
Modeling Myotonic Dystrophy Type 2 Using Drosophila melanogaster

 , , and
, , and 
Abstract
1. Introduction
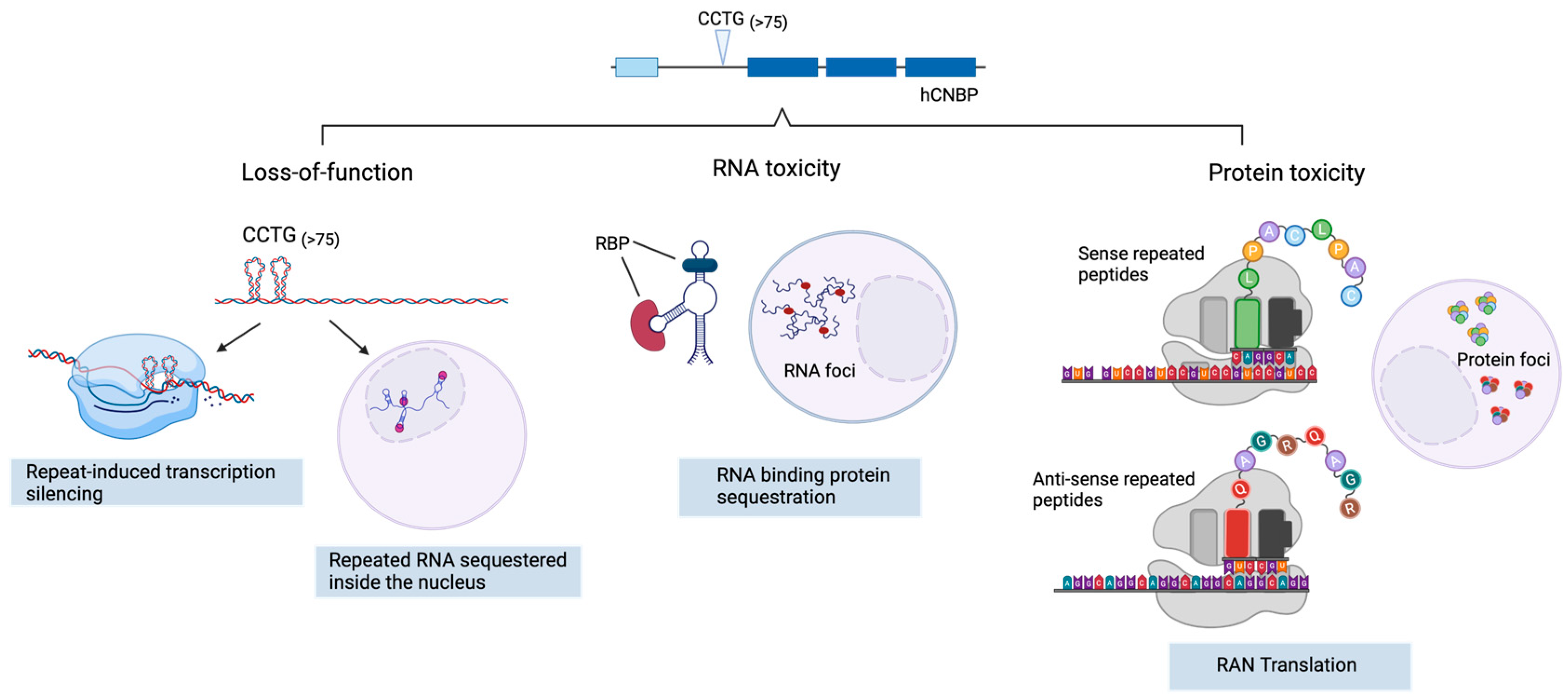
2. DM2 Pathogenesis
2.1. CNBP Protein Loss of Function
2.2. Toxic Gain of Function mRNA from Expanded Repeats
2.3. Tetrapeptide-Repeat Rrotein (TPR)-Mediated Toxicity
3. Drosophila melanogaster as a Tool to Study Neuromuscular Disorders
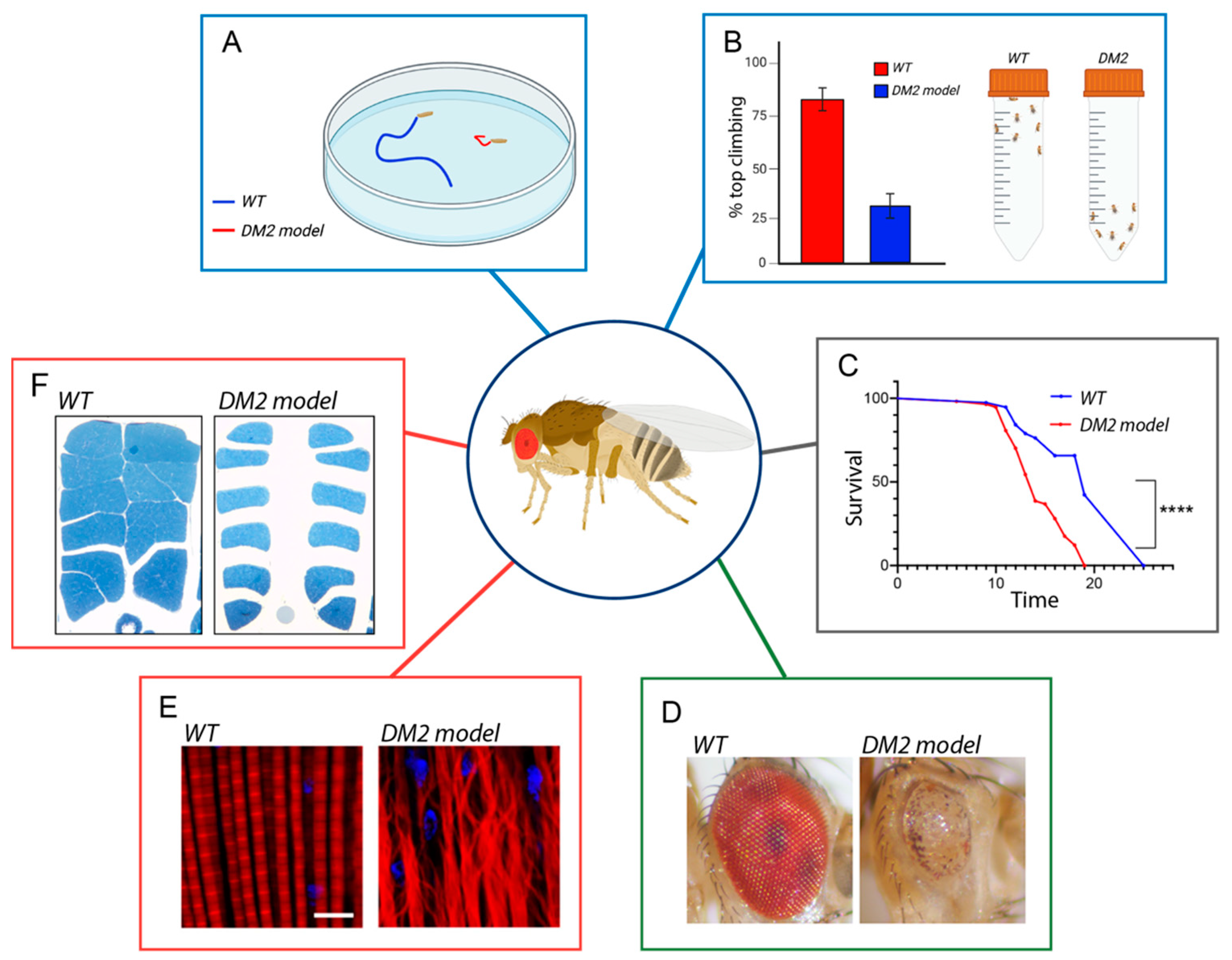
4. DM2 Pathogenesis Using Drosophila as a Study Model
4.1. CNBP Protein Downregulation
4.2. Toxic Gain of Function of RNAs—Bi-Directional Antisense Transcription
4.3. RAN Translation-Protein Toxicity
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meola, G.; Cardani, R. Myotonic Dystrophy Type 2: An Update on Clinical Aspects, Genetic and Pathomolecular Mechanism. J. Neuromuscul. Dis. 2015, 2, S59–S71. [Google Scholar] [CrossRef]
- Meola, G. Myotonic Dystrophy Type 2: The 2020 Update. Acta Myol. 2020, 39, 222–234. [Google Scholar] [CrossRef]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef]
- Bachinski, L.L.; Czernuszewicz, T.; Ramagli, L.S.; Suominen, T.; Shriver, M.D.; Udd, B.; Siciliano, M.J.; Krahe, R. Premutation Allele Pool in Myotonic Dystrophy Type 2. Neurology 2009, 72, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.E. Myotonic Muscular Dystrophies. Continuum 2019, 25, 1682–1695. [Google Scholar] [CrossRef] [PubMed]
- Peric, S.; Rakocevic-Stojanovic, V.; Meola, G. Cerebral Involvement and Related Aspects in Myotonic Dystrophy Type 2. Neuromuscul. Disord. 2021, 31, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Raheem, O.; Olufemi, S.-E.; Bachinski, L.L.; Vihola, A.; Sirito, M.; Holmlund-Hampf, J.; Haapasalo, H.; Li, Y.-P.; Udd, B.; Krahe, R. Mutant (CCTG)n Expansion Causes Abnormal Expression of Zinc Finger Protein 9 (ZNF9) in Myotonic Dystrophy Type 2. Am. J. Pathol. 2010, 177, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Huichalaf, C.; Schoser, B.; Schneider-Gold, C.; Jin, B.; Sarkar, P.; Timchenko, L. Reduction of the Rate of Protein Translation in Patients with Myotonic Dystrophy 2. J. Neurosci. 2009, 29, 9042. [Google Scholar] [CrossRef]
- Salisbury, E.; Schoser, B.; Schneider-Gold, C.; Wang, G.-L.; Huichalaf, C.; Jin, B.; Sirito, M.; Sarkar, P.; Krahe, R.; Timchenko, N.A.; et al. Expression of RNA CCUG Repeats Dysregulates Translation and Degradation of Proteins in Myotonic Dystrophy 2 Patients. Am. J. Pathol. 2009, 175, 748–762. [Google Scholar] [CrossRef]
- Schneider-Gold, C.; Timchenko, L.T. CCUG Repeats Reduce the Rate of Global Protein Synthesis in Myotonic Dystrophy Type 2. Rev. Neurosci. 2010, 21, 19–28. [Google Scholar] [CrossRef]
- Wei, C.; Stock, L.; Schneider-Gold, C.; Sommer, C.; Timchenko, N.A.; Timchenko, L. Reduction of Cellular Nucleic Acid Binding Protein Encoded by a Myotonic Dystrophy Type 2 Gene Causes Muscle Atrophy. Mol. Cell. Biol. 2018, 38, e00649-17. [Google Scholar] [CrossRef]
- Edwards, S.F.; Sirito, M.; Krahe, R.; Sinden, R.R. A Z-DNA Sequence Reduces Slipped-Strand Structure Formation in the Myotonic Dystrophy Type 2 (CCTG)·(CAGG) Repeat. Proc. Natl. Acad. Sci. USA 2009, 106, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Coni, S.; Falconio, F.A.; Marzullo, M.; Munafò, M.; Zuliani, B.; Mosti, F.; Fatica, A.; Ianniello, Z.; Bordone, R.; Macone, A.; et al. Translational Control of Polyamine Metabolism by CNBP Is Required for Drosophila Locomotor Function. eLife 2021, 10, e69269. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of Autophagy by Spermidine Promotes Longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, N.B.; Armas, P.; Weiner, A.M.J.; Borgognone, M. CNBP: A Multifunctional Nucleic Acid Chaperone Involved in Cell Death and Proliferation Control. IUBMB Life 2010, 62, 707–714. [Google Scholar] [CrossRef]
- Armas, P.; Nasif, S.; Calcaterra, N.B. Cellular Nucleic Acid Binding Protein Binds G-Rich Single-Stranded Nucleic Acids and May Function as a Nucleic Acid Chaperone. J. Cell. Biochem. 2008, 103, 1013–1036. [Google Scholar] [CrossRef]
- Benhalevy, D.; Gupta, S.K.; Danan, C.H.; Ghosal, S.; Sun, H.-W.; Kazemier, H.G.; Paeschke, K.; Hafner, M.; Juranek, S.A. The Human CCHC-Type Zinc Finger Nucleic Acid-Binding Protein Binds G-Rich Elements in Target MRNA Coding Sequences and Promotes Translation. Cell Rep. 2017, 18, 2979–2990. [Google Scholar] [CrossRef]
- David, A.P.; Pipier, A.; Pascutti, F.; Binolfi, A.; Weiner, A.M.J.; Challier, E.; Heckel, S.; Calsou, P.; Gomez, D.; Calcaterra, N.B.; et al. CNBP Controls Transcription by Unfolding DNA G-Quadruplex Structures. Nucleic Acids Res. 2019, 47, 7901–7913. [Google Scholar] [CrossRef]
- Iadevaia, V.; Caldarola, S.; Tino, E.; Amaldi, F.; Loreni, F. All Translation Elongation Factors and the e, f, and h Subunits of Translation Initiation Factor 3 Are Encoded by 5′-Terminal Oligopyrimidine (TOP) MRNAs. RNA 2008, 14, 1730–1736. [Google Scholar] [CrossRef]
- Leipheimer, J.; Bloom, A.L.M.; Baumstark, T.; Panepinto, J.C. CNBP Homologues Gis2 and Znf9 Interact with a Putative G-Quadruplex-Forming 3′ Untranslated Region, Altering Polysome Association and Stress Tolerance in Cryptococcus Neoformans. mSphere 2018, 3. [Google Scholar] [CrossRef]
- Zu, T.; Cleary, J.D.; Liu, Y.; Bañez-Coronel, M.; Bubenik, J.L.; Ayhan, F.; Ashizawa, T.; Xia, G.; Clark, H.B.; Yachnis, A.T.; et al. RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron 2017, 95, 1292–1305.e5. [Google Scholar] [CrossRef]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular Mechanisms Underlying Nucleotide Repeat Expansion Disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 589–607. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Shin, J.; Yuan, Y.; Beattie, S.G.; Wheeler, T.M.; Thornton, C.A.; Swanson, M.S. Reversal of RNA Missplicing and Myotonia after Muscleblind Overexpression in a Mouse Poly(CUG) Model for Myotonic Dystrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11748–11753. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Goodwin, M.; Swanson, M.S. RNA–Protein Interactions in Unstable Microsatellite Diseases. Brain Res. 2014, 1584, 3–14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sznajder, Ł.J.; Thomas, J.D.; Carrell, E.M.; Reid, T.; McFarland, K.N.; Cleary, J.D.; Oliveira, R.; Nutter, C.A.; Bhatt, K.; Sobczak, K.; et al. Intron Retention Induced by Microsatellite Expansions as a Disease Biomarker. Proc. Natl. Acad. Sci. USA 2018, 115, 4234–4239. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG–Initiated Translation Directed by Microsatellite Expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Pattamatta, A.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation in Neurological Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033019. [Google Scholar] [CrossRef] [PubMed]
- Tusi, S.K.; Nguyen, L.; Thangaraju, K.; Li, J.; Cleary, J.D.; Zu, T.; Ranum, L.P.W. The Alternative Initiation Factor EIF2A Plays Key Role in RAN Translation of Myotonic Dystrophy Type 2 CCUG•CAGG Repeats. Hum. Mol. Genet. 2021, 30, 1020–1029. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.-P.; Hudson, T.; et al. Molecular Basis of Myotonic Dystrophy: Expansion of a Trinucleotide (CTG) Repeat at the 3′ End of a Transcript Encoding a Protein Kinase Family Member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Timchenko, L. Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2. Int. J. Mol. Sci. 2022, 23, 10491. [Google Scholar] [CrossRef]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A Systematic Analysis of Human Disease-Associated Gene Sequences in Drosophila Melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Bosso, G.; Cipressa, F.; Moroni, M.L.; Pennisi, R.; Albanesi, J.; Brandi, V.; Cugusi, S.; Renda, F.; Ciapponi, L.; Polticelli, F.; et al. NBS1 Interacts with HP1 to Ensure Genome Integrity. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bier, E. Drosophila, the Golden Bug, Emerges as a Tool for Human Genetics. Nat. Rev. Genet. 2005, 6, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila Tools and Assays for the Study of Human Diseases. Dis. Model. Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an In Vivo Model for Human Neurodegenerative Disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef]
- Deng, J.; Guan, X.-X.; Zhu, Y.-B.; Deng, H.-T.; Li, G.-X.; Guo, Y.-C.; Jin, P.; Duan, R.-H.; Huang, W. Reducing the Excess Activin Signaling Rescues Muscle Degeneration in Myotonic Dystrophy Type 2 Drosophila Model. J. Pers. Med. 2022, 12, 385. [Google Scholar] [CrossRef]
- Sellier, C.; Cerro-Herreros, E.; Blatter, M.; Freyermuth, F.; Gaucherot, A.; Ruffenach, F.; Sarkar, P.; Puymirat, J.; Udd, B.; Day, J.W.; et al. RbFOX1/MBNL1 Competition for CCUG RNA Repeats Binding Contributes to Myotonic Dystrophy Type 1/Type 2 Differences. Nat. Commun. 2018, 9, 2009. [Google Scholar] [CrossRef]
- Loveless, J.; Lagogiannis, K.; Webb, B. Modelling the Mechanics of Exploration in Larval Drosophila. PLoS Comput. Biol. 2019, 15, e1006635. [Google Scholar] [CrossRef]
- Fushiki, A.; Zwart, M.F.; Kohsaka, H.; Fetter, R.D.; Cardona, A.; Nose, A. A Circuit Mechanism for the Propagation of Waves of Muscle Contraction in Drosophila. Elife 2016, 5, e13253. [Google Scholar] [CrossRef]
- Marzullo, M.; Romano, G.; Pellacani, C.; Riccardi, F.; Ciapponi, L.; Feiguin, F. Su(Var)3-9 Mediates Age-Dependent Increase in H3K9 Methylation on TDP-43 Promoter Triggering Neurodegeneration. Cell Death and Discovery 2023. [Google Scholar] [CrossRef]
- Madabattula, S.T.; Strautman, J.C.; Bysice, A.M.; O’Sullivan, J.A.; Androschuk, A.; Rosenfelt, C.; Doucet, K.; Rouleau, G.; Bolduc, F. Quantitative Analysis of Climbing Defects in a Drosophila Model of Neurodegenerative Disorders. J. Vis. Exp. 2015, 100, e52741. [Google Scholar] [CrossRef]
- Taylor, M.J.; Tuxworth, R.I. Continuous Tracking of Startled Drosophila as an Alternative to the Negative Geotaxis Climbing Assay. J. Neurogenet. 2019, 33, 190–198. [Google Scholar] [CrossRef]
- D’Ercole, C.; D’Angelo, P.; Ruggieri, V.; Proietti, D.; Virtanen, L.; Parisi, C.; Riera, C.S.; Renzini, A.; Macone, A.; Marzullo, M.; et al. Spatially Resolved Transcriptomics Reveals Innervation-Responsive Functional Clusters in Skeletal Muscle. Cell Rep. 2022, 41, 111861. [Google Scholar] [CrossRef] [PubMed]
- Dues, D.J.; Andrews, E.K.; Schaar, C.E.; Bergsma, A.L.; Senchuk, M.M.; Van Raamsdonk, J.M. Aging Causes Decreased Resistance to Multiple Stresses and a Failure to Activate Specific Stress Response Pathways. Aging 2016, 8, 777–795. [Google Scholar] [CrossRef] [PubMed]
- da Costa, J.P.; Vitorino, R.; Silva, G.M.; Vogel, C.; Duarte, A.C.; Rocha-Santos, T. A Synopsis on Aging-Theories, Mechanisms and Future Prospects. Ageing Res. Rev. 2016, 29, 90–112. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Liu, C.; Lu, M.; Dong, Q.; Wang, Z.; Wang, Z.; Xiong, W.; Zhang, N.; Zhou, J.; Liu, Q.; et al. Calorie Restriction Is the Most Reasonable Anti-Ageing Intervention: A Meta-Analysis of Survival Curves. Sci. Rep. 2018, 8, 5779. [Google Scholar] [CrossRef] [PubMed]
- De Rose, F.; Marotta, R.; Talani, G.; Catelani, T.; Solari, P.; Poddighe, S.; Borghero, G.; Marrosu, F.; Sanna, E.; Kasture, S.; et al. Differential Effects of Phytotherapic Preparations in the HSOD1 Drosophila Melanogaster Model of ALS. Sci. Rep. 2017, 7, 41059. [Google Scholar] [CrossRef] [PubMed]
- Iliadi, K.G.; Gluscencova, O.B.; Iliadi, N.; Boulianne, G.L. Mutations in the Drosophila Homolog of Human PLA2G6 Give Rise to Age-Dependent Loss of Psychomotor Activity and Neurodegeneration. Sci. Rep. 2018, 8, 2939. [Google Scholar] [CrossRef]
- Ready, D.F.; Hanson, T.E.; Benzer, S. Development of the Drosophila Retina, a Neurocrystalline Lattice. Dev. Biol. 1976, 53, 217–240. [Google Scholar] [CrossRef]
- Maccallini, P.; Bavasso, F.; Scatolini, L.; Bucciarelli, E.; Noviello, G.; Lisi, V.; Palumbo, V.; D’Angeli, S.; Cacchione, S.; Cenci, G.; et al. Intimate Functional Interactions between TGS1 and the Smn Complex Revealed by an Analysis of the Drosophila Eye Development. PLoS Genet. 2020, 16, e1008815. [Google Scholar] [CrossRef]
- Ciapponi, L.; Jackson, D.B.; Mlodzik, M.; Bohmann, D. Drosophila Fos Mediates ERK and JNK Signals via Distinct Phosphorylation Sites. Genes. Dev. 2001, 15, 1540–1553. [Google Scholar] [CrossRef]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovičić, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila Screen Connects Nuclear Transport Genes to DPR Pathology in C9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef] [PubMed]
- Goodman, L.D.; Prudencio, M.; Srinivasan, A.R.; Rifai, O.M.; Lee, V.M.-Y.; Petrucelli, L.; Bonini, N.M. EIF4B and EIF4H Mediate GR Production from Expanded G4C2 in a Drosophila Model for C9orf72-Associated ALS. Acta Neuropathol. Commun. 2019, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted Gene Expression as a Means of Altering Cell Fates and Generating Dominant Phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef]
- Kroksmark, A.-K.; Ekström, A.-B.; Björck, E.; Tulinius, M. Myotonic Dystrophy: Muscle Involvement in Relation to Disease Type and Size of Expanded CTG-Repeat Sequence. Dev. Med. Child. Neurol. 2005, 47, 478–485. [Google Scholar] [CrossRef]
- Dark, C.; Cheung, S.; Cheng, L.Y. Analyzing Cachectic Phenotypes in the Muscle and Fat Body of Drosophila Larvae. STAR Protoc. 2022, 3, 101230. [Google Scholar] [CrossRef] [PubMed]
- Strah, N.; Romano, G.; Introna, C.; Klima, R.; Marzullo, M.; Ciapponi, L.; Megighian, A.; Nizzardo, M.; Feiguin, F. TDP-43 Promotes the Formation of Neuromuscular Synapses through the Regulation of Disc-Large Expression in Drosophila Skeletal Muscles. BMC Biol. 2020, 18, 34. [Google Scholar] [CrossRef]
- Cerro-Herreros, E.; Chakraborty, M.; Pérez-Alonso, M.; Artero, R.; Llamusí, B. Expanded CCUG Repeat RNA Expression in Drosophila Heart and Muscle Trigger Myotonic Dystrophy Type 1-like Phenotypes and Activate Autophagocytosis Genes. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Abe, Y.; Cheney, L.; Udd, B.; Li, Y.-P. Haploinsuffciency for Znf9 in Znf9+/− Mice Is Associated with Multiorgan Abnormalities Resembling Myotonic Dystrophy. J. Mol. Biol. 2007, 368, 8–17. [Google Scholar] [CrossRef]
- Botta, A.; Caldarola, S.; Vallo, L.; Bonifazi, E.; Fruci, D.; Gullotta, F.; Massa, R.; Novelli, G.; Loreni, F. Effect of the [CCTG]n Repeat Expansion on ZNF9 Expression in Myotonic Dystrophy Type II (DM2). Biochim. Biophys. Acta 2006, 1762, 329–334. [Google Scholar] [CrossRef]
- Massa, R.; Panico, M.B.; Caldarola, S.; Fusco, F.R.; Sabatelli, P.; Terracciano, C.; Botta, A.; Novelli, G.; Bernardi, G.; Loreni, F. The Myotonic Dystrophy Type 2 (DM2) Gene Product Zinc Finger Protein 9 (ZNF9) Is Associated with Sarcomeres and Normally Localized in DM2 Patients’ Muscles. Neuropathol. Appl. Neurobiol. 2010, 36, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.; Hamel, F.; Beaulieu, D.; Patry, L.; Haineault, C.; Tarnopolsky, M.; Schoser, B.; Puymirat, J. Absence of a Differentiation Defect in Muscle Satellite Cells from DM2 Patients. Neurobiol. Dis. 2009, 36, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Stewart, T.M.; Pegg, A.E. Polyamine Metabolism and Cancer: Treatments, Challenges and Opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Coni, S.; Di Magno, L.; Serrao, S.M.; Kanamori, Y.; Agostinelli, E.; Canettieri, G. Polyamine Metabolism as a Therapeutic Target in Hedgehog-Driven Basal Cell Carcinoma and Medulloblastoma. Cells 2019, 8, 150. [Google Scholar] [CrossRef]
- Wallace The Physiological Role of the Polyamines. Eur. J. Clin. Investig. 2000, 30, 1–3. [CrossRef] [PubMed]
- Coni, S.; Bordone, R.; Ivy, D.M.; Yurtsever, Z.N.; Di Magno, L.; D’Amico, R.; Cesaro, B.; Fatica, A.; Belardinilli, F.; Bufalieri, F.; et al. Combined Inhibition of Polyamine Metabolism and EIF5A Hypusination Suppresses Colorectal Cancer Growth through a Converging Effect on MYC Translation. Cancer Lett. 2023, 559, 216120. [Google Scholar] [CrossRef]
- Benhamou, R.I.; Angelbello, A.J.; Wang, E.T.; Disney, M.D. A Toxic RNA Catalyzes the Cellular Synthesis of Its Own Inhibitor, Shunting It to Endogenous Decay Pathways. Cell Chem. Biol. 2020, 27, 223–231.e4. [Google Scholar] [CrossRef]
- Yu, Z.; Goodman, L.D.; Shieh, S.-Y.; Min, M.; Teng, X.; Zhu, Y.; Bonini, N.M. A Fly Model for the CCUG-Repeat Expansion of Myotonic Dystrophy Type 2 Reveals a Novel Interaction with MBNL1. Hum. Mol. Genet. 2015, 24, 954–962. [Google Scholar] [CrossRef]
- Yenigun, V.B.; Sirito, M.; Amcheslavky, A.; Czernuszewicz, T.; Colonques-Bellmunt, J.; García-Alcover, I.; Wojciechowska, M.; Bolduc, C.; Chen, Z.; Castel, A.L.; et al. (CCUG)n RNA Toxicity in a Drosophila Model of Myotonic Dystrophy Type 2 (DM2) Activates Apoptosis. Dis. Models Mech. 2017, 10, 993–1003. [Google Scholar] [CrossRef]
- Souidi, A.; Zmojdzian, M.; Jagla, K. Dissecting Pathogenetic Mechanisms and Therapeutic Strategies in Drosophila Models of Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2018, 19, 4104. [Google Scholar] [CrossRef]
- Jin, Y.; Suzuki, H.; Maegawa, S.; Endo, H.; Sugano, S.; Hashimoto, K.; Yasuda, K.; Inoue, K. A Vertebrate RNA-Binding Protein Fox-1 Regulates Tissue-Specific Splicing via the Pentanucleotide GCAUG. EMBO J. 2003, 22, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P.; Lapasset, L.; Gadea, G.; Fort, P.; Klinck, R.; Irimia, M.; Vignal, E.; Thibault, P.; Prinos, P.; Chabot, B.; et al. MBNL1 and RBFOX2 Cooperate to Establish a Splicing Programme Involved in Pluripotent Stem Cell Differentiation. Nat. Commun. 2013, 4, 2480. [Google Scholar] [CrossRef] [PubMed]
- Damianov, A.; Ying, Y.; Lin, C.-H.; Lee, J.-A.; Tran, D.; Vashisht, A.A.; Bahrami-Samani, E.; Xing, Y.; Martin, K.C.; Wohlschlegel, J.A.; et al. Rbfox Proteins Regulate Splicing as Part of a Large Multiprotein Complex LASR. Cell 2016, 165, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense Transcripts of the Expanded C9ORF72 Hexanucleotide Repeat Form Nuclear RNA Foci and Undergo Repeat-Associated Non-ATG Translation in C9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.D.; Ranum, L.P.W. Repeat Associated Non-ATG (RAN) Translation: New Starts in Microsatellite Expansion Disorders. Curr. Opin. Genet. Dev. 2014, 26, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Almeida, S.; Moore, J.; Gendron, T.F.; Chalasani, U.; Lu, Y.; Du, X.; Nickerson, J.A.; Petrucelli, L.; Weng, Z.; et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 2015, 87, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates That Initiate in Vitro and in Vivo Neuronal Death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pathogenic Mechanism | Drosophila Model | Affected Tissue | Phenotype | Ref |
|---|---|---|---|---|
| Loss of function | UAS-CNBPRNAi | Muscle (c179-G4; How24B-G4; Mef-G4, Mhc-G4) | Locomotion Climbing | [13] |
| RNA toxicity | UAS-CCTG16 | Eye (GMR-G4), Muscle (How24B-G4), Nervous system (elav-G4) | Eye degeneration | |
| UAS-CCTG200 | Muscle RNA foci | [68] | ||
| UAS-CCTG475 | ||||
| UAS-CCTG525 | ||||
| UAS-CCTG700 | ||||
| UAS-CCTG720 | ||||
| Missplicing | [69] | |||
| UAS-CCTG106 | Muscle (Mhc-G4) Eye (GMR-G4) | Apoptotic response | ||
| UAS-CCTG480 | ||||
| Autophagy | ||||
| UAS-CCTG20 | Muscle (Mhc-G4) Heart (GMH5-G4) | Muscle degeneration | [58] | |
| UAS-CCTG1100 | Reduced survival | |||
| Locomotor defects |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzullo, M.; Coni, S.; De Simone, A.; Canettieri, G.; Ciapponi, L. Modeling Myotonic Dystrophy Type 2 Using Drosophila melanogaster. Int. J. Mol. Sci. 2023, 24, 14182. https://doi.org/10.3390/ijms241814182
Marzullo M, Coni S, De Simone A, Canettieri G, Ciapponi L. Modeling Myotonic Dystrophy Type 2 Using Drosophila melanogaster. International Journal of Molecular Sciences. 2023; 24(18):14182. https://doi.org/10.3390/ijms241814182
Chicago/Turabian StyleMarzullo, Marta, Sonia Coni, Assia De Simone, Gianluca Canettieri, and Laura Ciapponi. 2023. "Modeling Myotonic Dystrophy Type 2 Using Drosophila melanogaster" International Journal of Molecular Sciences 24, no. 18: 14182. https://doi.org/10.3390/ijms241814182
APA StyleMarzullo, M., Coni, S., De Simone, A., Canettieri, G., & Ciapponi, L. (2023). Modeling Myotonic Dystrophy Type 2 Using Drosophila melanogaster. International Journal of Molecular Sciences, 24(18), 14182. https://doi.org/10.3390/ijms241814182