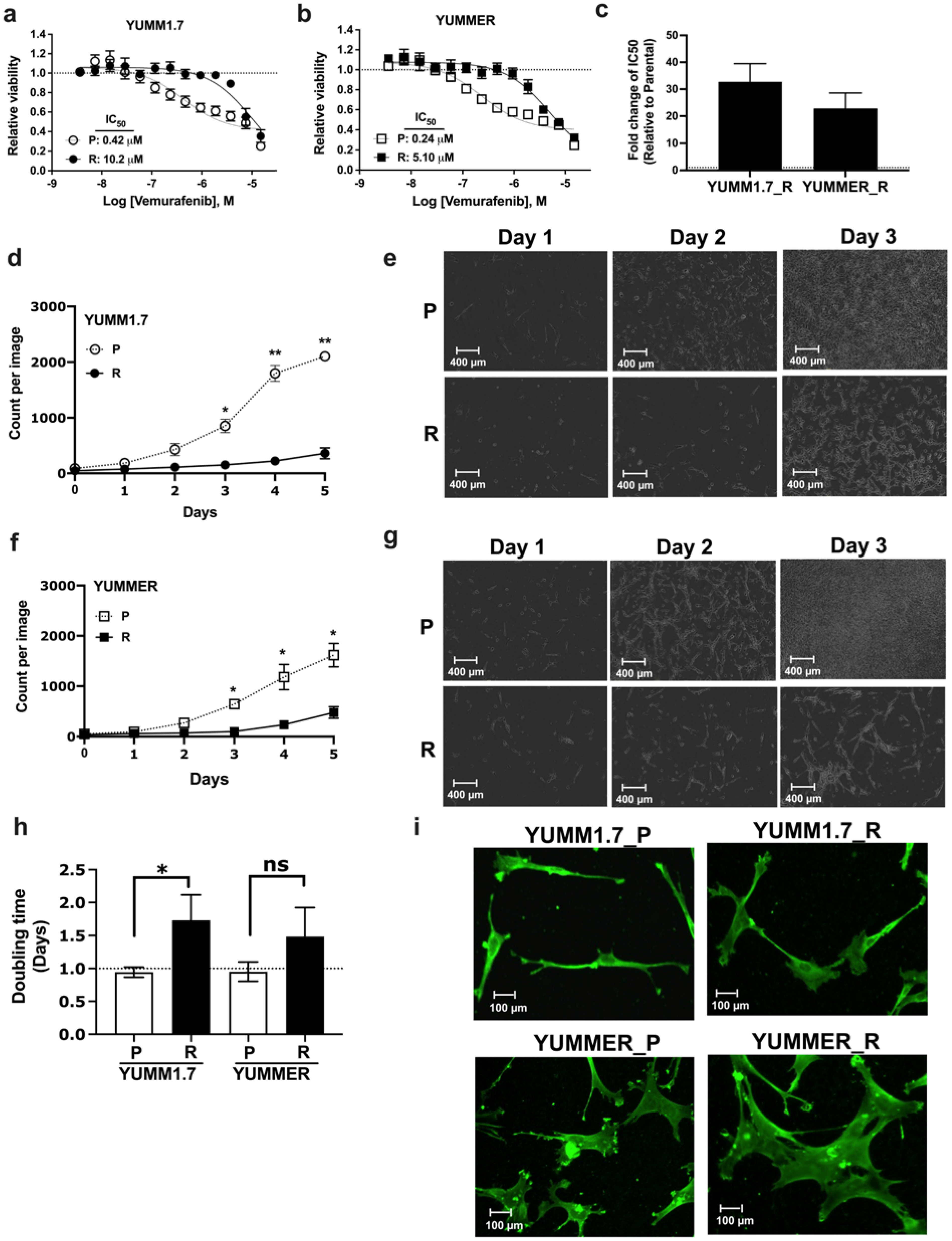
2.1. Generating Vem–Resistant Mouse Melanoma Lines
2.2. Resistant Cells Have Reduced Proliferation Rates and Altered Morphology
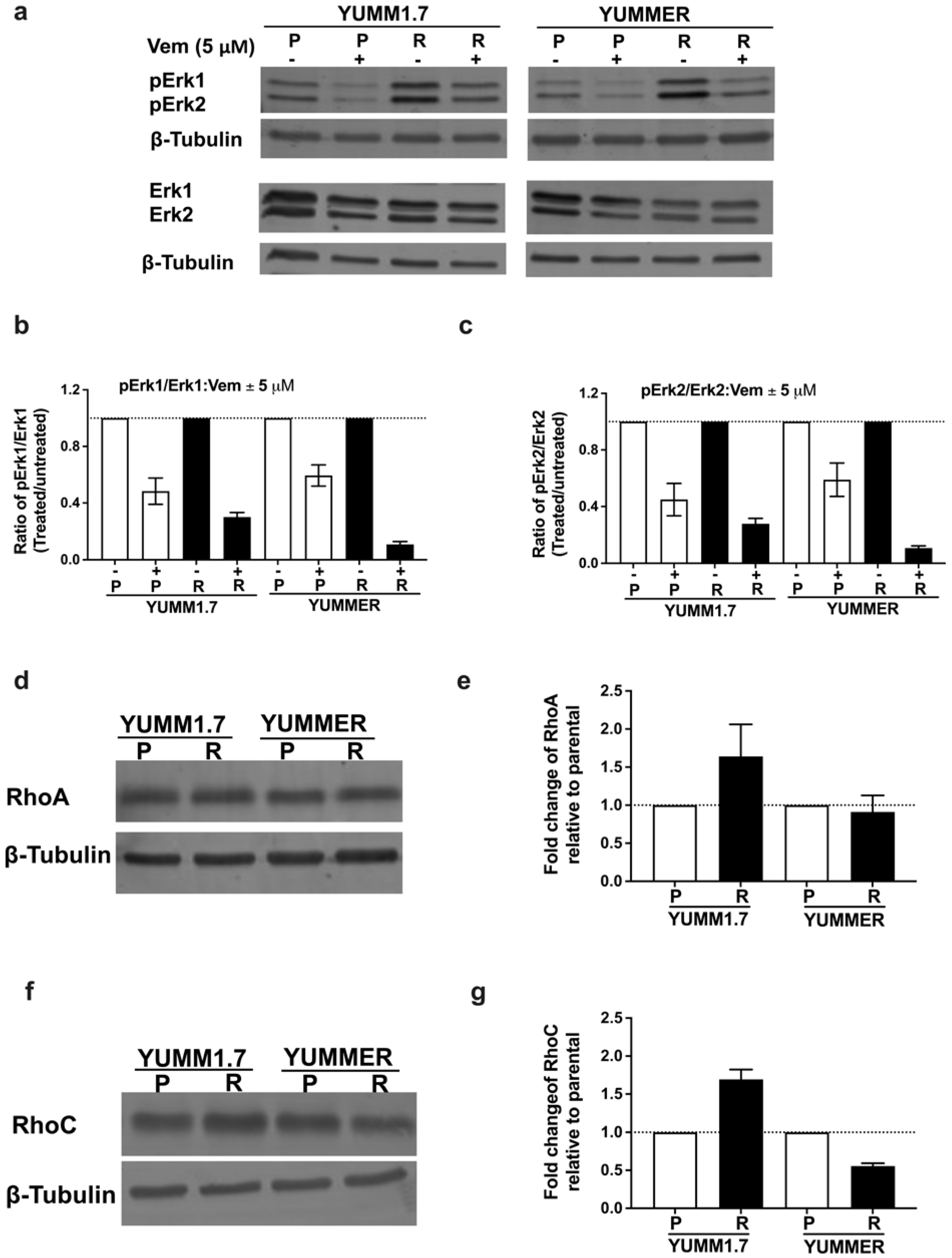
2.3. Vem Insensitivity of Resistant Cells Is Independent of MAPK Reactivation
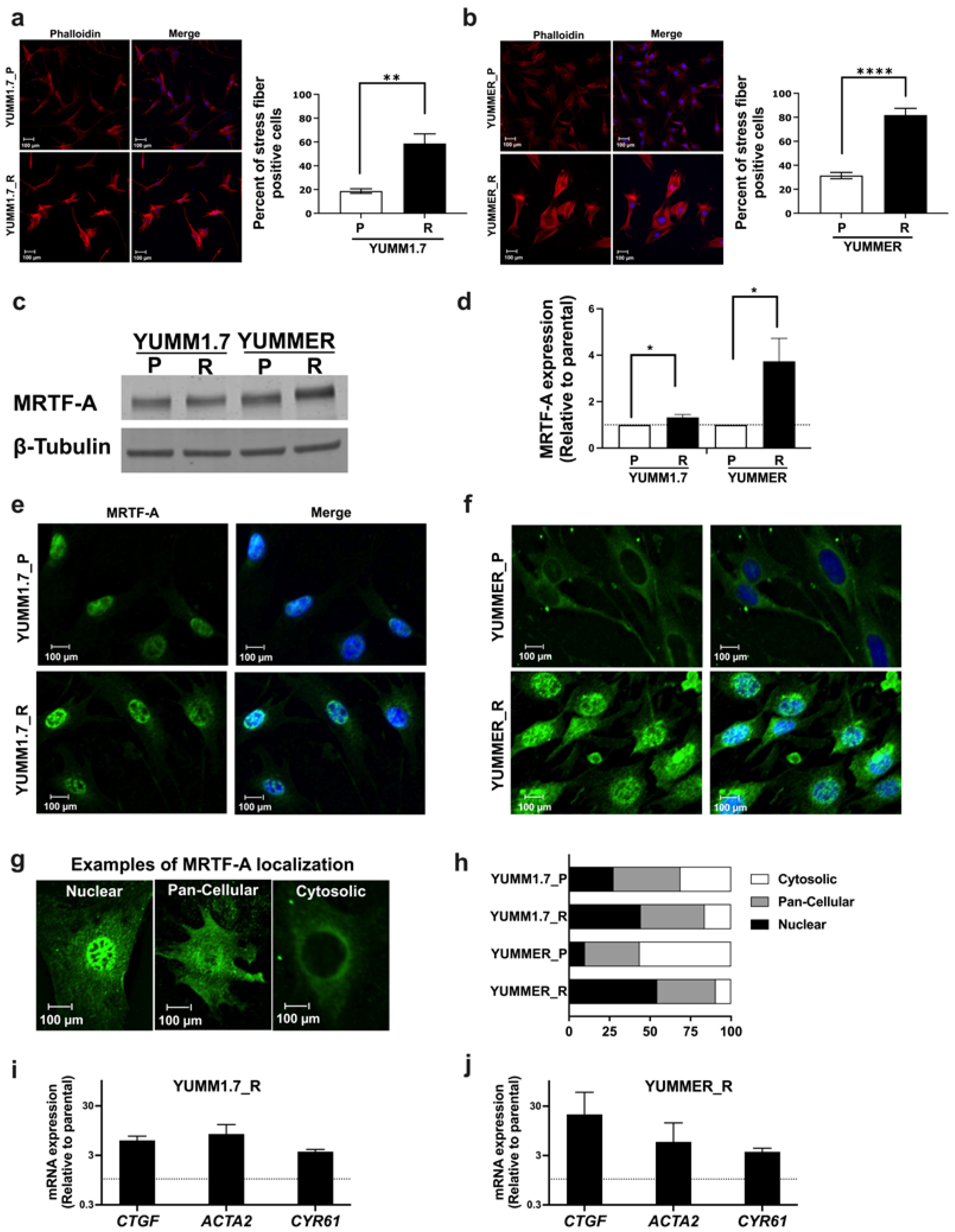
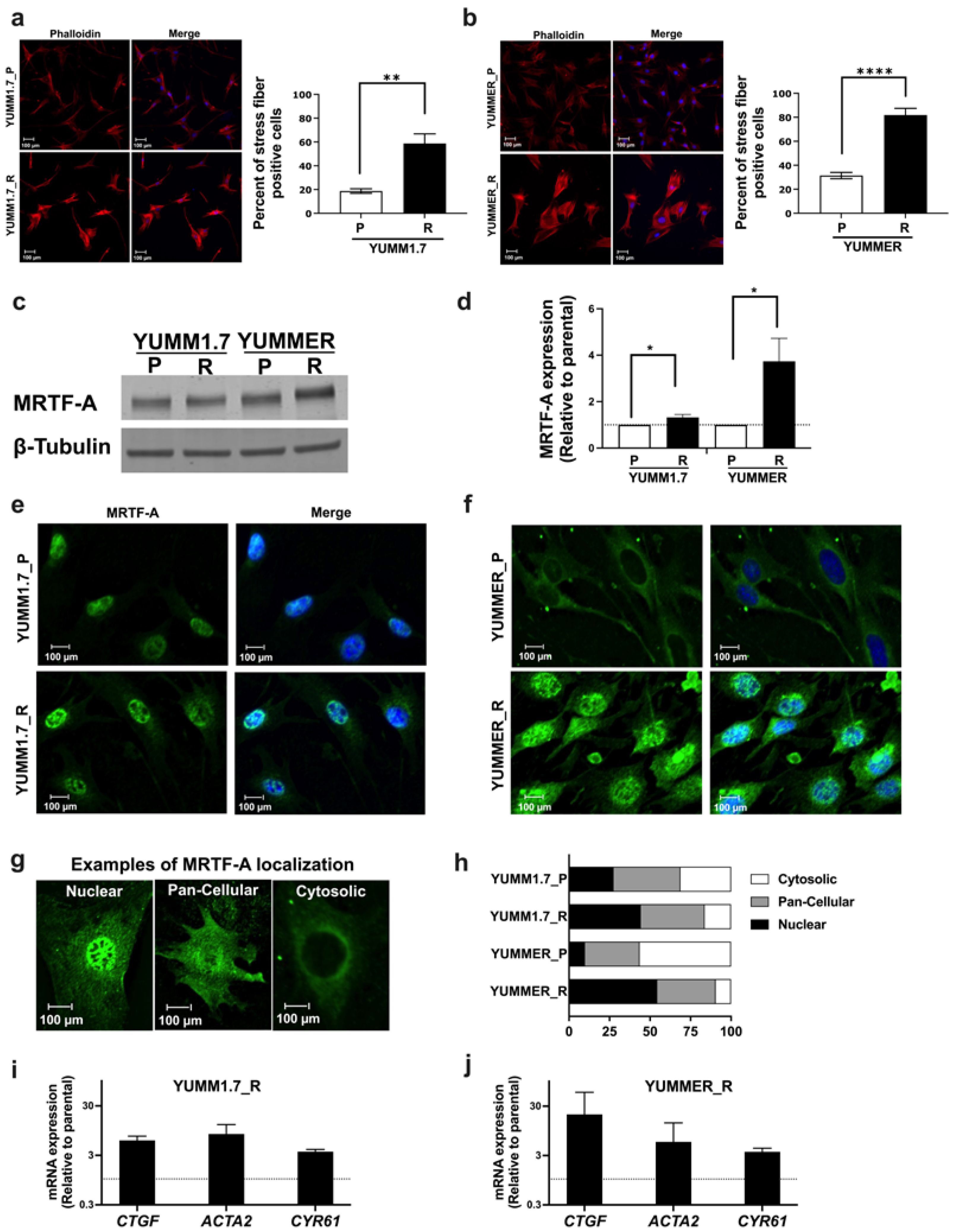
2.4. The Rho/MRTF Pathway Is Activated in Vem–Resistant YUMM Cells
2.5. The MRTF Pathway Is Upregulated in Resistant YUMM Cells
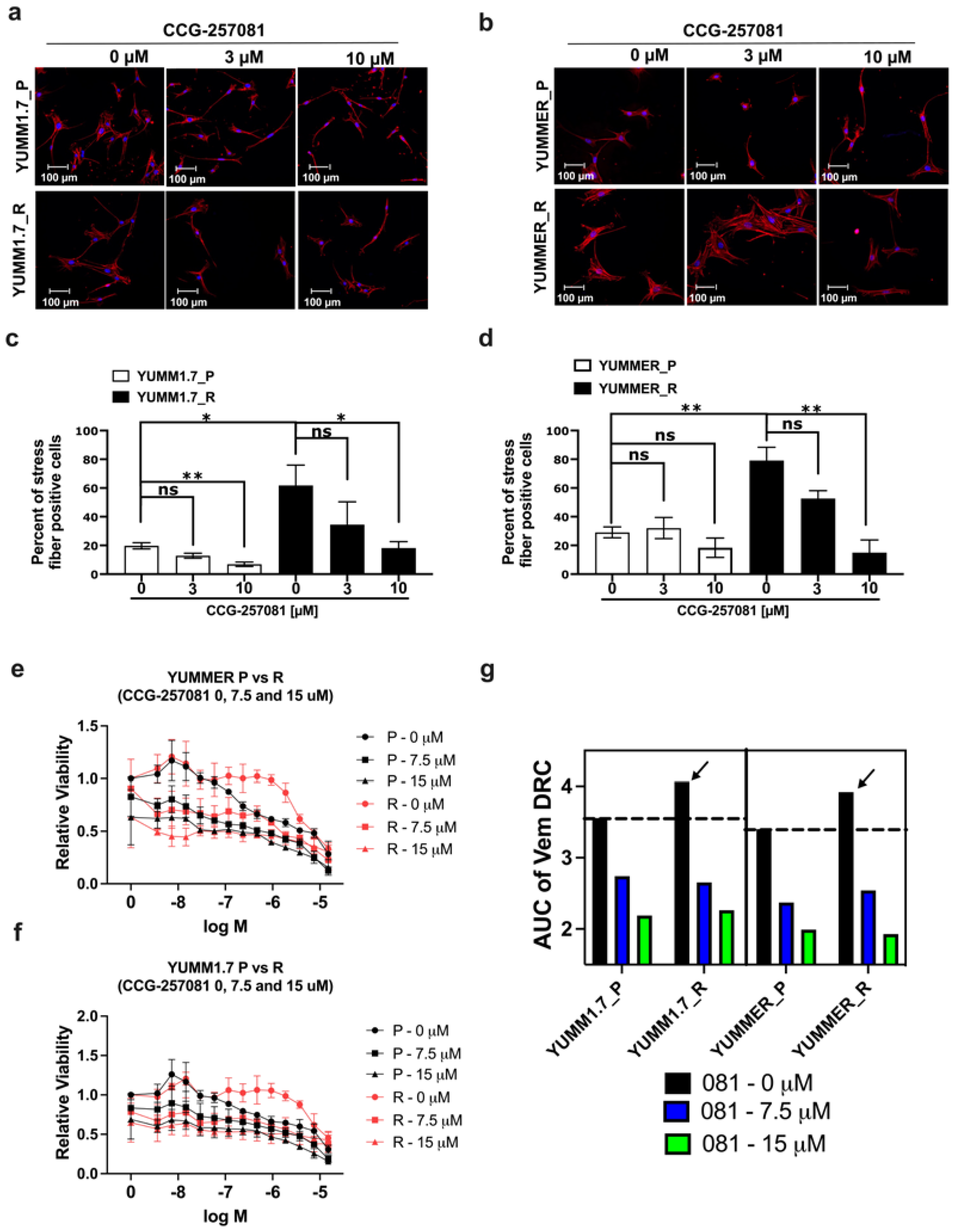
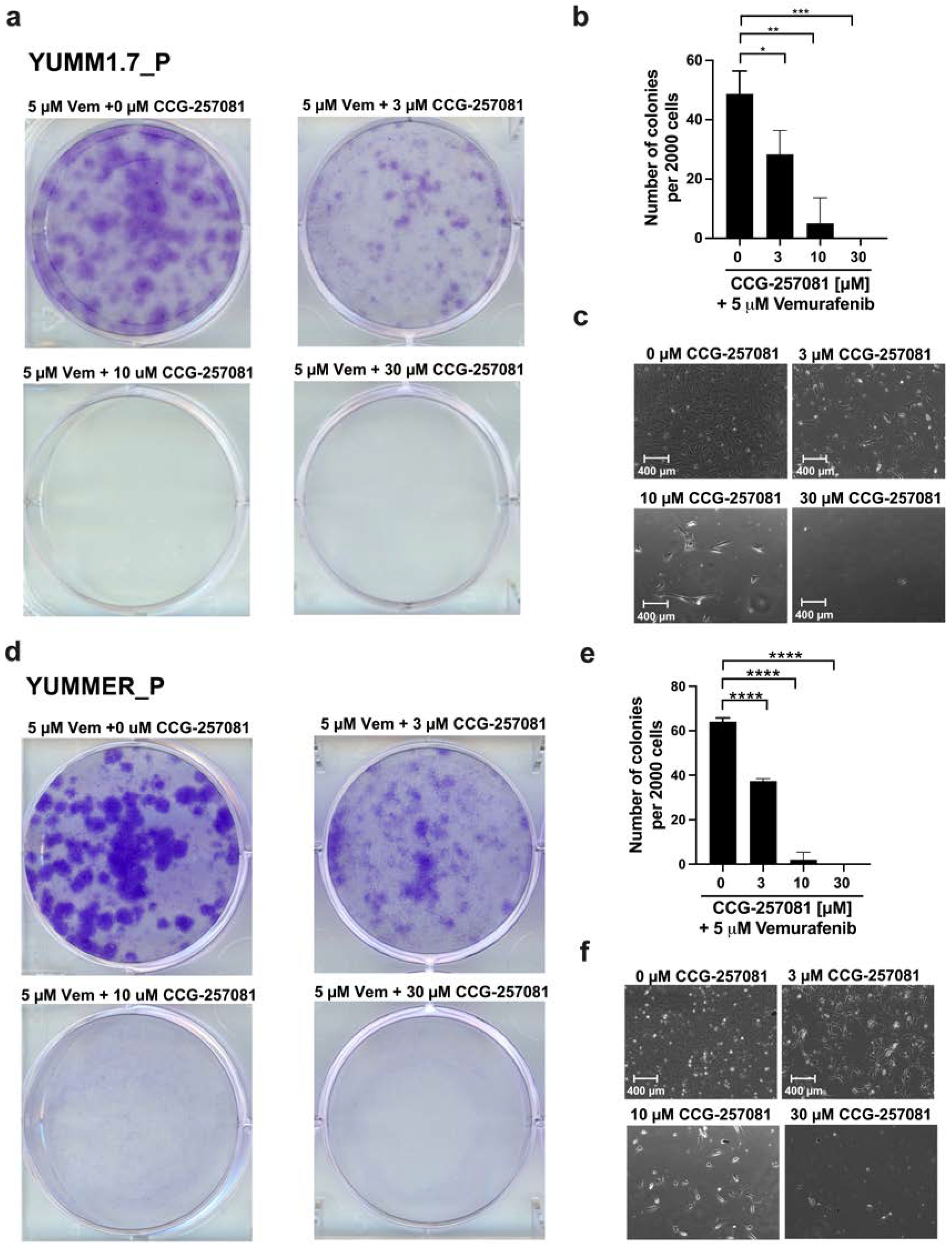
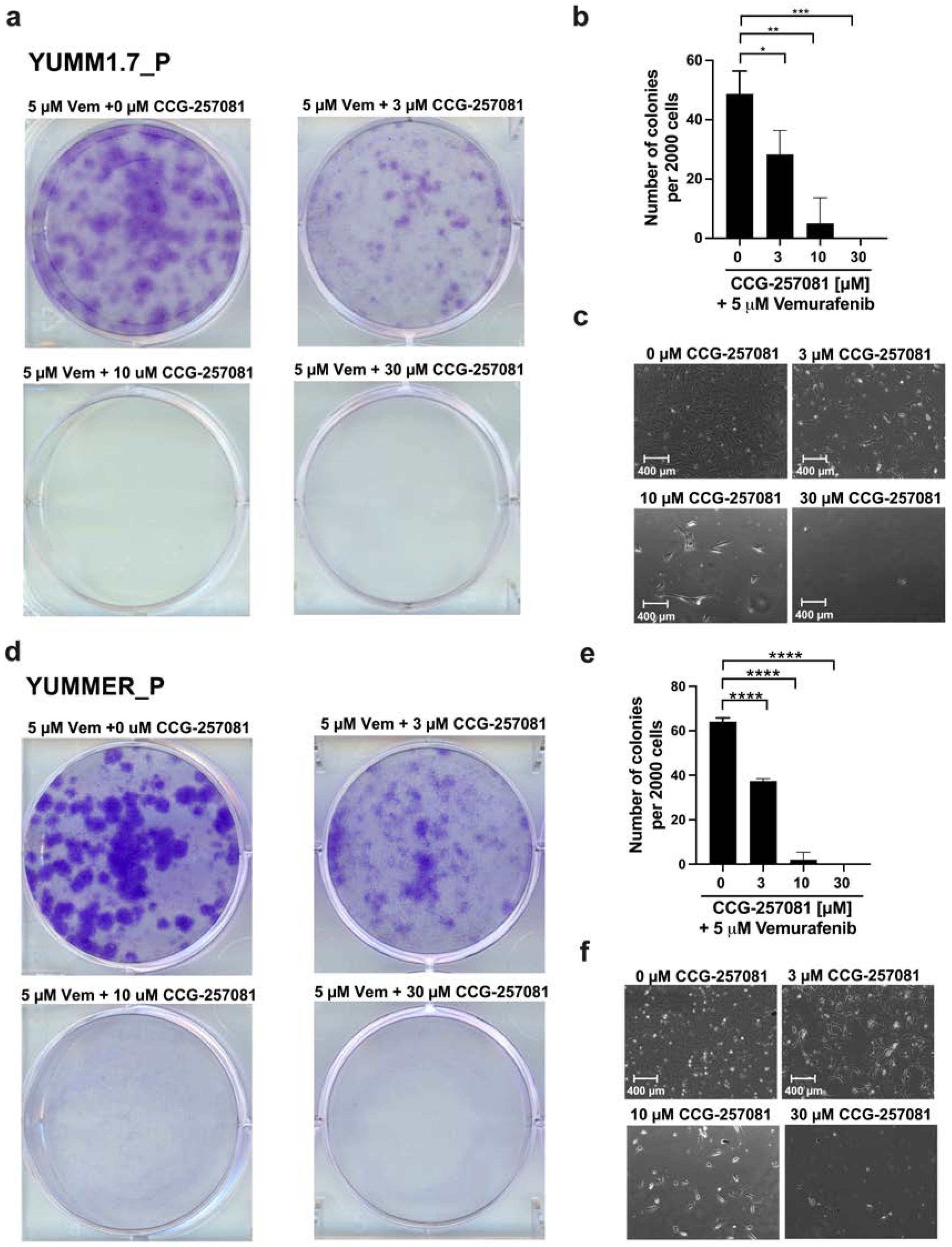
2.6. Inhibition of the Rho/MRTF Pathway Enhances Vem Sensitivity
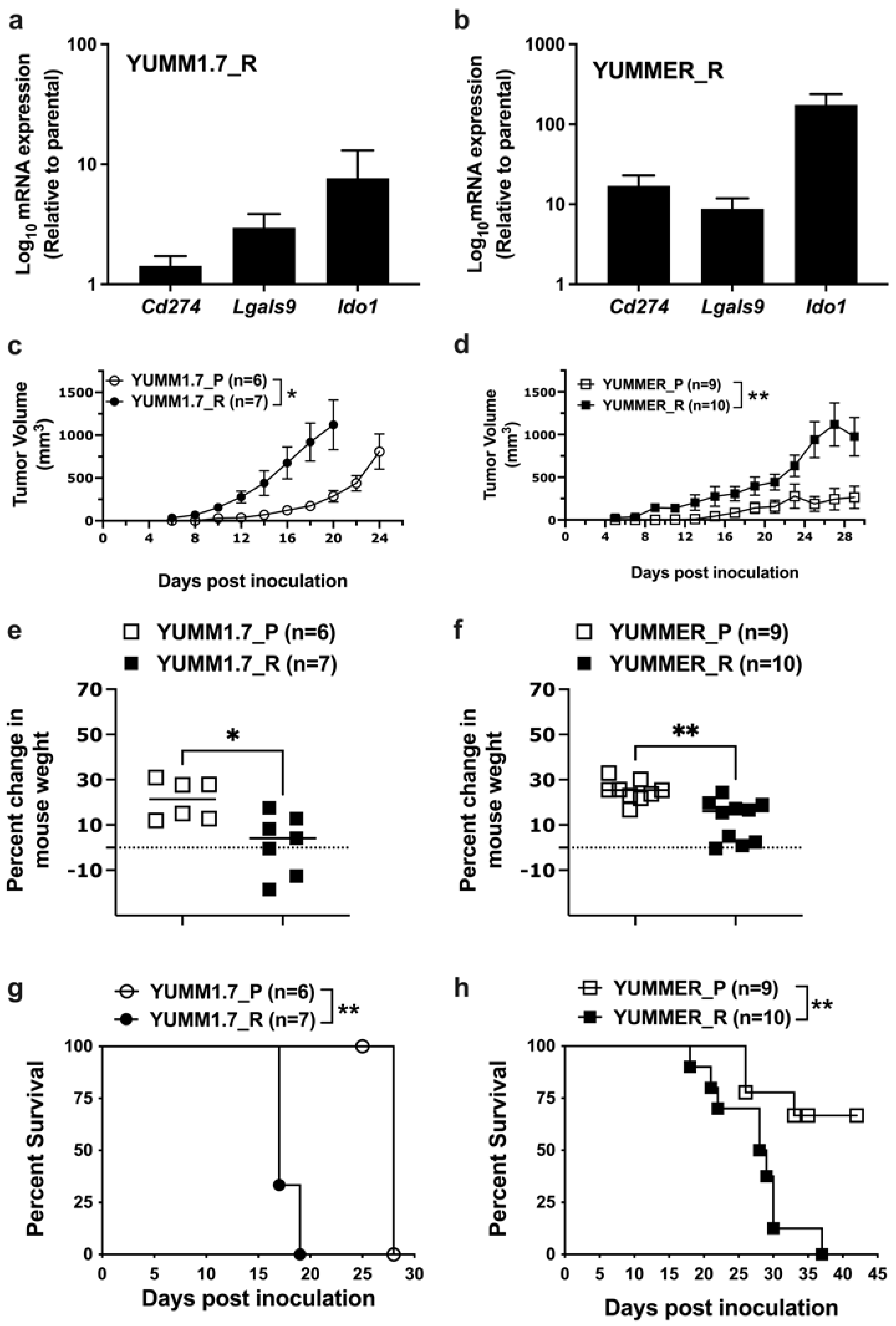
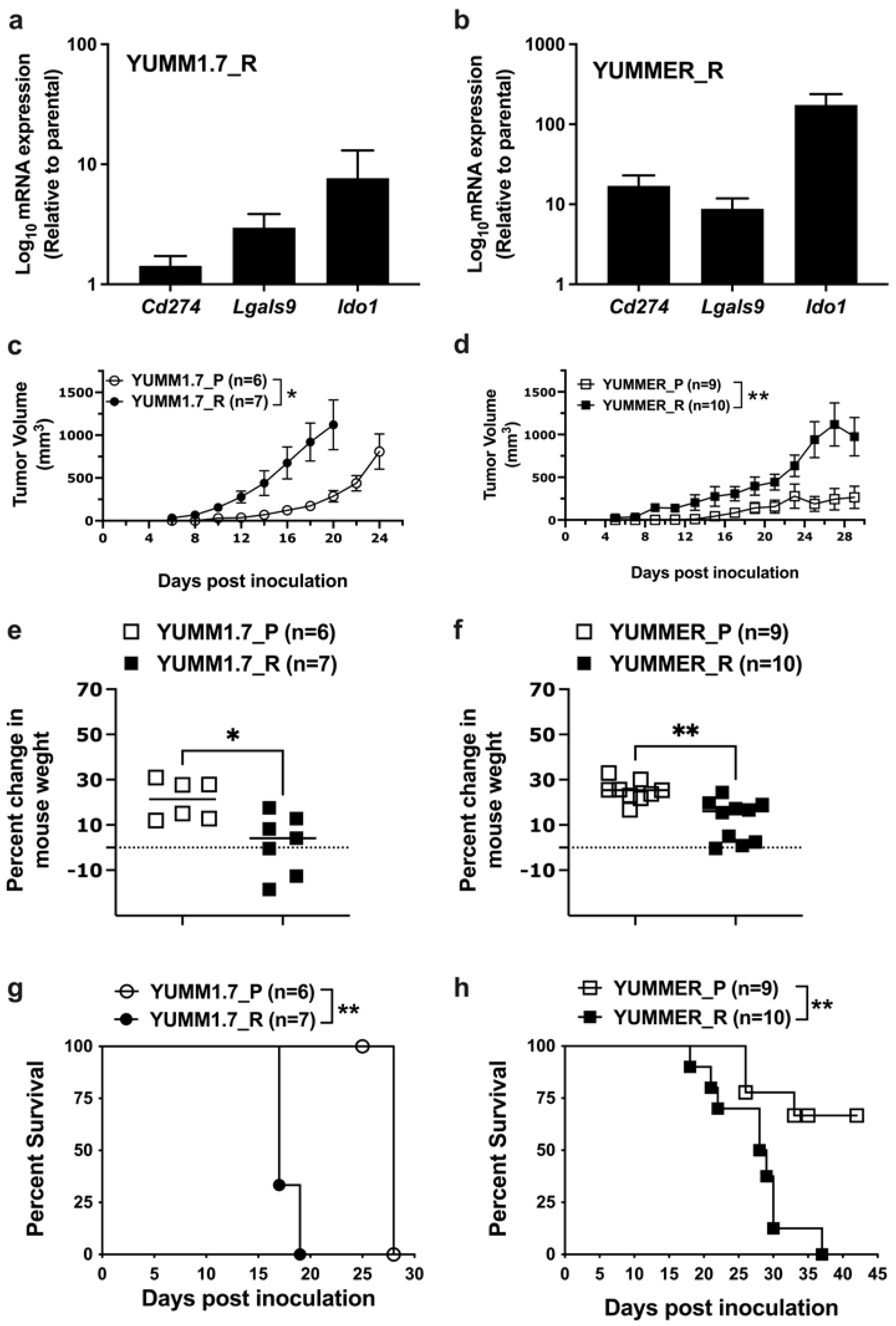
2.7. Effect of Targeted Therapy Resistance on Immune Checkpoint Gene Expression
2.8. Enhanced In Vivo Tumor Growth by Vem–Resistant Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Selection of Vem–Resistant Populations
4.2. Cell Viability
4.3. Compounds and Antibodies
4.4. RNA Extraction, cDNA Synthesis, and qRT-PCR Analysis
4.5. Western Blot Analysis
4.6. Fluorescence Microscopy
4.6.1. Immunofluorescence
4.6.2. Staining Actin Stress Fibers
4.7. Incucyte Live-Cell Imaging
4.8. In Vitro Clonogenicity Assay
4.9. Mice and Tumor Engraftment
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| YUMM | Yale University Mouse Melanoma |
| YUMMER | YUMM exposed to radiation |
| Vem | Vemurafenib |
| MRTF | Myocardin related transcription factor |
| MAPK | Mitogen-activated protein kinase |
| ICIs | Immune checkpoint inhibitor proteins |
| ICT | Immune checkpoint therapy |
| IC50 | half maximal inhibitory concentration |
| SRF | Serum response factor |
References
- Appleton, K.M.; Palsuledesai, C.C.; Misek, S.A.; Blake, M.; Zagorski, J.; Gallo, K.A.; Dexheimer, T.S.; Neubig, R.R. Inhibition of the Myocardin-Related Transcription Factor Pathway Increases Efficacy of Trametinib in NRAS-Mutant Melanoma Cell Lines. Cancers 2021, 13, 2012. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Curiel-Lewandrowski, C.; Fisher, D.E.; Swetter, S.M.; Tsao, H.; Aguirre-Ghiso, J.A.; Soengas, M.S.; Weeraratna, A.T.; Flaherty, K.T.; Herlyn, M.; et al. The State of Melanoma: Emergent Challenges and Opportunities. Clin. Cancer Res. 2021, 27, 2678–2697. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk with Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, Y.C.; Sharma, R.; Yadav, S.; Dwivedi, S.; Sharma, A.; Awasthi, S. The Non-ABC Drug Transporter RLIP76 (RALBP-1) Plays a Major Role in the Mechanisms of Drug Resistance. Curr. Drug Metab. 2007, 8, 315–323. [Google Scholar] [CrossRef]
- Ball, D.; Rachfal, A.; Kemper, S.; Brigstock, D.R. The heparin-binding 10 kDa fragment of connective tissue growth factor (CTGF) containing module 4 alone stimulates cell adhesion. J. Endocrinol. 2003, 176, R1–R7. [Google Scholar] [CrossRef]
- Basu, S.; Dong, Y.; Kumar, R.; Jeter, C.; Tang, D.G. Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin. Cancer Biol. 2022, 78, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Calvani, M.; Cavallini, L.; Tondo, A.; Spinelli, V.; Ricci, L.; Pasha, A.; Bruno, G.; Buonvicino, D.; Bigagli, E.; Vignoli, M.; et al. β3-Adrenoreceptors Control Mitochondrial Dormancy in Melanoma and Embryonic Stem Cells. Oxidative Med. Cell. Longev. 2018, 2018, 6816508. [Google Scholar] [CrossRef]
- Clark, E.A.; Golub, T.R.; Lander, E.S.; Hynes, R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000, 406, 532–535. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef]
- Dratkiewicz, E.; Simiczyjew, A.; Pietraszek-Gremplewicz, K.; Mazurkiewicz, J.; Nowak, D. Characterization of Melanoma Cell Lines Resistant to Vemurafenib and Evaluation of Their Responsiveness to EGFR- and MET-Inhibitor Treatment. Int. J. Mol. Sci. 2019, 21, 113. [Google Scholar] [CrossRef]
- Ephraim, R.; Fraser, S.; Nurgali, K.; Apostolopoulos, V. Checkpoint Markers and Tumor Microenvironment: What Do We Know? Cancers 2022, 14, 3788. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef]
- Evelyn, C.R.; Wade, S.M.; Wang, Q.; Wu, M.; Iñiguez-Lluhí, J.A.; Merajver, S.D.; Neubig, R.R. CCG-1423: A small-molecule inhibitor of RhoA transcriptional signaling. Mol. Cancer Ther. 2007, 6, 2249–2260. [Google Scholar] [CrossRef]
- Gao, R.; Brigstock, D.R. A novel integrin 5 1 binding domain in module 4 of connective tissue growth factor (CCN2/CTGF) promotes adhesion and migration of activated pancreatic stellate cells. Gut 2006, 55, 856–862. [Google Scholar] [CrossRef]
- Gau, D.; Chawla, P.; Eder, I.; Roy, P. Myocardin-related transcription factor’s interaction with serum-response factor is critical for outgrowth initiation, progression, and metastatic colonization of breast cancer cells. FASEB BioAdvances 2022, 4, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Haak, A.J.; Appleton, K.M.; Lisabeth, E.M.; Misek, S.A.; Ji, Y.; Wade, S.M.; Bell, J.L.; Rockwell, C.E.; Airik, M.; Krook, M.A.; et al. Pharmacological Inhibition of Myocardin-related Transcription Factor Pathway Blocks Lung Metastases of RhoC-Overexpressing Melanoma. Mol. Cancer Ther. 2017, 16, 193–204. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S.; Wynne, J.; Treisman, R. The Rho family GTPases RhoA, Racl, and CDC42Hsregulate transcriptional activation by SRF. Cell 1995, 81, 1159–1170. [Google Scholar] [CrossRef]
- Hutchings, K.M.; Lisabeth, E.M.; Rajeswaran, W.; Wilson, M.W.; Sorenson, R.J.; Campbell, P.L.; Ruth, J.H.; Amin, A.; Tsou, P.-S.; Leipprandt, J.R.; et al. Pharmacokinetic optimitzation of CCG-203971: Novel inhibitors of the Rho/MRTF/SRF transcriptional pathway as potential antifibrotic therapeutics for systemic scleroderma. Bioorganic Med. Chem. Lett. 2017, 27, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.U.; Shehzad, A.; Sonn, J.K.; Lee, Y.S. PRPF overexpression induces drug resistance through actin cytoskeleton rearrangement and epithelial-mesenchymal transition. Oncotarget 2017, 8, 56659–56671. [Google Scholar] [CrossRef]
- Jin, H.; Wang, L.; Bernards, R. Rational combinations of targeted cancer therapies: Background, advances and challenges. Nat. Rev. Drug Discov. 2023, 22, 213–234. [Google Scholar] [CrossRef] [PubMed]
- Juárez, P.; Mohammad, K.S.; Yin, J.J.; Fournier, P.G.J.; McKenna, R.C.; Davis, H.W.; Peng, X.H.; Niewolna, M.; Javelaud, D.; Chirgwin, J.M.; et al. Halofuginone Inhibits the Establishment and Progression of Melanoma Bone Metastases. Cancer Res. 2012, 72, 6247–6256. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Sensenbach, S.; Angardi, V.; Orman, M.A. BRAF-Inhibitor-Induced Metabolic Alterations in A375 Melanoma Cells. Metabolites 2021, 11, 777. [Google Scholar] [CrossRef]
- Ketchen, S.E.; Gamboa-Esteves, F.O.; Lawler, S.E.; Nowicki, M.O.; Rohwedder, A.; Knipp, S.; Prior, S.; Short, S.C.; Ladbury, J.E.; Brüning-Richardson, A. Drug Resistance in Glioma Cells Induced by a Mesenchymal–Amoeboid Migratory Switch. Biomedicines 2021, 10, 9. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.H.; Lee, J.-K.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef]
- Kümper, S.; Mardakheh, F.K.; McCarthy, A.; Yeo, M.; Stamp, G.W.; Paul, A.; Worboys, J.; Sadok, A.; Jørgensen, C.; Guichard, S.; et al. Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. eLife 2016, 5, 12203. [Google Scholar] [CrossRef]
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1P29S Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e9. [Google Scholar] [CrossRef]
- Lisabeth, E.M.; Kahl, D.; Gopallawa, I.; Haynes, S.E.; Misek, S.A.; Campbell, P.L.; Dexheimer, T.S.; Khanna, D.; Fox, D.A.; Jin, X.; et al. Identification of Pirin as a Molecular Target of the CCG-1423/CCG-203971 Series of Antifibrotic and Antimetastatic Compounds. ACS Pharmacol. Transl. Sci. 2019, 2, 92–100. [Google Scholar] [CrossRef]
- Liu, A.-X.; Rane, N.; Liu, J.-P.; Prendergast, G.C. RhoB Is Dispensable for Mouse Development, but It Modifies Susceptibility to Tumor Formation as Well as Cell Adhesion and Growth Factor Signaling in Transformed Cells. Mol. Cell. Biol. 2001, 21, 6906–6912. [Google Scholar] [CrossRef]
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal Cells Present in the Melanoma Niche Affect Tumor Invasiveness and Its Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 529. [Google Scholar] [CrossRef] [PubMed]
- Medjkane, S.; Perez-Sanchez, C.; Gaggioli, C.; Sahai, E.; Treisman, R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat. Cell Biol. 2009, 11, 257–268. [Google Scholar] [CrossRef]
- Meeth, K.; Wang, J.X.; Micevic, G.; Damsky, W.; Bosenberg, M.W. The YUMM lines: A series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res. 2016, 29, 590–597. [Google Scholar] [CrossRef]
- Misek, S.A.; Appleton, K.M.; Dexheimer, T.S.; Lisabeth, E.M.; Lo, R.S.; Larsen, S.D.; Gallo, K.A.; Neubig, R.R. Rho-mediated signaling promotes BRAF inhibitor resistance in de-differentiated melanoma cells. Oncogene 2020, 39, 1466–1483. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Simizu, S.; Okumura, H.; Takagi, S.; Osada, H. A small-molecule inhibitor shows that pirin regulates migration of melanoma cells. Nat. Chem. Biol. 2010, 6, 667–673. [Google Scholar] [CrossRef]
- Mohammad, K.S.; Javelaud, D.; Fournier, P.; Niewolna, M.; McKenna, C.R.; Peng, X.H.; Duong, V.; Dunn, L.K.; Mauviel, A.; Guise, T.A. TGF-β-RI Kinase Inhibitor SD-208 Reduces the Development and Progression of Melanoma Bone Metastases. Cancer Res. 2011, 71, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S.; Perona, R.; Saniger, L.; Lacal, J.C.; Ritty, T.M.; Broekelmann, T.; Tisdale, C.; Milewicz, D.M.; Mecham, R.P. Activation of Serum Response Factor by RhoA Is Mediated by the Nuclear Factor-κB and C/EBP Transcription Factors. J. Biol. Chem. 1999, 274, 8506–8515. [Google Scholar] [CrossRef]
- Orgaz, J.L.; Crosas-Molist, E.; Sadok, A.; Perdrix-Rosell, A.; Maiques, O.; Rodriguez-Hernandez, I.; Monger, J.; Mele, S.; Georgouli, M.; Bridgeman, V.; et al. Myosin II Reactivation and Cytoskeletal Remodeling as a Hallmark and a Vulnerability in Melanoma Therapy Resistance. Cancer Cell 2020, 37, 85–103.e9. [Google Scholar] [CrossRef]
- Pavlick, A.C.; Zhao, R.; Lee, C.-H.; Ritchings, C.; Rao, S. First-line immunotherapy versus targeted therapy in patients with BRAF-mutant advanced melanoma: A real-world analysis. Future Oncol. 2021, 17, 689–699. [Google Scholar] [CrossRef]
- Penas, C.; Arroyo-Berdugo, Y.; Apraiz, A.; Rasero, J.; Muñoa-Hoyos, I.; Andollo, N.; Cancho-Galán, G.; Izu, R.; Gardeazabal, J.; Ezkurra, P.A.; et al. Pirin is a prognostic marker of human melanoma that dampens the proliferation of malignant cells by downregulating JARID1B/KDM5B expression. Sci. Rep. 2023, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef]
- Reijers, I.L.M.; Rozeman, E.A.; van Wilgenhof, S.; Thienen, J.V.; Haanen, J.B.A.G.; Blank, C.U. Switch to checkpoint inhibition after targeted therapy at time of progression or during ongoing response: A retrospective single-centre experience in patients with BRAF-mutated melanoma. Pigment Cell Melanoma Res. 2020, 33, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1Bhigh Cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef]
- Rozeman, E.A.; Blank, C.U. Combining checkpoint inhibition and targeted therapy in melanoma. Nat. Med. 2019, 25, 879–882. [Google Scholar] [CrossRef]
- Sakai, N.; Chun, J.; Duffield, J.S.; Wada, T.; Luster, A.D.; Tager, A.M. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013, 27, 1830–1846. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-W.; Zhou, Y.-D.; Chen, H.-Z.; Luan, X.; Zhang, W.-D. Targeting CTGF in Cancer: An Emerging Therapeutic Opportunity. Trends Cancer 2021, 7, 511–524. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Suleman, M.; Qamar, M.T.U.; Saleem, S.; Ahmad, S.; Ali, S.S.; Khan, H.; Akbar, F.; Khan, W.; Alblihy, A.; Alrumaihi, F.; et al. Mutational Landscape of Pirin and Elucidation of the Impact of Most Detrimental Missense Variants That Accelerate the Breast Cancer Pathways: A Computational Modelling Study. Front. Mol. Biosci. 2021, 8, 692835. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hamid, O.; Gonzalez, R.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Lewis, K.D.; Tawbi, H.A.; Hernandez, G.; Wongchenko, M.J.; et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat. Med. 2019, 25, 929–935. [Google Scholar] [CrossRef]
- Torre, E.; Dueck, H.; Shaffer, S.; Gospocic, J.; Gupte, R.; Bonasio, R.; Kim, J.; Murray, J.; Raj, A. Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH. Cell Syst. 2018, 6, 171–179.e5. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Peeters, M.; Siena, S.; Humblet, Y.; Hendlisz, A.; Neyns, B.; Canon, J.-L.; Van Laethem, J.-L.; Maurel, J.; Richardson, G.; et al. Open-Label Phase III Trial of Panitumumab Plus Best Supportive Care Compared with Best Supportive Care Alone in Patients with Chemotherapy-Refractory Metastatic Colorectal Cancer. J. Clin. Oncol. 2007, 25, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, L.S.; Sutherland, L.B.; Liu, Z.; Grinnell, F.; Kamm, K.E.; Schneider, J.W.; Olson, E.N.; Small, E.M. Activation of MRTF-A–dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc. Natl. Acad. Sci. USA 2013, 110, 16850–16855. [Google Scholar] [CrossRef]
- Wang, J.; Perry, C.J.; Meeth, K.; Thakral, D.; Damsky, W.; Micevic, G.; Kaech, S.; Blenman, K.; Bosenberg, M. UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment Cell Melanoma Res. 2017, 30, 428–435. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.; Zhang, Z.; Chen, X. Identification ACTA2 and KDR as key proteins for prognosis of PD-1/PD-L1 blockade therapy in melanoma. Anim. Model. Exp. Med. 2021, 4, 138–150. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Yang, Z.; Algazi, A.P.; Lomeli, S.H.; Wang, Y.; Othus, M.; Hong, A.; Wang, X.; Randolph, C.E.; et al. Anti-PD-1/L1 lead-in before MAPK inhibitor combination maximizes antitumor immunity and efficacy. Cancer Cell 2021, 39, 1375–1387.e6. [Google Scholar] [CrossRef]
- Qiao, Z.; Wang, D.; Hahn, J.; Ai, J.; Wang, Z. Pirin down-regulates the EAF2/U19 protein and alleviates its growth inhibition in prostate cancer cells. Prostate 2014, 74, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF–MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef]
- Wong, C.C.-L.; Wong, C.-M.; Tung, E.K.-K.; Man, K.; Ng, I.O.-L. Rho-kinase 2 is frequently overexpressed in hepatocellular carcinoma and involved in tumor invasion. Hepatology 2009, 49, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.-M. α-Smooth Muscle Actin and ACTA2 Gene Expressions in Vasculopathies. Rev. Bras. Cir. Cardiovasc. 2015, 30, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Qin, J.; Sun, L.; Gui, L.; Zhang, C.; Huang, Y.; Deng, W.; Huang, A.; Sun, D.; Luo, M. Intrahepatic upregulation of MRTF-A signaling contributes to increased hepatic vascular resistance in cirrhotic rats with portal hypertension. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 303–310. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foda, B.M.; Neubig, R.R. Role of Rho/MRTF in Aggressive Vemurafenib-Resistant Murine Melanomas and Immune Checkpoint Upregulation. Int. J. Mol. Sci. 2023, 24, 13785. https://doi.org/10.3390/ijms241813785
Foda BM, Neubig RR. Role of Rho/MRTF in Aggressive Vemurafenib-Resistant Murine Melanomas and Immune Checkpoint Upregulation. International Journal of Molecular Sciences. 2023; 24(18):13785. https://doi.org/10.3390/ijms241813785
Chicago/Turabian StyleFoda, Bardees M., and Richard R. Neubig. 2023. "Role of Rho/MRTF in Aggressive Vemurafenib-Resistant Murine Melanomas and Immune Checkpoint Upregulation" International Journal of Molecular Sciences 24, no. 18: 13785. https://doi.org/10.3390/ijms241813785
APA StyleFoda, B. M., & Neubig, R. R. (2023). Role of Rho/MRTF in Aggressive Vemurafenib-Resistant Murine Melanomas and Immune Checkpoint Upregulation. International Journal of Molecular Sciences, 24(18), 13785. https://doi.org/10.3390/ijms241813785