
Functional Insight into and Refinement of the Genomic Boundaries of the JARID2-Neurodevelopmental Disorder Episignature

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. JARID2 Molecular and Clinical Information
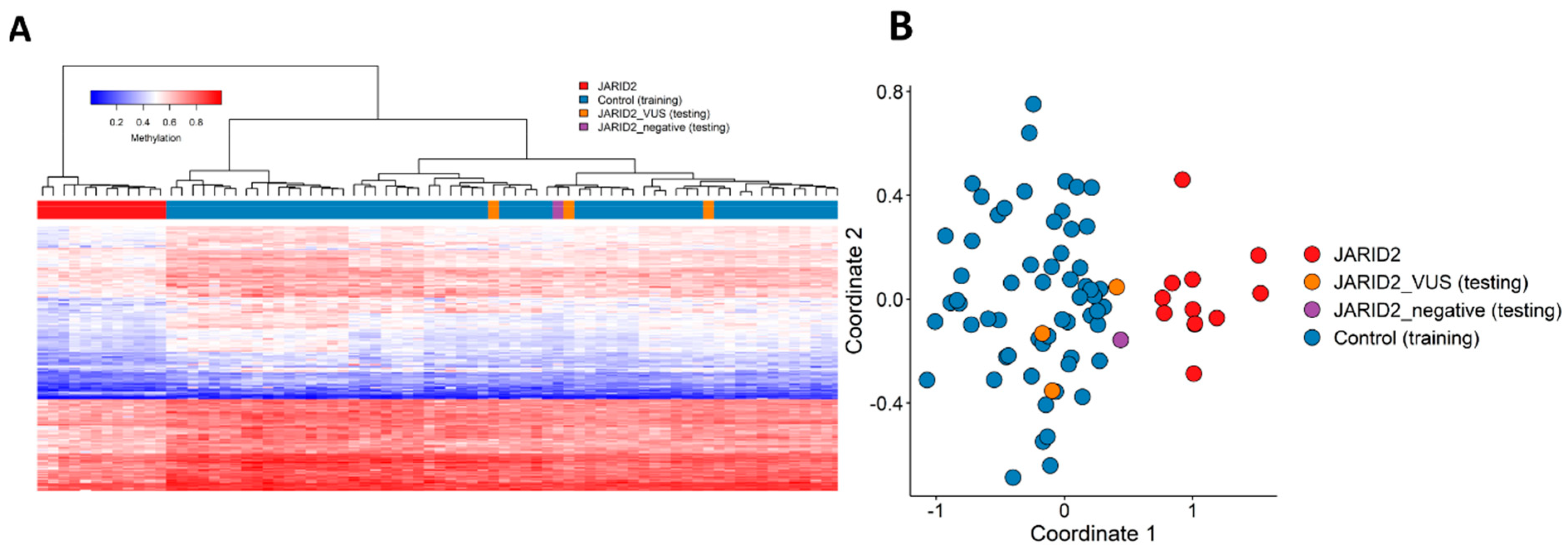
2.2. Identification and Assessment of an Episignature for JARID2
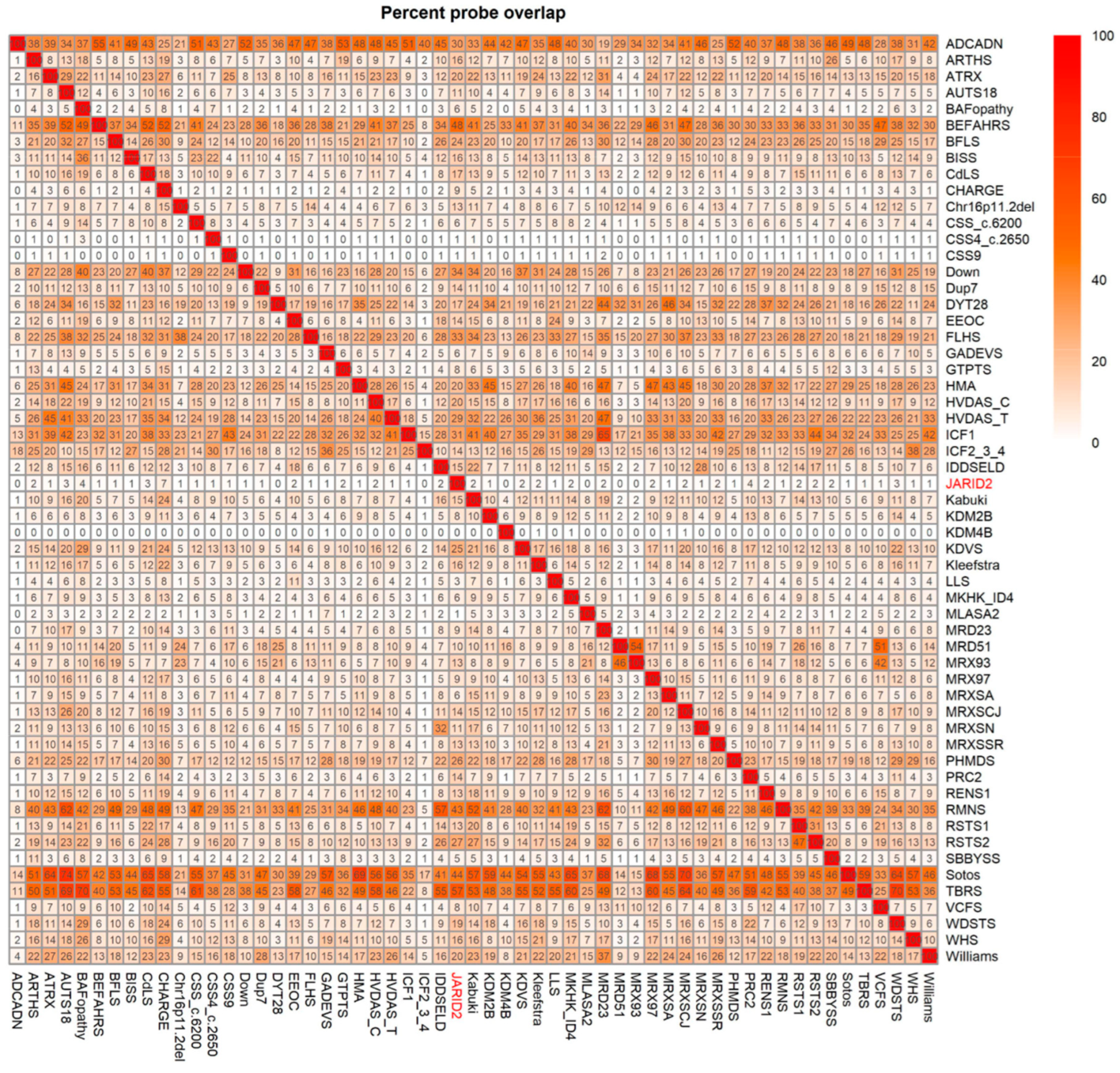
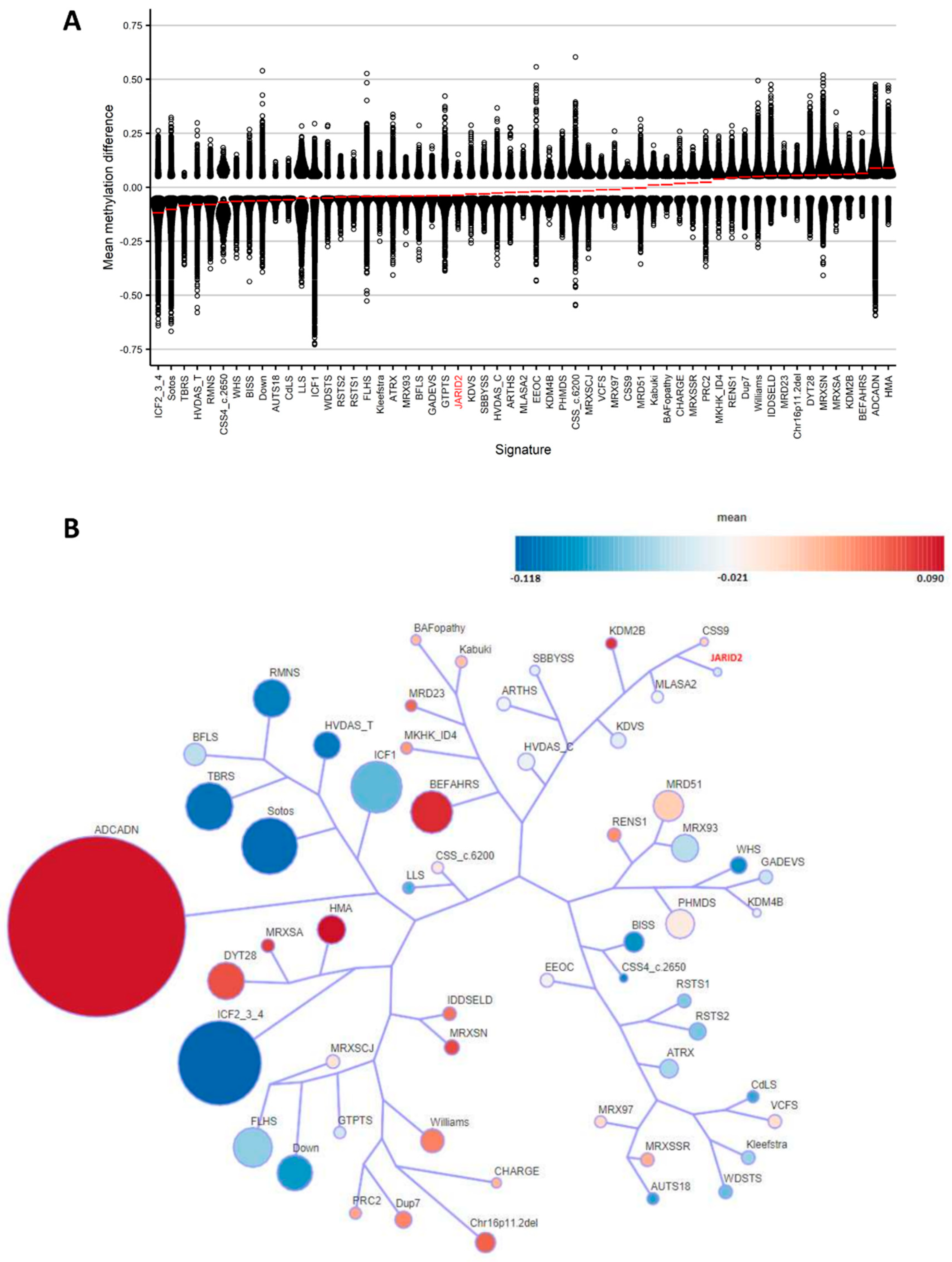
2.3. Annotation of the Global JARID2 DNA Methylation Profile and Correlation to the 56 Neurodevelopmental Disorder Episignatures on EpiSign™
2.4. Detection of Differentially Methylated Regions
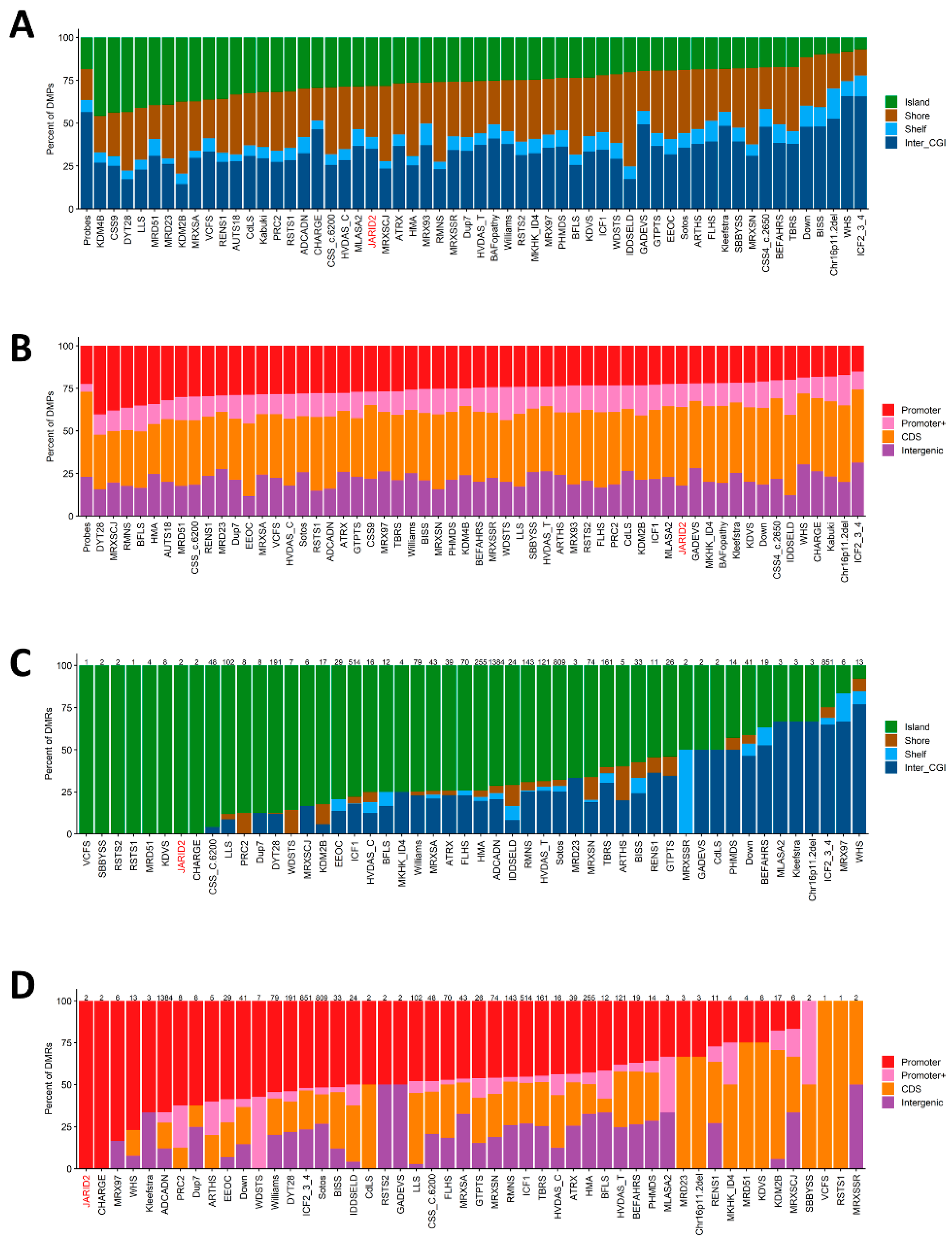
2.5. Genomic Location of Classifying DMPs and DMRs
3. Discussion
4. Materials and Methods
4.1. Subjects and Study Cohort
4.2. Sample Processing
4.3. Probe Selection and Episignature Classifier Construction
4.4. Annotation of the Global JARID2 DNA Methylation Profile and Correlation to the 56 Neurodevelopmental Disorder Episignatures on EpiSign™
4.5. Differentially Methylated Regions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cadieux-Dion, M.; Farrow, E.; Thiffault, I.; Cohen, A.S.A.; Welsh, H.; Bartik, L.; Schwager, C.; Engleman, K.; Zhou, D.; Zhang, L.; et al. Phenotypic expansion and variable expressivity in individuals with JARID2-related intellectual disability: A case series. Clin. Genet. 2022, 102, 136–141. [Google Scholar] [CrossRef]
- Verberne, E.A.; Goh, S.; England, J.; van Ginkel, M.; Rafael-Croes, L.; Maas, S.; Polstra, A.; Zarate, Y.A.; Bosanko, K.A.; Pechter, K.B.; et al. JARID2 haploinsufficiency is associated with a clinically distinct neurodevelopmental syndrome. Genet. Med. 2021, 23, 374–383. [Google Scholar] [CrossRef]
- Barøy, T.; Misceo, D.; Strømme, P.; Stray-Pedersen, A.; Holmgren, A.; Rødningen, O.K.; Blomhoff, A.; Helle, J.R.; Stormyr, A.; Tvedt, B.; et al. Haploinsufficiency of two histone modifier genes on 6p22.3, ATXN1 and JARID2, is associated with intellectual disability. Orphanet J. Rare Dis. 2013, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Kasinath, V.; Beck, C.; Sauer, P.; Poepsel, S.; Kosmatka, J.; Faini, M.; Toso, D.; Aebersold, R.; Nogales, E. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science 2021, 371, eabc3393. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Cloos, P.A.; Walfridsson, J.; Olsson, L.; Bukowski, J.P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef]
- Verberne, E.A.; van der Laan, L.; Haghshenas, S.; Rooney, K.; Levy, M.A.; Alders, M.; Maas, S.M.; Jansen, S.; Lieden, A.; Anderlid, B.-M.; et al. DNA Methylation Signature for JARID2-Neurodevelopmental Syndrome. Int. J. Mol. Sci. 2022, 23, 8001. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, L.; Rooney, K.; Trooster, T.M.; Mannens, M.M.; Sadikovic, B.; Henneman, P. DNA methylation episignatures: Insight into copy number variation. Epigenomics 2022, 14, 1373–1388. [Google Scholar] [CrossRef]
- Rooney, K.; van der Laan, L.; Trajkova, S.; Haghshenas, S.; Relator, R.; Lauffer, P.; Vos, N.; Levy, M.A.; Brunetti-Pierri, N.; Terrone, G.; et al. DNA methylation episignature and comparative epigenomic profiling of HNRNPU-related neurodevelopmental disorder. Genet. Med. 2023, 25, 100871. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Levy, M.A.; Relator, R.; McConkey, H.; Pranckeviciene, E.; Kerkhof, J.; Barat-Houari, M.; Bargiacchi, S.; Biamino, E.; Bralo, M.P.; Cappuccio, G.; et al. Functional correlation of genome-wide DNA methylation profiles in genetic neurodevelopmental disorders. Hum. Mutat. 2022, 43, 1609–1628. [Google Scholar] [CrossRef]
- Levy, M.A.; McConkey, H.; Kerkhof, J.; Barat-Houari, M.; Bargiacchi, S.; Biamino, E.; Bralo, M.P.; Cappuccio, G.; Ciolfi, A.; Clarke, A.; et al. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Adv. 2022, 3, 100075. [Google Scholar] [CrossRef]
- Austenaa, L.M.I.; Piccolo, V.; Russo, M.; Prosperini, E.; Polletti, S.; Polizzese, D.; Ghisletti, S.; Barozzi, I.; Diaferia, G.R.; Natoli, G. A first exon termination checkpoint preferentially suppresses extragenic transcription. Nat. Struct. Mol. Biol. 2021, 28, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Zahir, F.R.; Tucker, T.; Mayo, S.; Brown, C.J.; Lim, E.L.; Taylor, J.; Marra, M.A.; Hamdan, F.F.; Michaud, J.L.; Friedman, J.M. Intragenic CNVs for epigenetic regulatory genes in intellectual disability: Survey identifies pathogenic and benign single exon changes. Am. J. Med. Genet. A 2016, 170, 2916–2926. [Google Scholar] [CrossRef]
- Butcher, D.T.; Cytrynbaum, C.; Turinsky, A.L.; Siu, M.T.; Inbar-Feigenberg, M.; Mendoza-Londono, R.; Chitayat, D.; Walker, S.; Machado, J.; Caluseriu, O.; et al. CHARGE and Kabuki Syndromes: Gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am. J. Hum. Genet. 2017, 100, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.S.; Tuysuz, B.; Shen, Y.; Bhalla, S.K.; Jones, S.J.; Gibson, W.T. A novel mutation in EED associated with overgrowth. J. Hum. Genet. 2015, 60, 339–342. [Google Scholar] [CrossRef]
- Crawford, M.W.; Rohan, D. The upper airway in Weaver syndrome. Paediatr. Anaesth. 2005, 15, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Granadillo, J.L.; Wegner, D.J.; Paul, A.J.; Willing, M.; Sisco, K.; Tedder, M.L.; Sadikovic, B.; Wambach, J.A.; Baldridge, D.; Cole, F.S. Discovery of a novel CHD7 CHARGE syndrome variant by integrated omics analyses. Am. J. Med. Genet. A 2021, 185, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Machol, K.; Rousseau, J.; Ehresmann, S.; Garcia, T.; Nguyen, T.T.M.; Spillmann, R.C.; Sullivan, J.A.; Shashi, V.; Jiang, Y.H.; Stong, N.; et al. Expanding the Spectrum of BAF-Related Disorders: De Novo Variants in SMARCC2 Cause a Syndrome with Intellectual Disability and Developmental Delay. Am. J. Hum. Genet. 2019, 104, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Hempel, A.; Pagnamenta, A.T.; Blyth, M.; Mansour, S.; McConnell, V.; Kou, I.; Ikegawa, S.; Tsurusaki, Y.; Matsumoto, N.; Lo-Castro, A.; et al. Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin-Siris syndrome. J. Med. Genet. 2016, 53, 152–162. [Google Scholar] [CrossRef]
- Tsang, S.M.; Oliemuller, E.; Howard, B.A. Regulatory roles for SOX11 in development, stem cells and cancer. Semin. Cancer Biol. 2020, 67, 3–11. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Rodenhiser, D.I.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Hood, R.L.; Bulman, D.E.; Kernohan, K.D.; et al. Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am. J. Hum. Genet. 2018, 102, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.; Imai, K.; King, G.; Stuart, E.A. MatchIt: Nonparametric Preprocessing for Parametric Causal Inference. J. Stat. Softw. 2011, 42, 1–28. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Cavalcante, R.G.; Sartor, M.A. annotatr: Genomic regions in context. Bioinformatics 2017, 33, 2381–2383. [Google Scholar] [CrossRef]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Reginald, V.L.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient # | 1 | 2 | 3 | 4 | Summary of This Report | Verberne et al. (2022) [6] (n = 8) |
|---|---|---|---|---|---|---|
| Variant information | ||||||
| Variant type | Deletion | Deletion | Deletion | Deletion | ||
| Variant | 6p22.3 (14571015_16248244)x1 | 6p22.3 (14571015_16381865)x1 | 6p22.3–24.2 (11327614_15291611)x1 | 6p22.3–24.2 (9796651_19501625)x1 | ||
| Platform | WES and confirmed by Array-CGH | Array-CGH | Array-CGH | Array-CGH | ||
| Inheritance | Not present in their siblings, parents deceased | dn | dn | dn | ||
| Classification | LP | P | P | P | ||
| General information | ||||||
| Gender | M | M | M | M | ||
| Age (years) | 28 | 24 | 7 | 10 | ||
| Clinical information | ||||||
| Development/behavior | ||||||
| Intellectual disability | + | + | + | + | 4/4 (100%) | 6/7 (85.5%) |
| Developmental delay | + | + | + | + | 4/4 (100%) | 8/8 (100%) |
| Behavior abnormalities | + | + | + | + | 4/4 (100%) | 3/8 (37.5%) |
| Autistic features | + | + | + | + | 4/4 (100%) | 4/8 (50%) |
| ASD diagnosis | No formal ASD diagnosis | No formal ASD diagnosis, autistic traits | No formal ASD diagnosis | + | 2/4 (50%) | 2/8 (25%) |
| Neurologic | ||||||
| Hypotonia | + | − | + | − | 2/4 (50%) | 2/8 (25%) |
| Gait disturbance | − | − | − | − | 0/4 (0%) | 2/8 (25%) |
| MRI abnormalities | − | − | − | + | 1/4 (25%) | 3/3 (100%) |
| Dysmorphism | ||||||
| Broad forehead | + | − | − | + | 2/4 (50%) | 3/8 (37.5%) |
| High anterior hair line | − | + | − | − | 1/4 (25%) | 5/8 (62.5%) |
| Prominent supraorbital ridges | + | − | − | − | 1/4 (25%) | 1/8 (12.5%) |
| Deep set eyes | + | − | − | − | 1/4 (25%) | 4/8 (50%) |
| Infraorbital dark circles | + | + | − | − | 2/4 (50%) | 4/8 (50%) |
| Midface hypoplasia | + | - | + | − | 2/4 (50%) | 1/8 (12.5%) |
| Depressed nasal bridge | − | - | − | + | 1/4 (25%) | 2/8 (25%) |
| Bulbous nasal tip | − | + | + | + | 3/4 (75%) | 3/8 (37.5%) |
| Short philtrum | + | − | − | − | 1/4 (25%) | 3/8 (37.5%) |
| Full lips | + | − | + | + | 3/4 (75%) | 2/8 (25%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Laan, L.; Rooney, K.; Haghshenas, S.; Silva, A.; McConkey, H.; Relator, R.; Levy, M.A.; Valenzuela, I.; Trujillano, L.; Lasa-Aranzasti, A.; et al. Functional Insight into and Refinement of the Genomic Boundaries of the JARID2-Neurodevelopmental Disorder Episignature. Int. J. Mol. Sci. 2023, 24, 14240. https://doi.org/10.3390/ijms241814240
van der Laan L, Rooney K, Haghshenas S, Silva A, McConkey H, Relator R, Levy MA, Valenzuela I, Trujillano L, Lasa-Aranzasti A, et al. Functional Insight into and Refinement of the Genomic Boundaries of the JARID2-Neurodevelopmental Disorder Episignature. International Journal of Molecular Sciences. 2023; 24(18):14240. https://doi.org/10.3390/ijms241814240
Chicago/Turabian Stylevan der Laan, Liselot, Kathleen Rooney, Sadegheh Haghshenas, Ananília Silva, Haley McConkey, Raissa Relator, Michael A. Levy, Irene Valenzuela, Laura Trujillano, Amaia Lasa-Aranzasti, and et al. 2023. "Functional Insight into and Refinement of the Genomic Boundaries of the JARID2-Neurodevelopmental Disorder Episignature" International Journal of Molecular Sciences 24, no. 18: 14240. https://doi.org/10.3390/ijms241814240
APA Stylevan der Laan, L., Rooney, K., Haghshenas, S., Silva, A., McConkey, H., Relator, R., Levy, M. A., Valenzuela, I., Trujillano, L., Lasa-Aranzasti, A., Campos, B., Castells, N., Verberne, E. A., Maas, S., Alders, M., Mannens, M. M. A. M., van Haelst, M. M., Sadikovic, B., & Henneman, P. (2023). Functional Insight into and Refinement of the Genomic Boundaries of the JARID2-Neurodevelopmental Disorder Episignature. International Journal of Molecular Sciences, 24(18), 14240. https://doi.org/10.3390/ijms241814240