
P2 Receptor Antagonists Rescue Defective Heme Content in an In Vitro SLC25A38-Associated Congenital Sideroblastic Anemia Cell Model
 ,
,  ,
,  ,
,  , , , , ,
, , , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Phenotypic Characterization of the A38-Low Cell Line
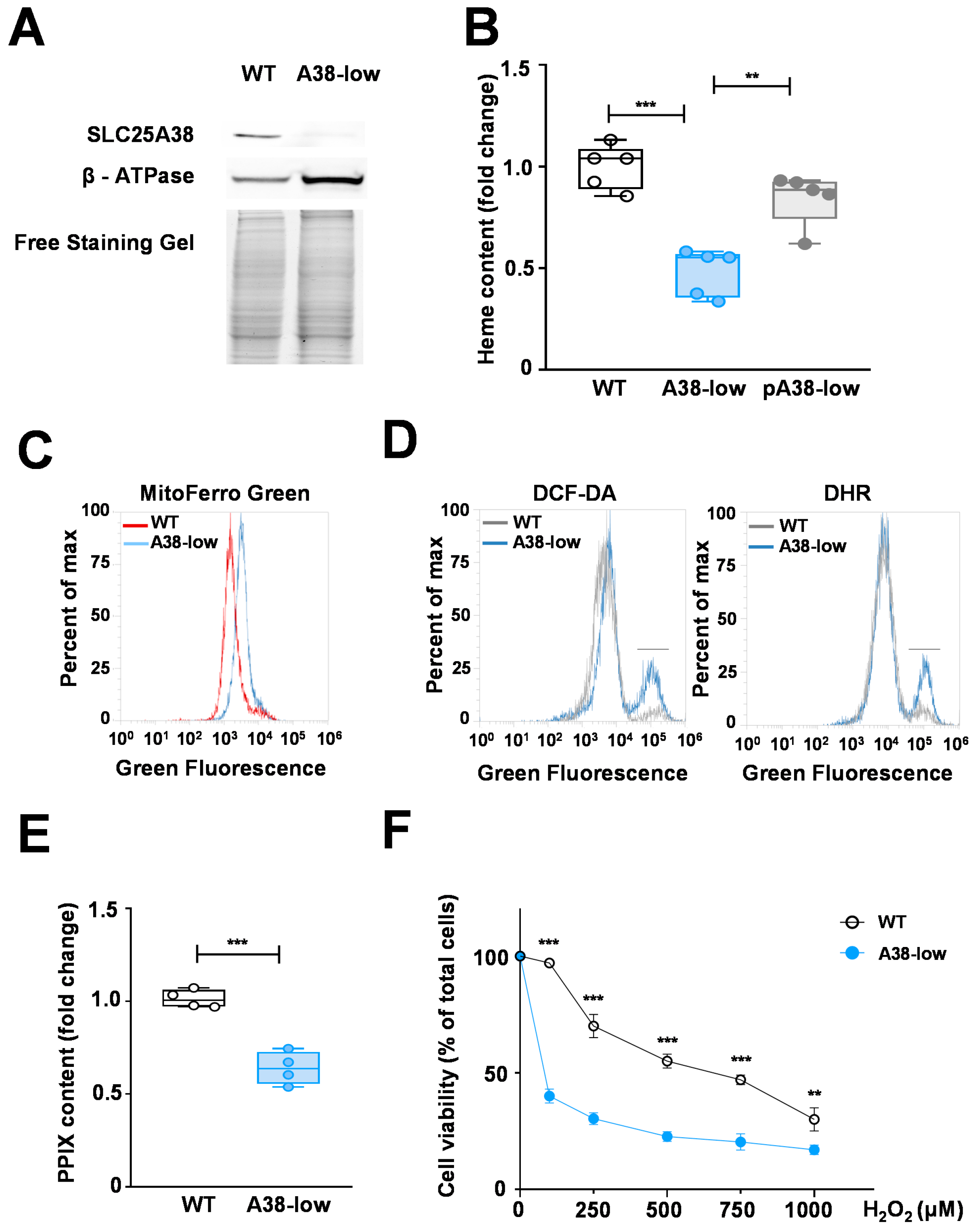
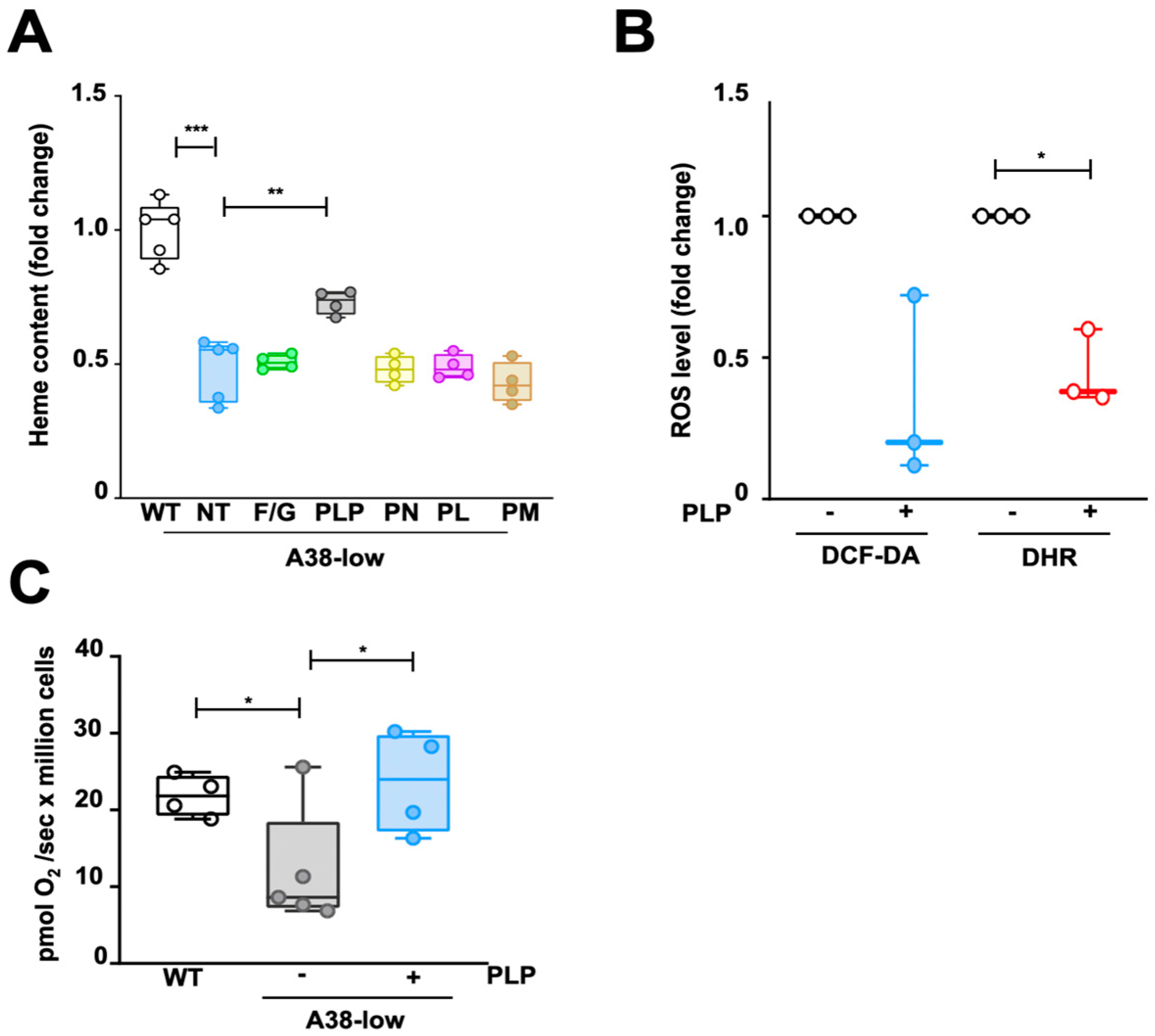
2.2. Impact of Folic Acid, Glycine, and B6 Vitamers on the Phenotypic Defects of A38-Low Cell Line
2.3. Decrease in PLP Content in A38-Low Cells and Its Restoration by Vitamin B6 Derivatives
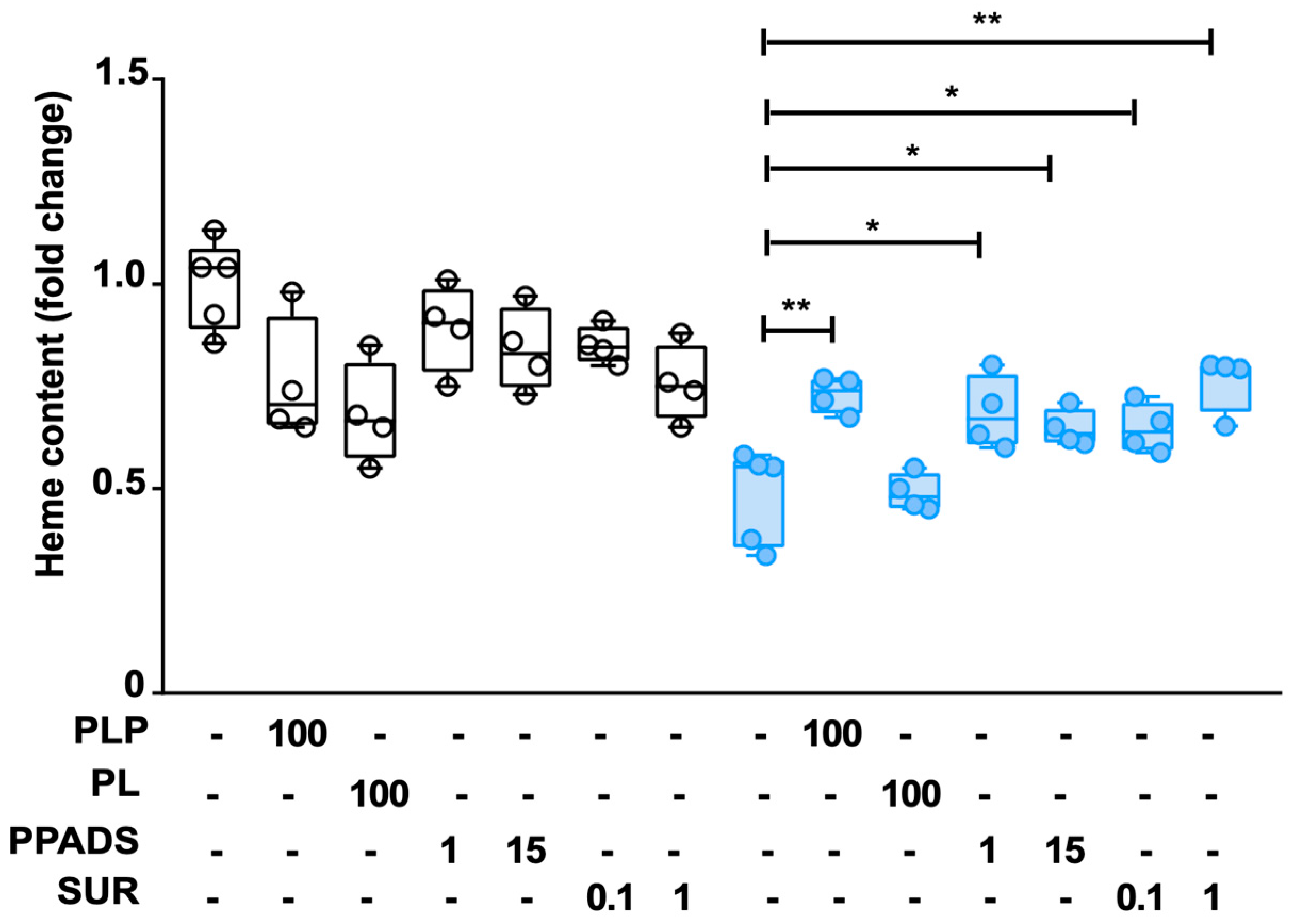
2.4. Effect of P2 Receptor Antagonists on WT and A38-Low Cell Lines
3. Discussion
4. Materials and Methods
4.1. Reagents, Cell Lines, and Culture Conditions
4.2. CRISPR/Cas9 Editing of the SLC25A38 Gene
4.3. SLC25A38 Gene Expression in A38-Low Cells
4.4. Western Blot Analysis
4.5. Cytotoxicity Assay
4.6. Protoporphyrin IX and Heme Content Assays
4.7. Flow Cytometry Analysis
4.8. Epifluorescence PLP Content Analysis
4.9. High-Resolution Respirometry
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fujiwara, T.; Harigae, H. Molecular pathophysiology and genetic mutations in congenital sideroblastic anemia. Free Radic. Biol. Med. 2019, 133, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, M.A.; Fouquet, C.; Leblanc, T.; Touati, M.; Fouyssac, F.; Vermylen, C.; Jäkel, N.; Guichard, J.F.; Maloum, K.; Toutain, F.; et al. Non syndromic childhood onset congenital sideroblastic anemia: A report of 13 patients identified with an ALAS2 or SLC25A38 mutation. Blood Cells Mol. Dis. 2017, 66, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, A.K.; Campagna, D.R.; McLoughlin, E.M.; Agarwal, S.; Fleming, S.D.; Bottomley, S.S.; Neufeld, E.J. Systematic molecular genetic analysis of congenital sideroblastic anemia: Evidence for genetic heterogeneity and identification of novel mutations. Pediatr. Blood Cancer. 2010, 54, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, S.S.; May, B.K.; Cox, T.C.; Cotter, P.D.; Bishop, D.F. Molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. J. Bioenerg. Biomembr. 1995, 27, 161–168. [Google Scholar] [CrossRef]
- Cox, T.C.; Bottomley, S.S.; Wiley, J.S.; Bawden, M.J.; Matthews, C.S.; May, B.K. X-linked pyridoxine-responsive sideroblastic anemia due to a Thr388-to-Ser substitution in erythroid 5-aminolevulinate synthase. N. Engl. J. Med. 1994, 330, 675–679. [Google Scholar] [CrossRef]
- Ajioka, R.S.; Phillips, J.D.; Kushner, J.P. Biosynthesis of heme in mammals. Biochim. Biophys. Acta 2006, 1763, 723–736. [Google Scholar] [CrossRef]
- Bottomley, S.S.; Fleming, M.D. Sideroblastic anemia: Diagnosis and management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670. [Google Scholar] [CrossRef]
- Heeney, M.M.; Berhe, S.; Campagna, D.R.; Oved, J.H.; Kurre, P.; Shaw, P.J.; Teo, J.; Shanap, M.A.; Hassab, H.M.; Glader, B.E.; et al. SLC25A38 congenital sideroblastic anemia: Phenotypes and genotypes of 31 individuals from 24 families, including 11 novel mutations, and a review of the literature. Hum. Mutat. 2021, 42, 1367–1383. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Aspects Med. 2013, 34, 465–484. [Google Scholar] [CrossRef]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef]
- Lunetti, P.; Damiano, F.; De Benedetto, G.; Siculella, L.; Pennetta, A.; Muto, L.; Paradies, E.; Marobbio, C.M.T.; Dolce, V.; Capobianco, L. Characterization of human and yeast mitochondrial glycine carriers with implications for heme biosynthesis and anemia. J. Biol. Chem. 2016, 291, 19746–19759. [Google Scholar]
- Fernandez-Murray, J.P.; Prykhozhij, S.V.; Dufay, J.N.; Steele, S.L.; Gaston, D.; Nasrallah, G.K.; Coombs, A.J.; Liwski, R.S.; Fernandez, C.V.; Berman, J.N.; et al. Glycine and folate ameliorate models of congenital sideroblastic anemia. PLoS Genet. 2016, 12, e1005783. [Google Scholar]
- Ducamp, S.; Sendamarai, A.K.; Campagna, D.R.; Chin, D.W.L.; Fujiwara, Y.; Schmidt, P.J.; Fleming, M.D. Murine models of erythroid 5ALA synthesis disorders and their conditional synthetic lethal dependency on pyridoxine. Blood 2024, 144, 1418–1432. [Google Scholar] [CrossRef] [PubMed]
- Uminski, K.; Houston, D.S.; Hartley, J.N.; Liu, J.; Cuvelier, G.D.E.; Israels, S.J. Clinical characterization and hematopoietic stem cell transplant outcomes for congenital sideroblastic anemia caused by a novel pathogenic variant in SLC25A38. Pediatr. Blood Cancer 2020, 67, e28623. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, M.A.; Bettle, A.; Berman, J.N.; Price, V.E.; Pambrun, C.; Yu, Z.; Tiller, M.; McMaster, C.R.; Fernandez, C.V. Study of glycine and folic acid supplementation to ameliorate transfusion dependence in congenital SLC25A38 mutated sideroblastic anemia. Pediatr. Blood Cancer 2016, 63, 1307–1309. [Google Scholar] [CrossRef]
- Sha, Z.; Benkovic, S.J. Purinosomes spatially co-localize with mitochondrial transporters. J. Biol. Chem. 2024, 300, 107620. [Google Scholar]
- Hirayama, T.; Kadota, S.; Niwa, M.; Nagasawa, H. A mitochondria-targeted fluorescent probe for selective detection of mitochondrial labile Fe(ii). Metallomics 2018, 10, 794–801. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Krumschnabel, G.; Fontana-Ayoub, M.; Sumbalova, Z.; Heidler, J.; Gauper, K.; Fasching, M.; Gnaiger, E. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol. Biol. 2015, 1264, 245–261. [Google Scholar]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar]
- Novak, B.; Schulten, R.; Lubbert, H. δ-Aminolevulinic acid and its methyl ester induce the formation of Protoporphyrin IX in cultured sensory neurones. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 583–602. [Google Scholar] [CrossRef]
- Jun, Y.W.; Hebenbrock, M.; Kool, E.T. A fluorescent hydrazone exchange probe of pyridoxal phosphate for the assessment of vitamin B6 status. Chem. Commun. 2019, 56, 317–320. [Google Scholar] [CrossRef]
- Whittaker, J.W. Intracellular trafficking of the pyridoxal cofactor. Implications for health and metabolic disease. Arch. Biochem. Biophys. 2016, 592, 20–26. [Google Scholar] [CrossRef]
- Trezise, D.J.; Bell, N.J.; Khakh, B.S.; Michel, A.D.; Humphrey, P.A. P2 purinoceptor antagonist properties of pyridoxal-5-phosphate. Eur. J. Pharmacol. 1994, 259, 295–300. [Google Scholar] [CrossRef]
- Millart, H.; Alouane, L.; Oszust, F.; Chevallier, S.; Robinet, A. Involvement of P2Y receptors in pyridoxal-5′-phosphate-induced cardiac preconditioning. Fundam. Clin. Pharmacol. 2009, 23, 279–292. [Google Scholar] [CrossRef]
- Wang, X.; Dakshinamurti, K.; Musat, S.; Dhalla, N.S. Pyridoxal 5′-phosphate is an ATP-receptor antagonist in freshly isolated rat cardiomyocytes. J. Mol. Cell. Cardiol. 1999, 31, 1063–1072. [Google Scholar] [CrossRef]
- North, R.A. Molecular physiology of P2X receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef]
- Burnstock, G. Introduction: P2 receptors. Curr. Top. Med. Chem. 2004, 4, 793–803. [Google Scholar] [CrossRef]
- Lambrecht, G.; Braun, K.; Damer, M.; Ganso, M.; Hildebrandt, C.; Ullmann, H.; Kassack, M.U.; Nickel, P. Structure-activity relationships of suramin and pyridoxal-5′-phosphate derivatives as P2 receptor antagonists. Curr. Pharm. Des. 2002, 8, 2371–2399. [Google Scholar] [CrossRef]
- Crispin, A.; Guo, C.; Chen, C.; Campagna, D.R.; Schmidt, P.J.; Lichtenstein, D.; Cao, C.; Sendamarai, A.K.; Hildick-Smith, G.J.; Huston, N.C.; et al. Mutations in the iron-sulfur cluster biogenesis protein HSCB cause congenital sideroblastic anemia. J. Clin. Investig. 2020, 130, 5245–5256. [Google Scholar] [CrossRef]
- Ochi, T.; Tohru Fujiwara, T.; Ono, K.; Suzuki, C.; Nikaido, M.; Inoue, D.; Kato, H.; Onodera, K.; Ichikawa, S.; Fukuhara, N.; et al. Exploring the mechanistic link between SF3B1 mutation and ring sideroblast formation in myelodysplastic syndrome. Sci. Rep. 2022, 12, 14562. [Google Scholar] [CrossRef]
- Fujiwara, T.; Okamoto, K.; Niikuni, R.; Takahashi, K.; Okitsu, Y.; Fukuhara, N.; Onishi, Y.; Ishizawa, K.; Ichinohasama, R.; Nakamura, Y.; et al. Effect of 5-aminolevulinic acid on erythropoiesis: A preclinical in vitro characterization for the treatment of congenital sideroblastic anemia. Biochem. Biophys. Res. Commun. 2014, 454, 102–108. [Google Scholar] [CrossRef]
- Liu, G.; Wang, Y.; Anderson, G.J.; Camaschella, C.; Chang, Y.; Nie, G. Functional analysis of GLRX5 mutants reveals distinct functionalities of GLRX5 protein. J. Cell. Biochem. 2016, 117, 207–217. [Google Scholar] [CrossRef]
- Filippin, K.J.; de Souza, K.F.S.; de Araujo Jùnior, R.T.; Torquato, H.F.V.; Dias, D.A.; Parisotto, E.B.; Ferreira, A.T.; Paredes-Gamero, E.J. Involvement of P2 receptors in hematopoiesis and hematopoietic disorders, and as pharmacological targets. Purinergic Signal. 2020, 16, 1–15. [Google Scholar] [CrossRef]
- Wang, H.S.; Kuo, M.F.; Chou, M.L.; Hung, P.C.; Lin, K.L.; Hsieh, M.Y.; Chang, M.Y. Pyridoxal phosphate is better than pyridoxine for controlling idiopathic intractable epilepsy. Arch. Dis. Child. 2005, 90, 512–515. [Google Scholar] [CrossRef]
- Kuo, M.F.; Wang, H.S. Pyridoxal phosphate-responsive epilepsy with resistance to pyridoxine. Pediatr. Neurol. 2002, 26, 146–147. [Google Scholar] [CrossRef]
- Clayton, P.T.; Surtees, R.A.; DeVile, C.; Hyland, K.; Heales, S.J. Neonatal epileptic encephalopathy. Lancet 2003, 361, 1614. [Google Scholar] [CrossRef]
- Wiedemar, N.; Hauser, D.A.; Maser, P. 100 Years of suramin. Antimicrob. Agents Chemother. 2020, 64, e01168-19. [Google Scholar] [CrossRef]
- Pacheva, I.; Ivanov, I. Targeted biomedical treatment for autism spectrum disorders. Curr. Pharm. Des. 2019, 25, 4430–4453. [Google Scholar] [CrossRef]
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P.T. Disorders affecting vitamin B6 metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646. [Google Scholar] [CrossRef]
- Santoro, A.; Anjomani Virmouni, S.; Paradies, E.; Villalobos Coa, V.L.; Al-Mahdawi, S.; Khoo, M.; Porcelli, V.; Vozza, A.; Perrone, M.; Denora, N.; et al. Effect of diazoxide on Friedreich ataxia models. Hum. Mol. Genet. 2018, 27, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Shahroor, M.A.; Lasorsa, F.M.; Porcelli, V.; Dweikat, I.; Di Noia, M.A.; Gur, M.; Agostino, G.; Shaag, A.; Rinaldi, T.; Gasparre, G.; et al. PNC2 (SLC25A36) deficiency associated with the Hyperinsulinism/Hyperammonemia syndrome. J. Clin. Endocrinol. Metab. 2022, 107, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Seccia, R.; De Santis, S.; Di Noia, M.A.; Palmieri, F.; Miniero, D.V.; Marmo, R.; Paradies, E.; Santoro, A.; Pierri, C.L.; Palmieri, L.; et al. Citrate Regulates the Saccharomyces cerevisiae Mitochondrial GDP/GTP Carrier (Ggc1p) by Triggering Unidirectional Transport of GTP. J. Fungi 2022, 8, 795. [Google Scholar] [CrossRef] [PubMed]
- Giannuzzi, G.; Lobefaro, N.; Paradies, E.; Vozza, A.; Punzi, G.; Marobbio, C.M.T. Overexpression in E. coli and purification of the L. pneumophila Lpp2981 protein. Mol. Biotechnol. 2014, 56, 157–165. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Arduino, I.; Santoro, A.; De Santis, S.; Iacobazzi, R.M.; Lopedota, A.A.; Paradies, E.; Merla, G.; Anjomani Virmouni, S.; Palmieri, L.; Marobbio, C.M.T.; et al. Microfluidic formulation of diazoxide-loaded solid lipid nanoparticles as a novel approach for Friedreich’s ataxia treatment. J. Drug Deliv. Sci. Technol. 2024, 97, 105837. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoro, A.; De Santis, S.; Palmieri, F.; Vozza, A.; Agrimi, G.; Andolfo, I.; Russo, R.; Palazzo, A.; Storlazzi, C.T.; Ferrucci, A.; et al. P2 Receptor Antagonists Rescue Defective Heme Content in an In Vitro SLC25A38-Associated Congenital Sideroblastic Anemia Cell Model. Int. J. Mol. Sci. 2024, 25, 13314. https://doi.org/10.3390/ijms252413314
Santoro A, De Santis S, Palmieri F, Vozza A, Agrimi G, Andolfo I, Russo R, Palazzo A, Storlazzi CT, Ferrucci A, et al. P2 Receptor Antagonists Rescue Defective Heme Content in an In Vitro SLC25A38-Associated Congenital Sideroblastic Anemia Cell Model. International Journal of Molecular Sciences. 2024; 25(24):13314. https://doi.org/10.3390/ijms252413314
Chicago/Turabian StyleSantoro, Antonella, Silvia De Santis, Ferdinando Palmieri, Angelo Vozza, Gennaro Agrimi, Immacolata Andolfo, Roberta Russo, Antonio Palazzo, Clelia Tiziana Storlazzi, Arianna Ferrucci, and et al. 2024. "P2 Receptor Antagonists Rescue Defective Heme Content in an In Vitro SLC25A38-Associated Congenital Sideroblastic Anemia Cell Model" International Journal of Molecular Sciences 25, no. 24: 13314. https://doi.org/10.3390/ijms252413314
APA StyleSantoro, A., De Santis, S., Palmieri, F., Vozza, A., Agrimi, G., Andolfo, I., Russo, R., Palazzo, A., Storlazzi, C. T., Ferrucci, A., Jun, Y. W., Kool, E. T., Fiermonte, G., Iolascon, A., Paradies, E., Marobbio, C. M. T., & Palmieri, L. (2024). P2 Receptor Antagonists Rescue Defective Heme Content in an In Vitro SLC25A38-Associated Congenital Sideroblastic Anemia Cell Model. International Journal of Molecular Sciences, 25(24), 13314. https://doi.org/10.3390/ijms252413314