

The Role of Cellular Defense Systems of Ferroptosis in Parkinson’s Disease and Alzheimer’s Disease

Abstract
:1. Introduction
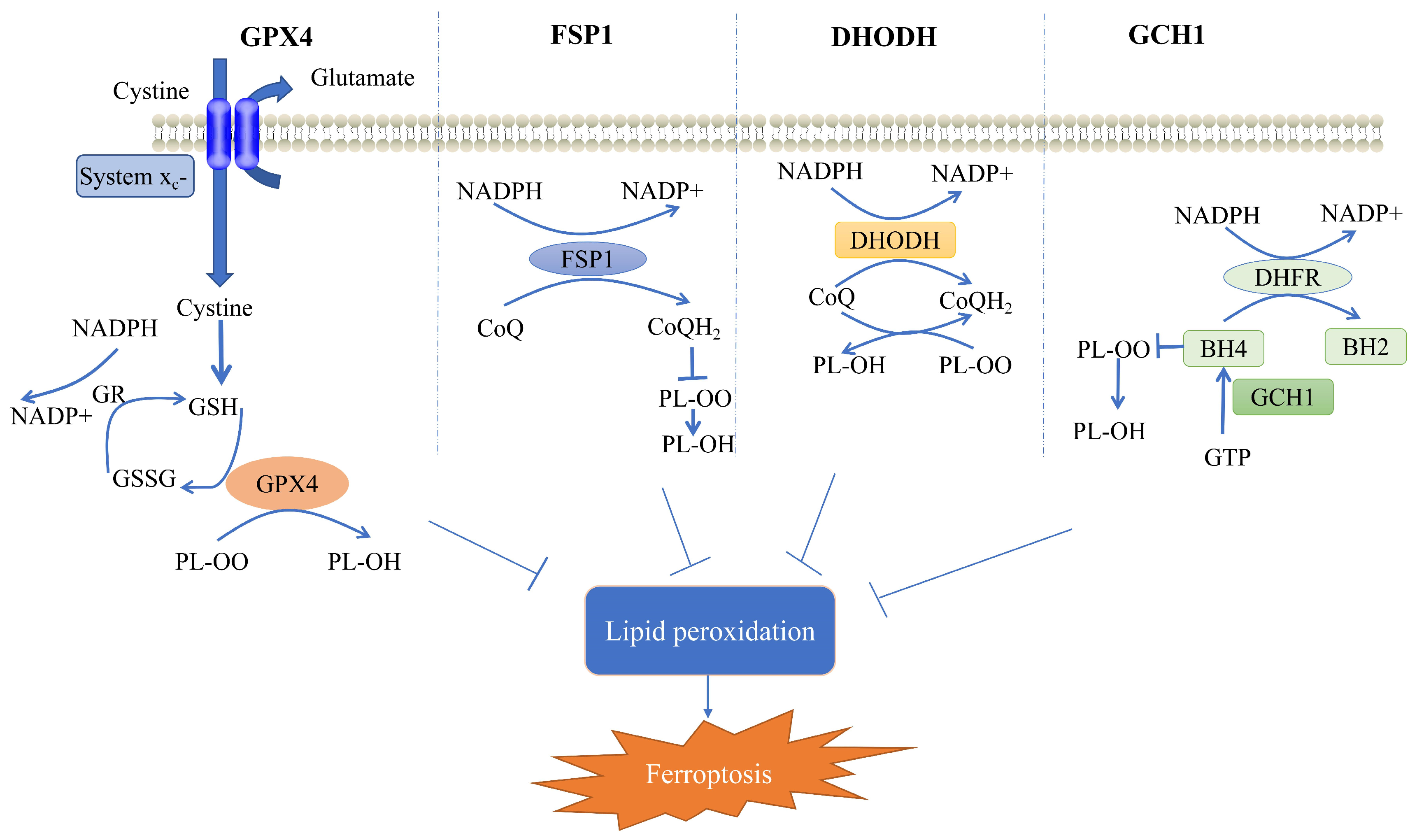
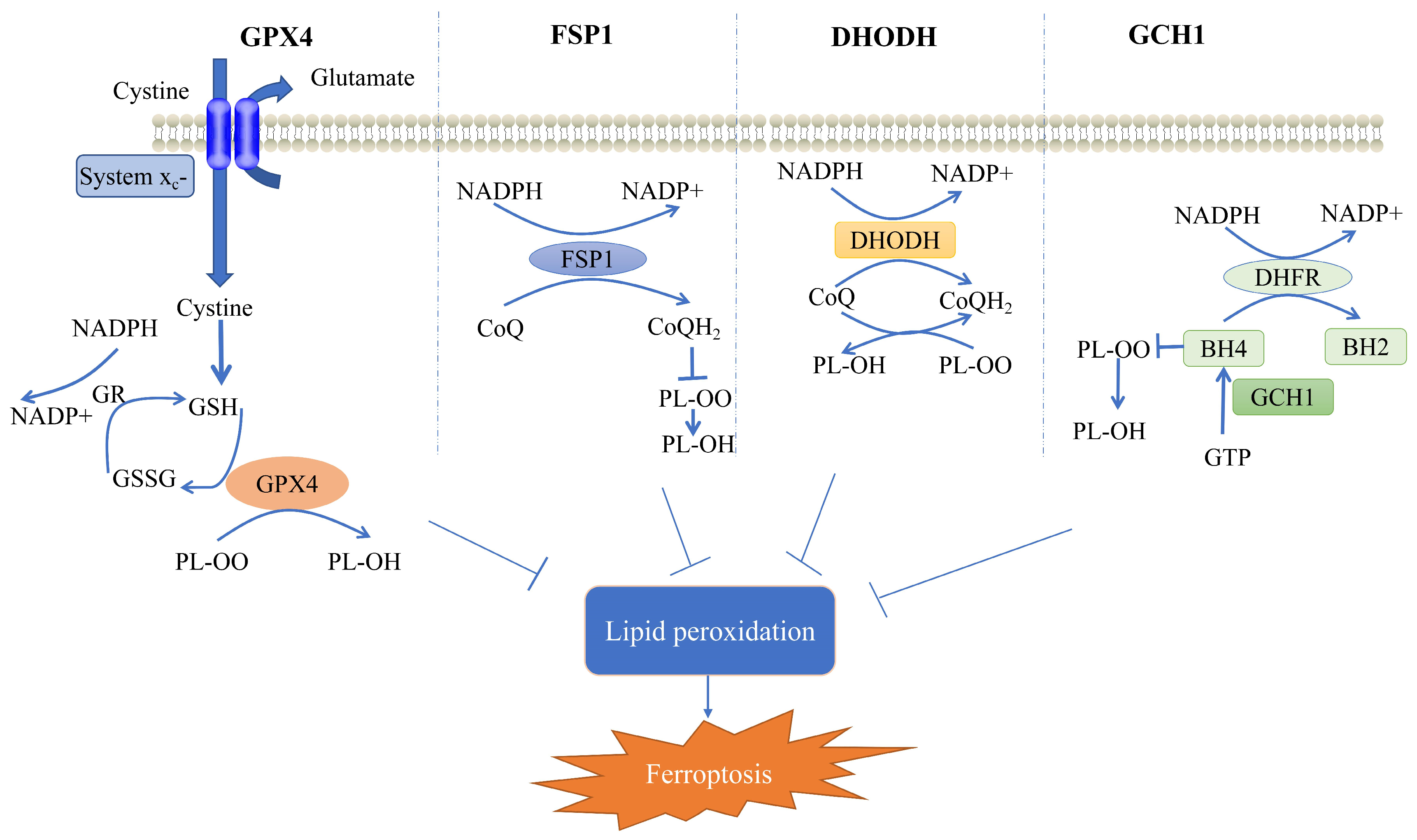
2. Cellular Defense System for Ferroptosis
2.1. GPX4-Mediated Cellular Defense System
2.2. FSP1-Mediated Cellular Defense System
2.3. DHODH-Mediated Cellular Defense System
2.4. GCH1-Mediated Cellular Defense System
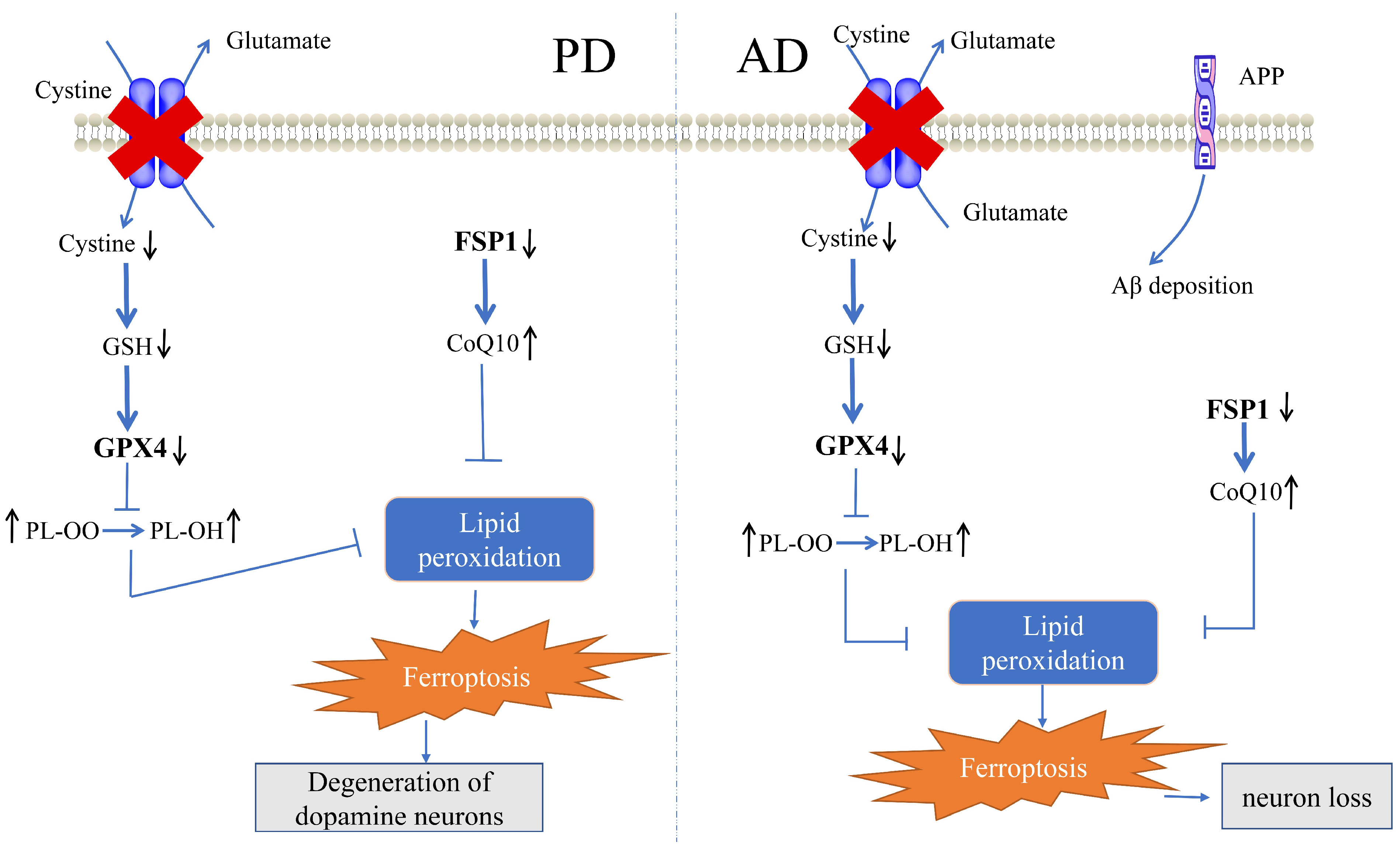
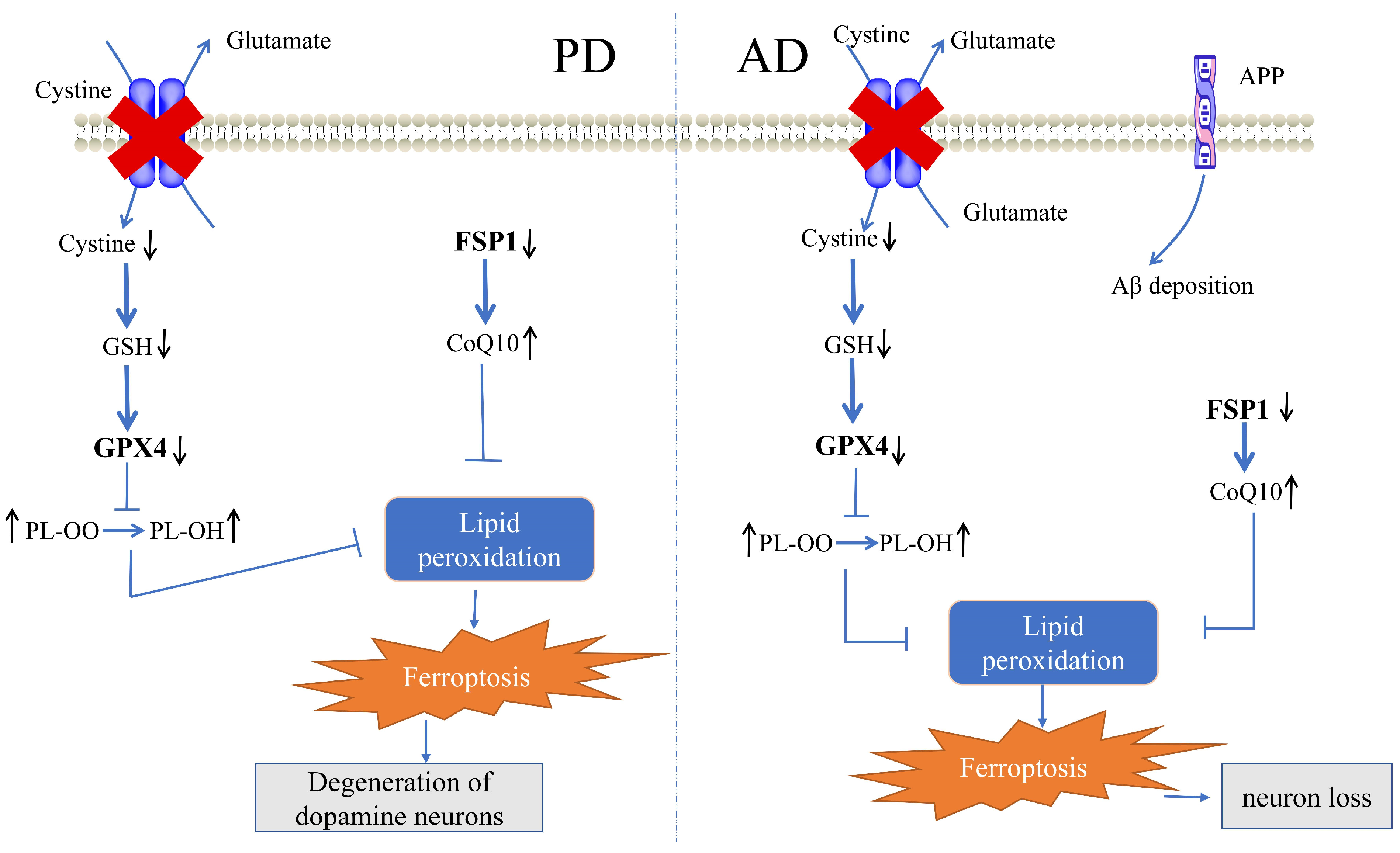
3. Cellular Defense System for Ferroptosis in PD
3.1. Role of the GPX4-Mediated Defense System in PD
3.2. Role of the FSP1-Mediated Defense System in PD
3.3. Role of the GCH1-Mediated Defense System in PD
4. Cellular Defense System for Ferroptosis in AD
4.1. Role of the GPX4-Mediated Defense System in AD
4.2. Role of the FSP1-Mediated Defense System in AD
4.3. Role of the GCH1-Mediated Defense System in AD
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Glossary
| Ferritin | ferritin is primarily recognized as an important intracellular iron storage protein, which is an essential component of iron homeostasis and is involved in a variety of physiological and pathological processes. |
| Phenylephrine | a drug for raising blood pressure. |
| Ferroptosis inducers | a compound or treatment that can induce ferroptosis by boosting ferroptosis-promoting mechanisms and/or suppressing ferroptosis defense mechanisms. |
| Ginsenoside Rg1 | one of the active components of ginseng. |
| MPP+ | 1-methyl-4-phenylpyridine, it has been proven to cause pathophysiological symptoms of PD and has been widely used in the creation of PD models. |
| 6-OHDA | 6-hydroxydopamine, it is mainly used in the establishment of PD models. |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, it is a compound that causes selective degeneration of the substantia nigra after systemic administration and is used in modeling PD. |
| APP/PS1 | APP/PS1 mice, which are commonly used AD animal models. |
| 5×FD | 5×FAD mice, it is a widely used mouse model of AD. |
| 4-hydroxynonenal | HNE, quantitatively one of the most important products of lipid peroxidation. |
| Forsythoside A | the main constituent of Forsythia suspensa. |
| Matrix metalloproteinase-2 | MMP-2, it can be involved in various intracellular mechanisms, including physiological and pathological processes, due to its proteolytic activity. |
| Malondialdehyde | MDA, which is a secondary product of free radical lipid peroxidation. |
References
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Vijiaratnam, N.; Simuni, T.; Bandmann, O.; Morris, H.R.; Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021, 20, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, Y.; Liu, T.; Xu, Y.; Zhao, X.; Wei, J. The Role of Exercise in Maintaining Mitochondrial Proteostasis in Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 7994. [Google Scholar] [CrossRef] [PubMed]
- Ta, M.; Blauwendraat, C.; Antar, T.; Leonard, H.L.; Singleton, A.B.; Nalls, M.A.; Iwaki, H.; Alzheimer’s Disease Neuroimaging Initiative (ADNI); the Fox Investigation for New Discovery of Biomarkers. Genome-Wide Meta-Analysis of Cerebrospinal Fluid Biomarkers in Alzheimer’s Disease and Parkinson’s Disease Cohorts. Mov. Disord. Off. J. Mov. Disord. Soc. 2023. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Mao, C.; Kondiparthi, L.; Poyurovsky, M.V.; Olszewski, K.; Gan, B. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc. Natl. Acad. Sci. USA 2022, 119, e2121987119. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Ito, J.; Wu, Z.J.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Xie, L.H.; Fefelova, N.; Pamarthi, S.H.; Gwathmey, J.K. Molecular Mechanisms of Ferroptosis and Relevance to Cardiovascular Disease. Cells 2022, 11, 2726. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.L. Unleashing Ferroptosis in Human Cancers: Targeting Ferroptosis Suppressor Protein 1 for Overcoming Therapy Resistance. Antioxidants 2023, 12, 1218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.L.; Yuan, L.; Li, W.; Li, J.Y. Ferroptosis in Parkinson’s disease: Glia-neuron crosstalk. Trends Mol. Med. 2022, 28, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Adameova, A.; Hill, J.A.; Baines, C.P.; Kang, P.M.; Downey, J.M.; Narula, J.; Takahashi, M.; Abbate, A.; Piristine, H.C.; et al. Guidelines for evaluating myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H891–H922. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Leak, L.; Dixon, S.J. Surveying the landscape of emerging and understudied cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119432. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Janelidze, S.; Kalinowski, P.; Palmqvist, S.; Belaidi, A.A.; Stomrud, E.; Roberts, A.; Roberts, B.; Hansson, O.; Bush, A.I. CSF ferritin in the clinicopathological progression of Alzheimer’s disease and associations with APOE and inflammation biomarkers. J. Neurol. Neurosurg. Psychiatry 2023, 94, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Libby, P.; Tuomilehto, J.; Lip, G.Y.H.; Penninger, J.M.; Richardson, D.R.; Tang, D.; Zhou, H.; Wang, S.; et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol. Metab. 2021, 32, 444–462. [Google Scholar] [CrossRef]
- Yang, B.; Pan, J.; Zhang, X.N.; Wang, H.; He, L.; Rong, X.; Li, X.; Peng, Y. NRF2 activation suppresses motor neuron ferroptosis induced by the SOD1(G93A) mutation and exerts neuroprotection in amyotrophic lateral sclerosis. Neurobiol. Dis. 2023, 184, 106210. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lin, X.M.; Lu, D.H.; Wang, M.; Li, K.; Li, S.R.; Li, Z.Q.; Zhu, C.J.; Zhang, Z.M.; Yan, C.Y.; et al. Midbrain dopamine oxidation links ubiquitination of glutathione peroxidase 4 to ferroptosis of dopaminergic neurons. J. Clin. Investig. 2023, 133, e165228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yu, X.; Xie, J.; Xu, H. New Insights into the Role of Ferritin in Iron Homeostasis and Neurodegenerative Diseases. Mol. Neurobiol. 2021, 58, 2812–2823. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, S.; Li, Q.; Sun, H.; Wang, H. Pharmacological Inhibition of Ferroptosis as a Therapeutic Target for Neurodegenerative Diseases and Strokes. Adv. Sci. 2023, 10, e2300325. [Google Scholar] [CrossRef]
- Mayneris-Perxachs, J.; Moreno-Navarrete, J.M.; Fernandez-Real, J.M. The role of iron in host-microbiota crosstalk and its effects on systemic glucose metabolism. Nat. Rev. Endocrinol. 2022, 18, 683–698. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, F. Iron homeostasis and tumorigenesis: Molecular mechanisms and therapeutic opportunities. Protein Cell 2015, 6, 88–100. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron: The cancer connection. Mol. Asp. Med. 2020, 75, 100860. [Google Scholar] [CrossRef] [PubMed]
- Holbein, B.E.; Lehmann, C. Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target. Antioxidants 2023, 12, 671. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Petrillo, S.; Fiorenza, M.T.; Bertini, E.S.; Piemonte, F. Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease. Biomolecules 2020, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Gordan, R.; Wongjaikam, S.; Gwathmey, J.K.; Chattipakorn, N.; Chattipakorn, S.C.; Xie, L.H. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: An update. Heart Fail. Rev. 2018, 23, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e310. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Liao, K.; Zhou, Y.; Wen, T.; Quan, G.; Pan, X.; Wu, C. Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials 2021, 277, 121110. [Google Scholar] [CrossRef]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Amos, A.; Amos, A.; Wu, L.; Xia, H. The Warburg effect modulates DHODH role in ferroptosis: A review. Cell Commun. Signal 2023, 21, 100. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Min, J. DHODH tangoing with GPX4 on the ferroptotic stage. Signal Transduct. Target. Ther. 2021, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, J.; Li, R.; Liu, Y.; Zhou, L.; Wang, C.; Lv, C.; Gao, L.; Cui, D. CircLRFN5 inhibits the progression of glioblastoma via PRRX2/GCH1 mediated ferroptosis. J. Exp. Clin. Cancer Res. 2022, 41, 307. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Zhang, Y.; Swanda, R.V.; Nie, L.; Liu, X.; Wang, C.; Lee, H.; Lei, G.; Mao, C.; Koppula, P.; Cheng, W.; et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 2021, 12, 1589. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kang, S.W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.H.; Kim, G.; Lee, P.; et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, Y.; Jiang, R.T.; Xue, R.; Yin, X.H.; Wu, M.C.; Meng, Q.H. Ferroptosis in liver disease: New insights into disease mechanisms. Cell Death Discov. 2021, 7, 276. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bi, J.B.; Ren, Y.F.; Du, Z.Q.; Li, T.; Wang, T.; Zhang, L.; Wang, M.Z.; Wei, S.S.; Lv, Y.; et al. Involvement of GPX4 in irisin’s protection against ischemia reperfusion-induced acute kidney injury. J. Cell Physiol. 2021, 236, 931–945. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dancik, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e895. [Google Scholar] [CrossRef]
- Jin, D.Y.; Chen, X.; Liu, Y.; Williams, C.M.; Pedersen, L.C.; Stafford, D.W.; Tie, J.K. A genome-wide CRISPR-Cas9 knockout screen identifies FSP1 as the warfarin-resistant vitamin K reductase. Nat. Commun. 2023, 14, 828. [Google Scholar] [CrossRef]
- Xavier da Silva, T.N.; Schulte, C.; Alves, A.N.; Maric, H.M.; Friedmann Angeli, J.P. Molecular characterization of AIFM2/FSP1 inhibition by iFSP1-like molecules. Cell Death Dis. 2023, 14, 281. [Google Scholar] [CrossRef]
- Hendricks, J.M.; Doubravsky, C.E.; Wehri, E.; Li, Z.; Roberts, M.A.; Deol, K.K.; Lange, M.; Lasheras-Otero, I.; Momper, J.D.; Dixon, S.J.; et al. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem. Biol. 2023, in press. [Google Scholar] [CrossRef]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Shi, J.; Liu, X.; Horbath, A.; Das, M.; et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar] [CrossRef]
- Nakamura, T.; Hipp, C.; Santos Dias Mourao, A.; Borggrafe, J.; Aldrovandi, M.; Henkelmann, B.; Wanninger, J.; Mishima, E.; Lytton, E.; Emler, D.; et al. Phase separation of FSP1 promotes ferroptosis. Nature 2023, 619, 371–377. [Google Scholar] [CrossRef]
- Li, W.; Liang, L.; Liu, S.; Yi, H.; Zhou, Y. FSP1: A key regulator of ferroptosis. Trends Mol. Med. 2023, 29, 753–764. [Google Scholar] [CrossRef]
- Wang, S.; Li, W.; Zhang, P.; Wang, Z.; Ma, X.; Liu, C.; Vasilev, K.; Zhang, L.; Zhou, X.; Liu, L.; et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J. Adv. Res. 2022, 41, 63–75. [Google Scholar] [CrossRef]
- Guo, J.; Wang, R.; Min, F. Ginsenoside Rg1 ameliorates sepsis-induced acute kidney injury by inhibiting ferroptosis in renal tubular epithelial cells. J. Leukoc. Biol. 2022, 112, 1065–1077. [Google Scholar] [CrossRef]
- Tonnus, W.; Meyer, C.; Steinebach, C.; Belavgeni, A.; von Massenhausen, A.; Gonzalez, N.Z.; Maremonti, F.; Gembardt, F.; Himmerkus, N.; Latk, M.; et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 2021, 12, 4402. [Google Scholar] [CrossRef]
- Li, L.; Ng, S.R.; Colón, C.I.; Drapkin, B.J.; Hsu, P.P.; Li, Z.; Nabel, C.S.; Lewis, C.A.; Romero, R.; Mercer, K.L.; et al. Identification of DHODH as a therapeutic target in small cell lung cancer. Sci. Transl. Med. 2019, 11, eaaw7852. [Google Scholar] [CrossRef]
- Madak, J.T.; Bankhead, A., 3rd; Cuthbertson, C.R.; Showalter, H.D.; Neamati, N. Revisiting the role of dihydroorotate dehydrogenase as a therapeutic target for cancer. Pharmacol. Ther. 2019, 195, 111–131. [Google Scholar] [CrossRef]
- Abt, E.R.; Rosser, E.W.; Durst, M.A.; Lok, V.; Poddar, S.; Le, T.M.; Cho, A.; Kim, W.; Wei, L.; Song, J.; et al. Metabolic Modifier Screen Reveals Secondary Targets of Protein Kinase Inhibitors within Nucleotide Metabolism. Cell Chem. Biol. 2020, 27, 197–205.e6. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.G.; Zhang, Y.L.; Lei, G.; Yan, Y.L.; Lee, H.M.; Koppula, P.; Wu, S.Q.; Zhuang, L.; Fang, B.L.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 596, 586–590. [Google Scholar] [CrossRef]
- Eichwald, T.; da Silva, L.B.; Staats Pires, A.C.; Niero, L.; Schnorrenberger, E.; Filho, C.C.; Espíndola, G.; Huang, W.L.; Guillemin, G.J.; Abdenur, J.E.; et al. Tetrahydrobiopterin: Beyond Its Traditional Role as a Cofactor. Antioxidants 2023, 12, 1037. [Google Scholar] [CrossRef]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef]
- Lv, Y.; Wu, M.; Wang, Z.; Wang, J. Ferroptosis: From regulation of lipid peroxidation to the treatment of diseases. Cell Biol. Toxicol. 2022, 39, 827–851. [Google Scholar] [CrossRef]
- Xue, J.; Yu, C.; Sheng, W.; Zhu, W.; Luo, J.; Zhang, Q.; Yang, H.; Cao, H.; Wang, W.; Zhou, J.; et al. The Nrf2/GCH1/BH4 Axis Ameliorates Radiation-Induced Skin Injury by Modulating the ROS Cascade. J. Investig. Derm. 2017, 137, 2059–2068. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Y.; Chen, X.; Zhong, H.; Wang, Y. Ferroptosis in life: To be or not to be. Biomed. Pharm. 2023, 159, 114241. [Google Scholar] [CrossRef]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. 2023, 18, 95–121. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Y.; Wang, C.; Han, T.; Liu, H.; Sun, L.; Hong, J.; Hashimoto, M.; Wei, J. The reciprocal interactions between microglia and T cells in Parkinson’s disease: A double-edged sword. J. Neuroinflamm. 2023, 20, 33. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef]
- Kwon, K.; Cho, H.; Lee, S.; Cho, E.J.; Yu, W.; Kok, C.Y.L.; Je, H.S.; Kim, J.I.; Cho, H.J.; Kwon, T. Adaptive cellular response of the substantia nigra dopaminergic neurons upon age-dependent iron accumulation. Aging Cell 2022, 21, e13694. [Google Scholar] [CrossRef]
- Sterling, J.K.; Kam, T.I.; Guttha, S.; Park, H.; Baumann, B.; Mehrabani-Tabari, A.A.; Schultz, H.; Anderson, B.; Alnemri, A.; Chou, S.C.; et al. Interleukin-6 triggers toxic neuronal iron sequestration in response to pathological α-synuclein. Cell Rep. 2022, 38, 110358. [Google Scholar] [CrossRef]
- Chung, S.J.; Lee, H.S.; Yoo, H.S.; Lee, Y.H.; Lee, P.H.; Sohn, Y.H. Patterns of striatal dopamine depletion in early Parkinson disease: Prognostic relevance. Neurology 2020, 95, e280–e290. [Google Scholar] [CrossRef]
- Xu, P.; Huang, S.; Krumm, B.E.; Zhuang, Y.; Mao, C.; Zhang, Y.; Wang, Y.; Huang, X.P.; Liu, Y.F.; He, X.; et al. Structural genomics of the human dopamine receptor system. Cell Res. 2023, 33, 604–616. [Google Scholar] [CrossRef]
- David, S.; Jhelum, P.; Ryan, F.; Jeong, S.Y.; Kroner, A. Dysregulation of Iron Homeostasis in the Central Nervous System and the Role of Ferroptosis in Neurodegenerative Disorders. Antioxid. Redox Signal 2022, 37, 150–170. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Ryan, S.K.; Zelic, M.; Han, Y.; Teeple, E.; Chen, L.; Sadeghi, M.; Shankara, S.; Guo, L.; Li, C.; Pontarelli, F.; et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 2023, 26, 12–26. [Google Scholar] [CrossRef]
- Depierreux, F.; Parmentier, E.; Mackels, L.; Baquero, K.; Degueldre, C.; Balteau, E.; Salmon, E.; Phillips, C.; Bahri, M.A.; Maquet, P.; et al. Parkinson’s disease multimodal imaging: F-DOPA PET, neuromelanin-sensitive and quantitative iron-sensitive MRI. NPJ Park. Dis. 2021, 7, 57. [Google Scholar] [CrossRef]
- Biondetti, E.; Santin, M.D.; Valabregue, R.; Mangone, G.; Gaurav, R.; Pyatigorskaya, N.; Hutchison, M.; Yahia-Cherif, L.; Villain, N.; Habert, M.O.; et al. The spatiotemporal changes in dopamine, neuromelanin and iron characterizing Parkinson’s disease. Brain 2021, 144, 3114–3125. [Google Scholar] [CrossRef]
- Sun, Y.; He, L.; Wang, T.; Hua, W.; Qin, H.; Wang, J.; Wang, L.; Gu, W.; Li, T.; Li, N.; et al. Activation of p62-Keap1-Nrf2 Pathway Protects 6-Hydroxydopamine-Induced Ferroptosis in Dopaminergic Cells. Mol. Neurobiol. 2020, 57, 4628–4641. [Google Scholar] [CrossRef]
- Ito, K.; Eguchi, Y.; Imagawa, Y.; Akai, S.; Mochizuki, H.; Tsujimoto, Y. MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells. Cell Death Discov. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Song, L.M.; Xiao, Z.X.; Zhang, N.; Yu, X.Q.; Cui, W.; Xie, J.X.; Xu, H.M. Apoferritin improves motor deficits in MPTP-treated mice by regulating brain iron metabolism and ferroptosis. iScience 2021, 24, 102431. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; von Coelln, R.; Pihlstrøm, L.; Simón-Sánchez, J.; Schulte, C.; Sharma, M.; Krohn, L.; et al. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 866–875. [Google Scholar] [CrossRef]
- Artyukhova, M.A.; Tyurina, Y.Y.; Chu, C.T.; Zharikova, T.M.; Bayir, H.; Kagan, V.E.; Timashev, P.S. Interrogating Parkinson’s disease associated redox targets: Potential application of CRISPR editing. Free Radic. Biol. Med. 2019, 144, 279–292. [Google Scholar] [CrossRef]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remião, F.; Silva, R. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Metselaar, B.; Greenough, M.; Bush, A.I.; Ayton, S.J. Ferroptosis and NRF2: An emerging battlefield in the neurodegeneration of Alzheimer’s disease. Essays Biochem. 2021, 65, 925–940. [Google Scholar] [CrossRef]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef]
- Xie, Y.; Kang, R.; Klionsky, D.J.; Tang, D. GPX4 in cell death, autophagy, and disease. Autophagy 2023, 19, 2621–2638. [Google Scholar] [CrossRef]
- Proneth, B.; Conrad, M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019, 26, 14–24. [Google Scholar] [CrossRef]
- Bellinger, F.P.; Bellinger, M.T.; Seale, L.A.; Takemoto, A.S.; Raman, A.V.; Miki, T.; Manning-Boğ, A.B.; Berry, M.J.; White, L.R.; Ross, G.W. Glutathione Peroxidase 4 is associated with Neuromelanin in Substantia Nigra and Dystrophic Axons in Putamen of Parkinson’s brain. Mol. Neurodegener. 2011, 6, 8. [Google Scholar] [CrossRef]
- Vellingiri, B.; Suriyanarayanan, A.; Selvaraj, P.; Abraham, K.S.; Pasha, M.Y.; Winster, H.; Gopalakrishnan, A.V.; G, S.; Reddy, J.K.; Ayyadurai, N.; et al. Role of heavy metals (copper (Cu), arsenic (As), cadmium (Cd), iron (Fe) and lithium (Li)) induced neurotoxicity. Chemosphere 2022, 301, 134625. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, G.; Alimujiang, A.; Xie, H.; Yang, W.; Yin, F.; Huang, D. Protective Effects of Querectin against MPP(+)-Induced Dopaminergic Neurons Injury via the Nrf2 Signaling Pathway. Front. Biosci. 2023, 28, 42. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Yue, M.; Wei, J.; Chen, W.; Hong, D.; Chen, T.; Fang, X. Neurotrophic Role of the Next-Generation Probiotic Strain L. lactis MG1363-pMG36e-GLP-1 on Parkinson’s Disease via Inhibiting Ferroptosis. Nutrients 2022, 14, 4886. [Google Scholar] [CrossRef]
- Cronin, S.J.F.; Seehus, C.; Weidinger, A.; Talbot, S.; Reissig, S.; Seifert, M.; Pierson, Y.; McNeill, E.; Longhi, M.S.; Turnes, B.L.; et al. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 2018, 563, 564–568. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, Q.; Ma, S.R.; Zhao, Z.X.; Pan, L.B.; Cong, L.; Han, P.; Peng, R.; Yu, H.; Lin, Y.; et al. Oral berberine improves brain dopa/dopamine levels to ameliorate Parkinson’s disease by regulating gut microbiota. Signal Transduct. Target. Ther. 2021, 6, 77. [Google Scholar] [CrossRef]
- Pu, J.L.; Lin, Z.H.; Zheng, R.; Yan, Y.Q.; Xue, N.J.; Yin, X.Z.; Zhang, B.R. Association analysis of SYT11, FGF20, GCH1 rare variants in Parkinson’s disease. CNS Neurosci. 2022, 28, 175–177. [Google Scholar] [CrossRef]
- Jian, X.; Zhao, G.; Chen, H.; Wang, Y.; Li, J.; Xie, L.; Li, B. Revealing a novel contributing landscape of ferroptosis-related genes in Parkinson’s disease. Comput. Struct. Biotechnol. J. 2022, 20, 5218–5225. [Google Scholar] [CrossRef]
- Pan, H.X.; Zhao, Y.W.; Mei, J.P.; Fang, Z.H.; Wang, Y.; Zhou, X.; Zhou, Y.J.; Zhang, R.; Zhang, K.L.; Jiang, L.; et al. GCH1 variants contribute to the risk and earlier age-at-onset of Parkinson’s disease: A two-cohort case-control study. Transl. Neurodegener. 2020, 9, 31. [Google Scholar] [CrossRef]
- Xu, Q.; Li, K.; Sun, Q.; Ding, D.; Zhao, Y.; Yang, N.; Luo, Y.; Liu, Z.; Zhang, Y.; Wang, C.; et al. Rare GCH1 heterozygous variants contributing to Parkinson’s disease. Brain 2017, 140, e41. [Google Scholar] [CrossRef]
- Olazaran, J.; Carnero-Pardo, C.; Fortea, J.; Sanchez-Juan, P.; Garcia-Ribas, G.; Vinuela, F.; Martinez-Lage, P.; Boada, M. Prevalence of treated patients with Alzheimer’s disease: Current trends and COVID-19 impact. Alzheimer’s Res. Therapy 2023, 15, 130. [Google Scholar] [CrossRef]
- Kweon, O.J.; Youn, Y.C.; Lim, Y.K.; Lee, M.K.; Kim, H.R. Clinical utility of serum hepcidin and iron profile measurements in Alzheimer’s disease. J. Neurol. Sci. 2019, 403, 85–91. [Google Scholar] [CrossRef]
- Dysken, M.W.; Guarino, P.D.; Vertrees, J.E.; Asthana, S.; Sano, M.; Llorente, M.; Pallaki, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; et al. Vitamin E and memantine in Alzheimer’s disease: Clinical trial methods and baseline data. Alzheimer’s Dement. 2014, 10, 36–44. [Google Scholar] [CrossRef]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.D.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Wang, Y.M.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Y.T.; Zhang, J.H.; Han, K.; Zhang, X.W.; Bai, X.; You, L.H.; Yu, P.; Shi, Z.H.; Chang, Y.Z.; et al. Astrocyte hepcidin ameliorates neuronal loss through attenuating brain iron deposition and oxidative stress in APP/PS1 mice. Free Radic. Biol. Med. 2020, 158, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Gunn, A.P.; Wong, B.X.; Ayton, S.; Appukuttan, A.T.; Roberts, B.R.; Duce, J.A.; Bush, A.I. Marked Age-Related Changes in Brain Iron Homeostasis in Amyloid Protein Precursor Knockout Mice. Neurotherapeutics 2018, 15, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.; Mela, V.; Harty, C.; Minogue, A.M.; Costello, D.A.; Kerskens, C.; Lynch, M.A. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef] [PubMed]
- Tsatsanis, A.; Wong, B.X.; Gunn, A.P.; Ayton, S.; Bush, A.I.; Devos, D.; Duce, J.A. Amyloidogenic processing of Alzheimer’s disease β-amyloid precursor protein induces cellular iron retention. Mol. Psychiatry 2020, 25, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Svobodová, H.; Kosnáč, D.; Balázsiová, Z.; Tanila, H.; Miettinen, P.O.; Sierra, A.; Vitovič, P.; Wagner, A.; Polák, Š.; Kopáni, M. Elevated age-related cortical iron, ferritin and amyloid plaques in APP(swe)/PS1(deltaE9) transgenic mouse model of Alzheimer’s disease. Physiol. Res. 2019, 68, S445–S451. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Dar, N.J.; Na, R.; McLane, K.D.; Yoo, K.; Han, X.; Ran, Q. Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radic. Biol. Med. 2022, 180, 1–12. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Ge, Y.; Chen, J.; Ma, J.; Wang, C.; Sun, M.; Wang, L.; Yao, S.; Yao, C. β-amyloid protein induces mitophagy-dependent ferroptosis through the CD36/PINK/PARKIN pathway leading to blood-brain barrier destruction in Alzheimer’s disease. Cell Biosci. 2022, 12, 69. [Google Scholar] [CrossRef]
- Bulk, M.; Kenkhuis, B.; van der Graaf, L.M.; Goeman, J.J.; Natte, R.; van der Weerd, L. Postmortem T2*- Weighted MRI Imaging of Cortical Iron Reflects Severity of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 65, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.E.; Chen, L.J.; Na, R.; Liu, Y.H.; Rios, C.; Van Remmen, H.; Richardson, A.; Ran, Q.T. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biology Med. 2012, 52, 1820–1827. [Google Scholar] [CrossRef] [PubMed]
- Dar, N.J.; Na, R.; Ran, Q. Functional Deficits of 5×FAD Neural Stem Cells Are Ameliorated by Glutathione Peroxidase 4. Cells 2022, 11, 1770. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.T.; Wu, Q.L.; Wang, S.S.; Yang, S.W.; Li, X.; Chen, N.H.; Li, L.; Zhang, L. Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int. Immunopharmacol. 2021, 99, 108002. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Chen, S.S.; Guo, H.Y.; Jiang, H.B.; Liu, H.H.; Fu, H.R.; Wang, D. Forsythoside A Mitigates Alzheimer’s-like Pathology by Inhibiting Ferroptosis-mediated Neuroinflammation via Nrf2/GPX4 Axis Activation. Int. J. Biol. Sci. 2022, 18, 2075–2090. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, X.; Xiao, A.; Han, J.; Wang, Z.; Wen, M. Ketogenic diet prevents chronic sleep deprivation-induced Alzheimer’s disease by inhibiting iron dyshomeostasis and promoting repair via Sirt1/Nrf2 pathway. Front. Aging Neurosci. 2022, 14, 998292. [Google Scholar] [CrossRef]
- Zhou, X.P.; Chen, Y.; Mok, K.Y.; Zhao, Q.H.; Chen, K.L.; Chen, Y.W.; Hardy, J.; Li, Y.; Fu, A.K.Y.; Guo, Q.H.; et al. Identification of genetic risk factors in the Chinese population implicates a role of immune system in Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 1697–1706. [Google Scholar] [CrossRef]
- Li, K.; Wang, M.; Huang, Z.H.; Wang, M.; Sun, W.Y.; Kurihara, H.; Huang, R.T.; Wang, R.; Huang, F.; Liang, L.; et al. ALOX5 inhibition protects against dopaminergic neurons undergoing ferroptosis. Pharm. Res. 2023, 193, 106779. [Google Scholar] [CrossRef]
- Yu, X.; Yang, Y.; Zhang, B.; Han, G.; Yu, J.; Yu, Q.; Zhang, L. Ketone Body β-Hydroxybutyric Acid Ameliorates Dopaminergic Neuron Injury Through Modulating Zinc Finger Protein 36/Acyl-CoA Synthetase Long-Chain Family Member Four Signaling Axis-Mediated Ferroptosis. Neuroscience 2023, 509, 157–172. [Google Scholar] [CrossRef]
- Lin, Z.H.; Liu, Y.; Xue, N.J.; Zheng, R.; Yan, Y.Q.; Wang, Z.X.; Li, Y.L.; Ying, C.Z.; Song, Z.; Tian, J.; et al. Quercetin Protects against MPP(+)/MPTP-Induced Dopaminergic Neuron Death in Parkinson’s Disease by Inhibiting Ferroptosis. Oxid. Med. Cell Longev. 2022, 2022, 7769355. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.B.; Jiang, H.; Yang, Y.; Wang, G.H.; Ji, Q.H.; Jia, Z.Z.; Shen, L.H.; Luo, Q.Q. DL-3-n-butylphthalide alleviates motor disturbance by suppressing ferroptosis in a rat model of Parkinson’s disease. Neural Regen. Res. 2023, 18, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; He, L.; Wang, W.; Xie, Z.; Zhang, X.; Wang, P.; Wang, L.; Yan, C.; Liu, Z.; Zhao, J.; et al. Activation of Atg7-dependent autophagy by a novel inhibitor of the Keap1-Nrf2 protein-protein interaction from Penthorum chinense Pursh. attenuates 6-hydroxydopamine-induced ferroptosis in zebrafish and dopaminergic neurons. Food Funct. 2022, 13, 7885–7900. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; An, H.; Yu, F.; Yang, J.; Ding, H.; Bao, Y.; Xie, H.; Huang, D. The neuroprotective effects of paeoniflorin against MPP(+)-induced damage to dopaminergic neurons via the Akt/Nrf2/GPX4 pathway. J. Chem. Neuroanat. 2022, 122, 102103. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, S.; Wang, H. α-Lipoic acid alleviates ferroptosis in the MPP(+) -induced PC12 cells via activating the PI3K/Akt/Nrf2 pathway. Cell Biol. Int. 2021, 45, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; An, H.; Yu, F.; Wang, K.; Zheng, L.; Zhou, W.; Bao, Y.; Yang, J.; Shen, N.; Huang, D. Benefits of Iron Chelators in the Treatment of Parkinson’s Disease. Neurochem. Res. 2021, 46, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Avcı, B.; Günaydın, C.; Güvenç, T.; Yavuz, C.K.; Kuruca, N.; Bilge, S.S. Idebenone Ameliorates Rotenone-Induced Parkinson’s Disease in Rats Through Decreasing Lipid Peroxidation. Neurochem. Res. 2021, 46, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, L.; Zeng, Y.; Wang, Y.; Pei, T.; Xie, Z.; Xiong, Q.; Wei, H.; Li, W.; Li, J.; et al. Salidroside alleviates cognitive impairment by inhibiting ferroptosis via activation of the Nrf2/GPX4 axis in SAMP8 mice. Phytomedicine Int. J. Phytother. Phytopharm. 2023, 114, 154762. [Google Scholar] [CrossRef]
- Yang, S.; Xie, Z.; Pei, T.; Zeng, Y.; Xiong, Q.; Wei, H.; Wang, Y.; Cheng, W. Salidroside attenuates neuronal ferroptosis by activating the Nrf2/HO1 signaling pathway in Aβ(1-42)-induced Alzheimer’s disease mice and glutamate-injured HT22 cells. Chin. Med. 2022, 17, 82. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Li, B.; Yao, H.; Zarka, M.; Welch, J.; Sachdev, P.; Bridge, W.; Braidy, N. Supplementation with γ-glutamylcysteine (γ-GC) lessens oxidative stress, brain inflammation and amyloid pathology and improves spatial memory in a murine model of AD. Neurochem. Int. 2021, 144, 104931. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Type | Predisposing Factor | Morphological Features | Biochemical Features | Commonly Detected Indicators |
|---|---|---|---|---|
| Ferroptosis | Accumulation of iron ions | Cell membrane: lack of rupture and blebbing of the plasma membrane, rounding-up of the cell; Cytoplasm: small mitochondria with condensed mitochondrial membrane densities, reduction or vanishing of mitochondrial crista, as well as outer mitochondrial membrane rupture; Nucleus: normal nuclear size and lack of chronmatin condensation | Iron and ROS accumulation; Activation of MAPKs; Inhibition of system Xc- with decreased cystine uptake GSH depletion and increased NAPDH oxidation; Release of arachidonic acid mediators; Δψm dissipation | Iron glutathione MDA GPX4 ROS LPO LDH cytotoxicity |
| Apoptosis | Gene regulation under normal physiological conditions | Cell membrane: plasma membrane blebbing, rounding-up of the cell; Cytoplasm: retraction of pseudopods, reduction of cellular volume; Nucleus: reduction of neclear volume, nuclear fragmentation, chromatin condensation | Activation of caspase; Oligonucleosomal DNA fragmentation; Δψm dissipation; PS exposure | Caspas series TUNEL Bcl-2 Bax |
| Necroptosis | Activated by the death receptor ligands and pattern recognition receptors of the innate immune system | Cell membrane: rupture of plasma membrane; Cytoplasm: cytoplasmic swelling (oncosis), swelling of cytoplasmic organelles; Nucleus: moderate chromatin condensation | Drop in ATP levels; Activation of RIPK1, RIPK3, and MLKL; Release of DAMPs; PARP1 hyperactivation | Hexosaminidase Calcein-AM Annexin-V ATP |
| Autophagic cell death | Nutritional deficiencies or hormone induction | Cell membrane: lack of change; Cytoplasm: accumulation of double-membraned autophagic vacuoles; Nucleus: lack of chromatin condensation | LC3-I to LC3-Ⅱ conversion; Substrate (e.g., p62) degradation | LC3 ATG series proteins |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, J.; Li, J.; Sun, L.; Wei, J. The Role of Cellular Defense Systems of Ferroptosis in Parkinson’s Disease and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 14108. https://doi.org/10.3390/ijms241814108
Chu J, Li J, Sun L, Wei J. The Role of Cellular Defense Systems of Ferroptosis in Parkinson’s Disease and Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(18):14108. https://doi.org/10.3390/ijms241814108
Chicago/Turabian StyleChu, Jie, Jingwen Li, Lin Sun, and Jianshe Wei. 2023. "The Role of Cellular Defense Systems of Ferroptosis in Parkinson’s Disease and Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 18: 14108. https://doi.org/10.3390/ijms241814108
APA StyleChu, J., Li, J., Sun, L., & Wei, J. (2023). The Role of Cellular Defense Systems of Ferroptosis in Parkinson’s Disease and Alzheimer’s Disease. International Journal of Molecular Sciences, 24(18), 14108. https://doi.org/10.3390/ijms241814108