

Hydroxychloroquine-Loaded Chitosan Nanoparticles Induce Anticancer Activity in A549 Lung Cancer Cells: Design, BSA Binding, Molecular Docking, Mechanistic, and Biological Evaluation

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Synthesis and Characterization
2.2. Computational Finding
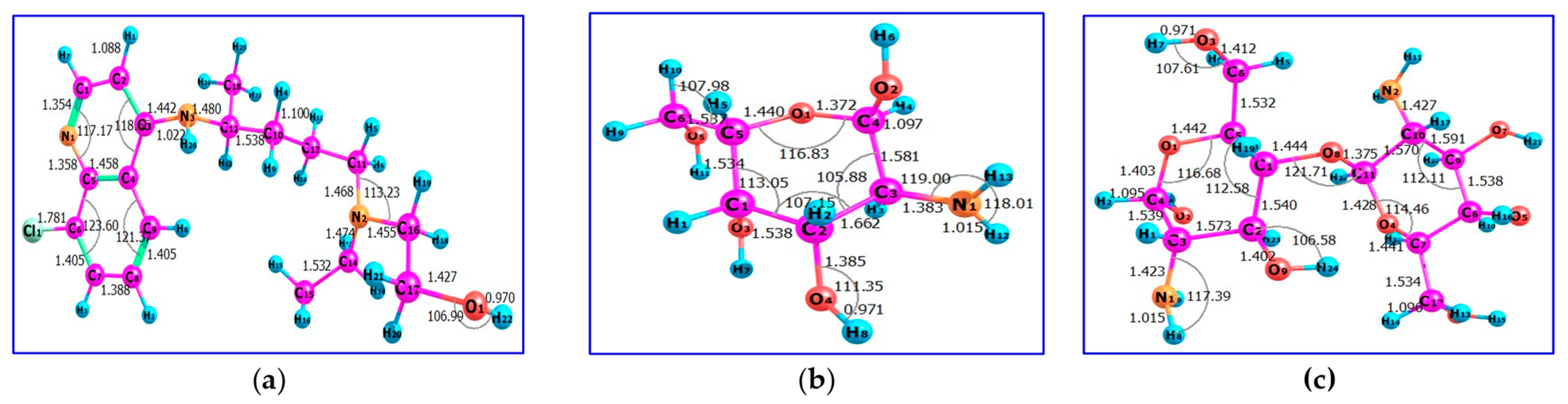
2.2.1. Quantum Chemical Parameters
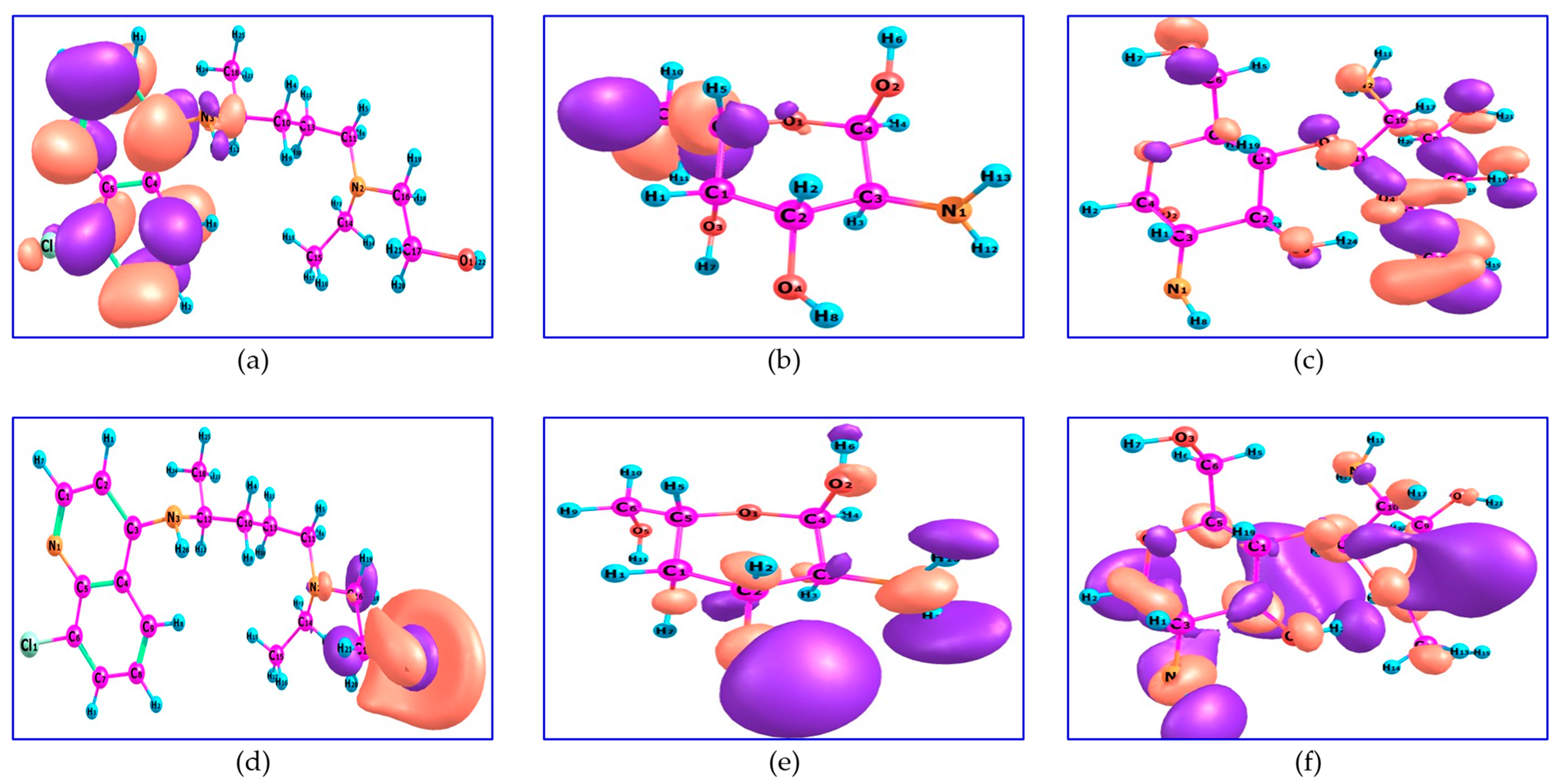
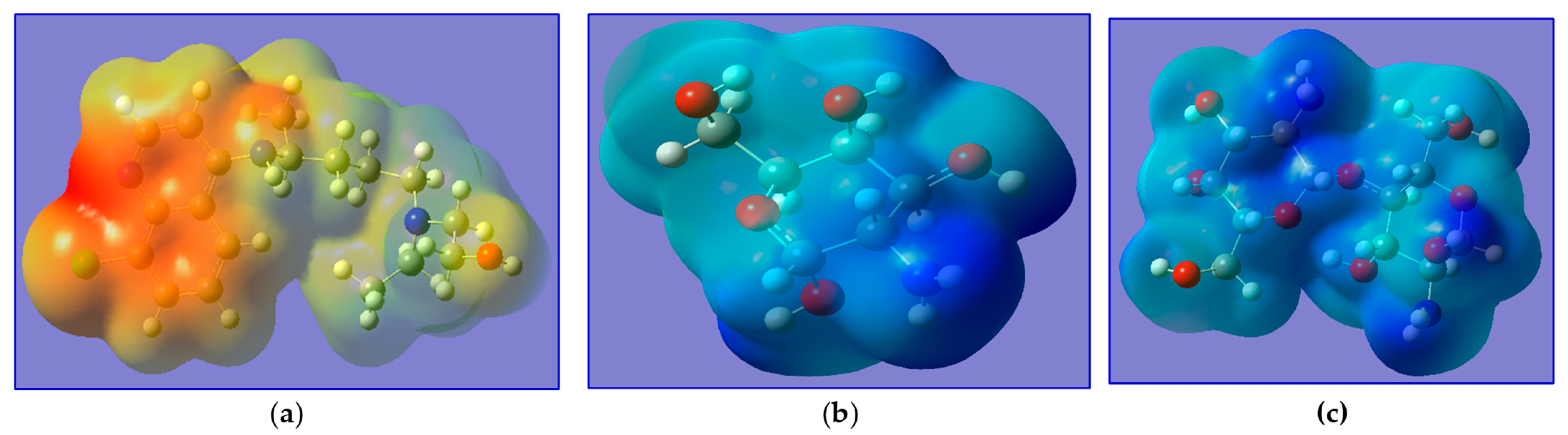
2.2.2. Frontier Molecular Orbitals and Chemical Reactivity
2.3. BSA Binding Studies
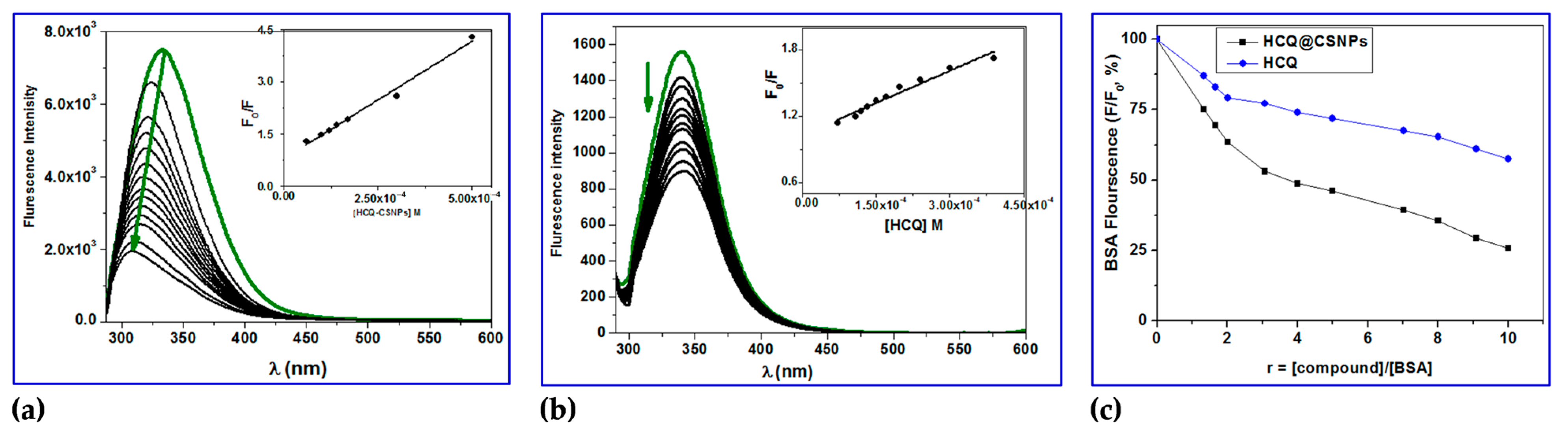
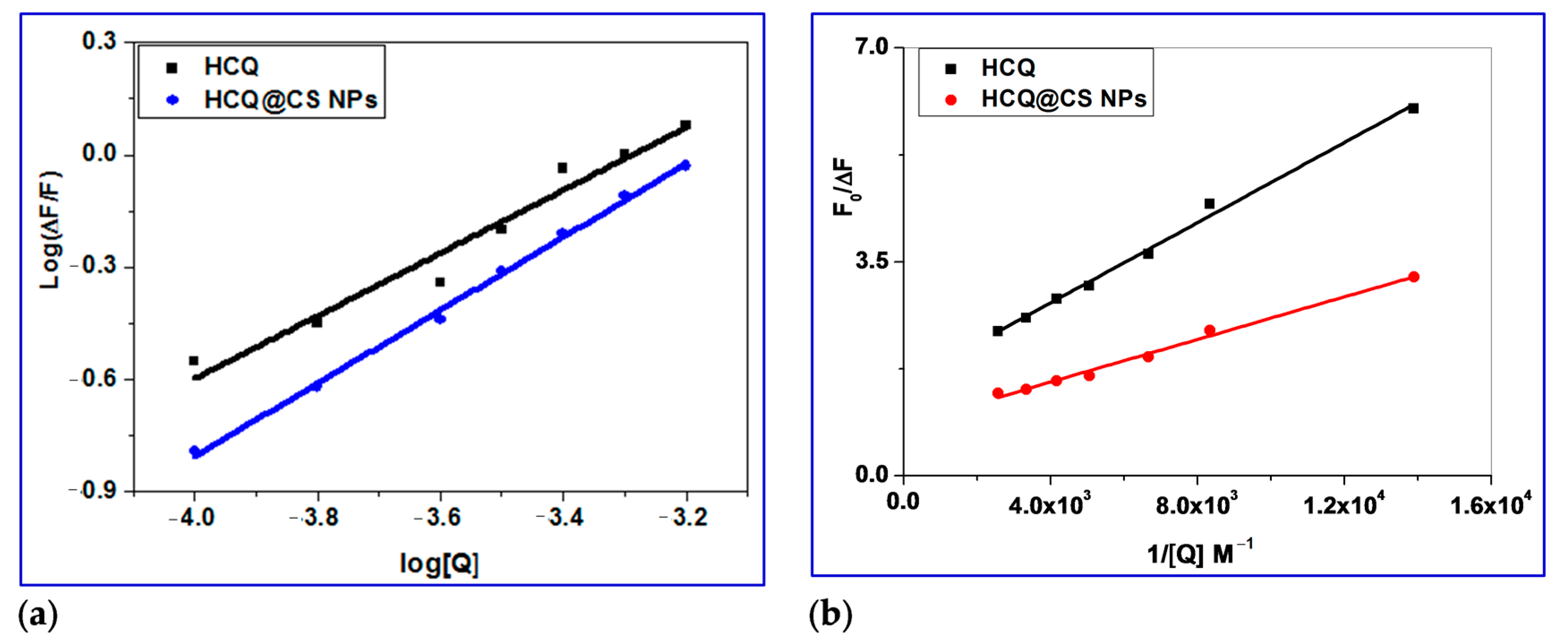
2.3.1. Fluorescence Studies
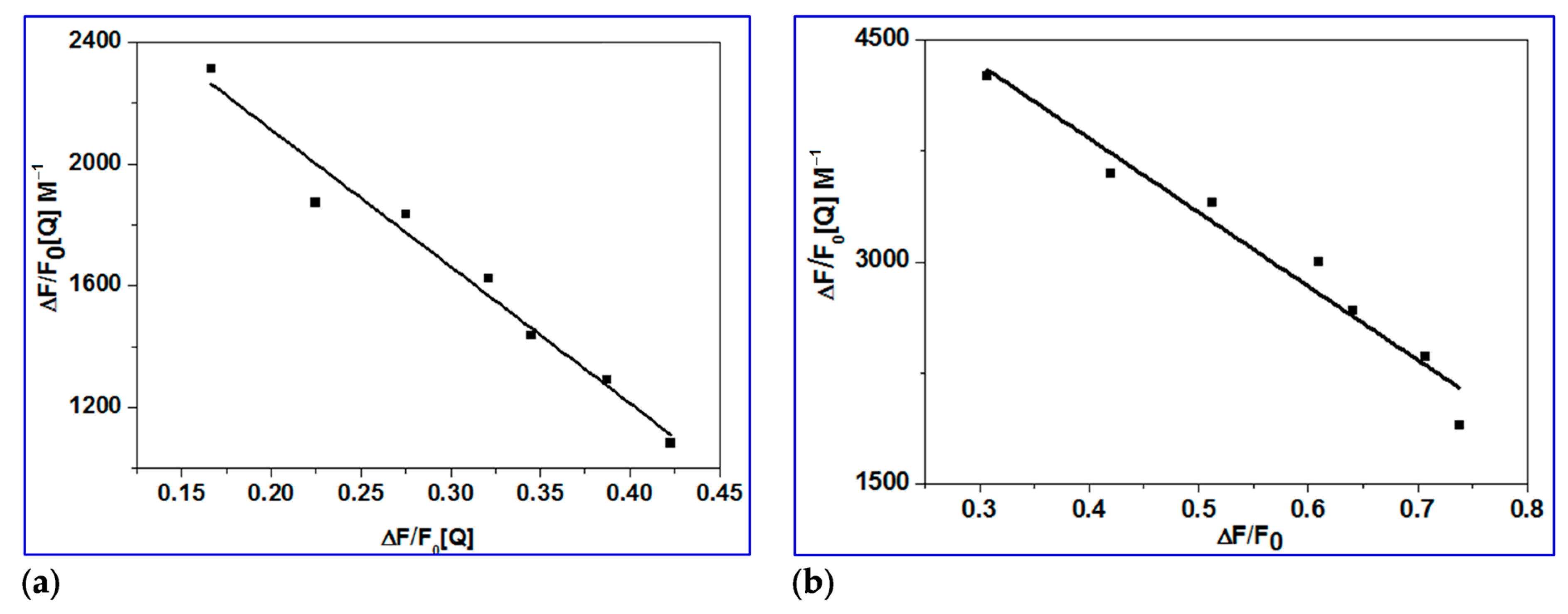
2.3.2. Determination of Binding Constant (Kbin), Number of Binding Sites (n) and ΔG
2.3.3. Determination of Accessible Fraction of the Fluorophore
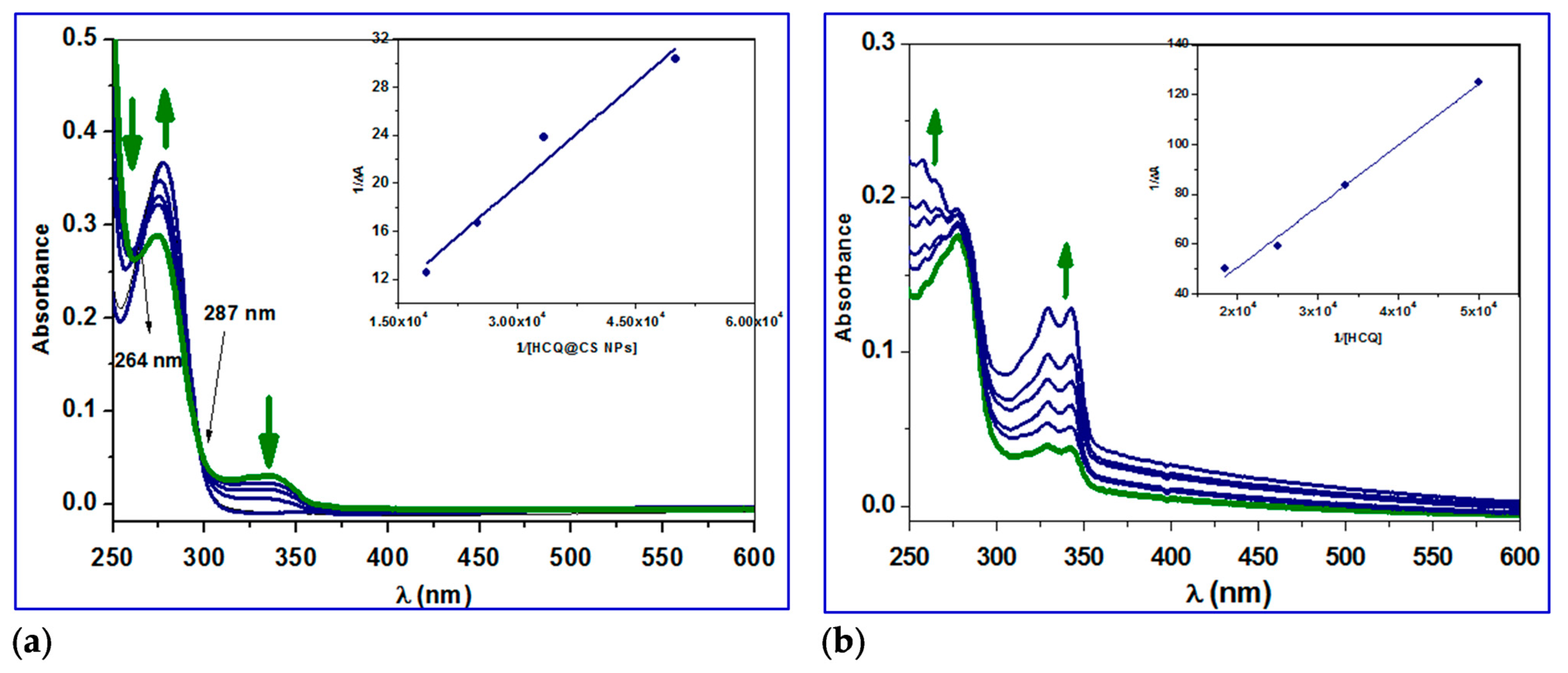
2.3.4. Conformation Investigation Using UV−Vis Spectroscopy
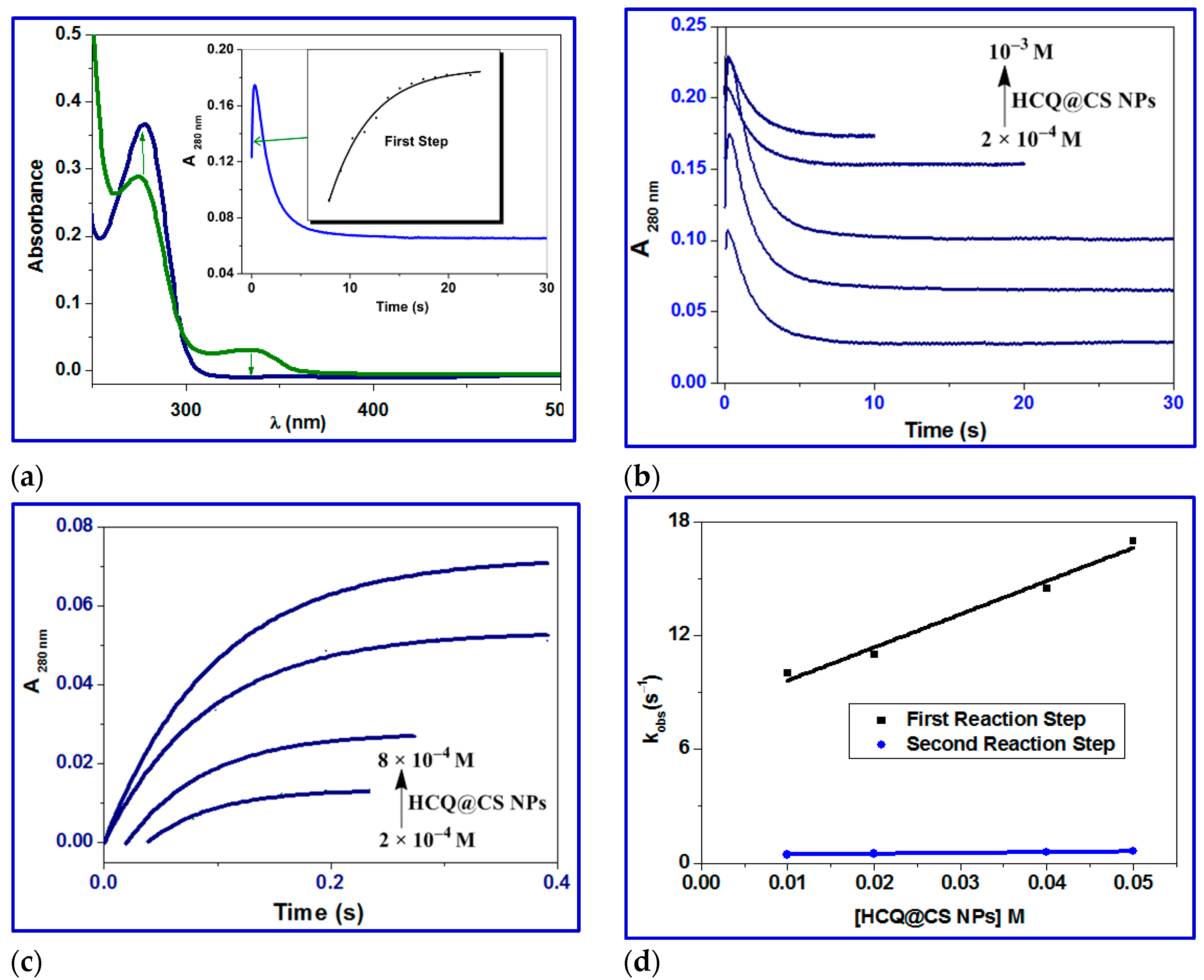
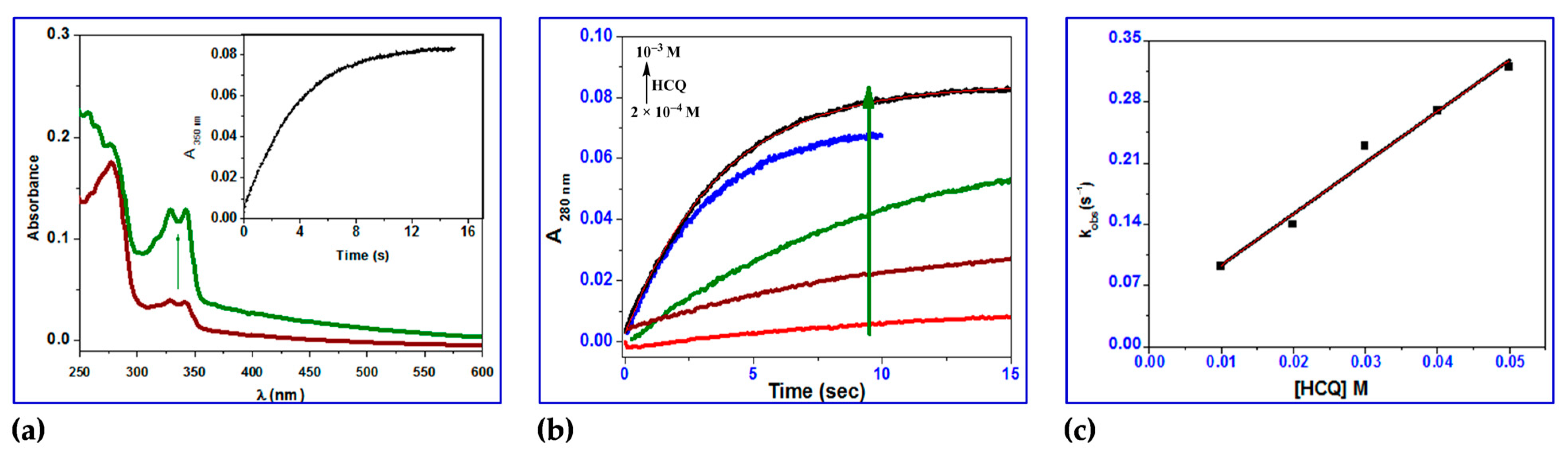
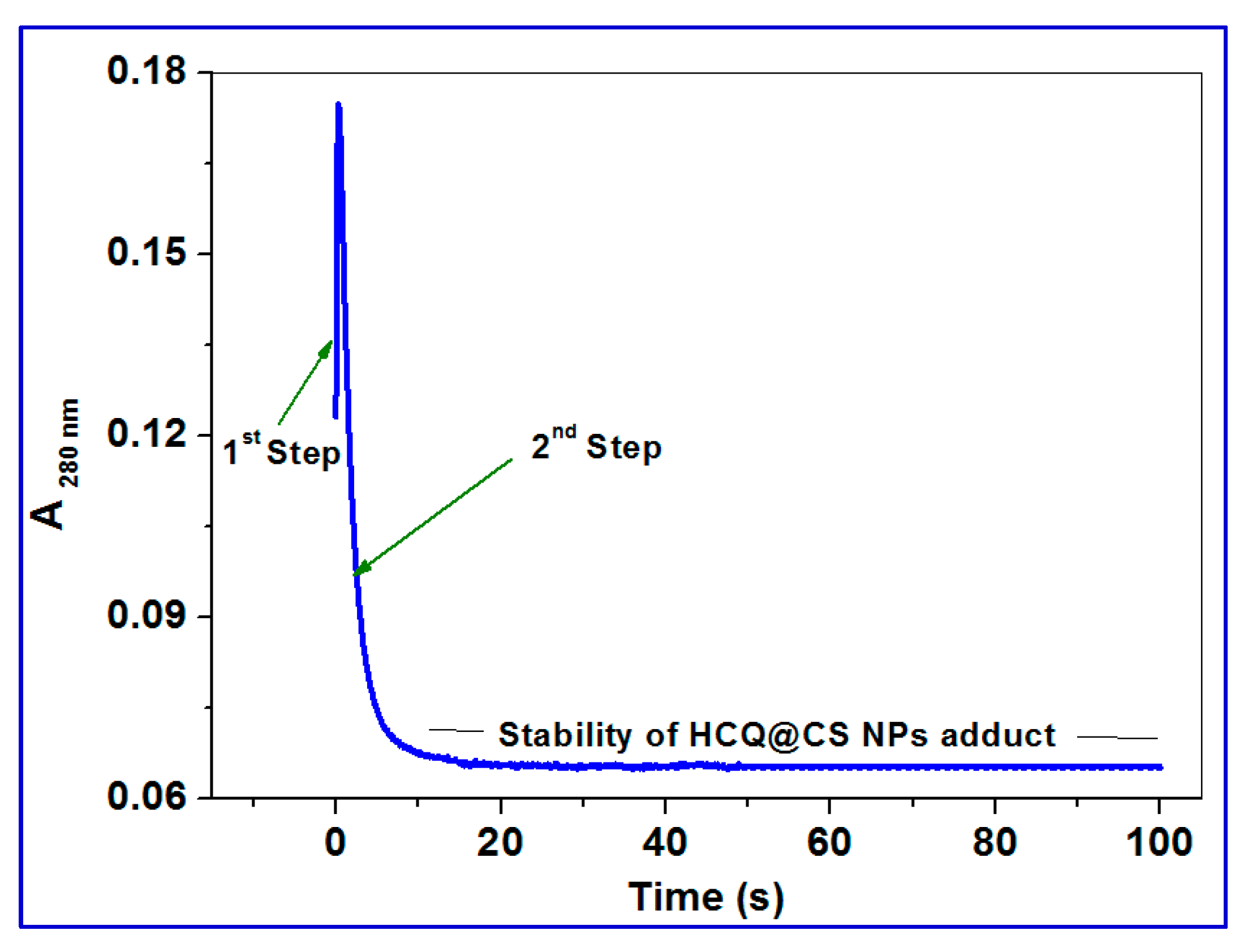
2.3.5. Stopped-Flow Binding Experiments and Kinetic Measurements
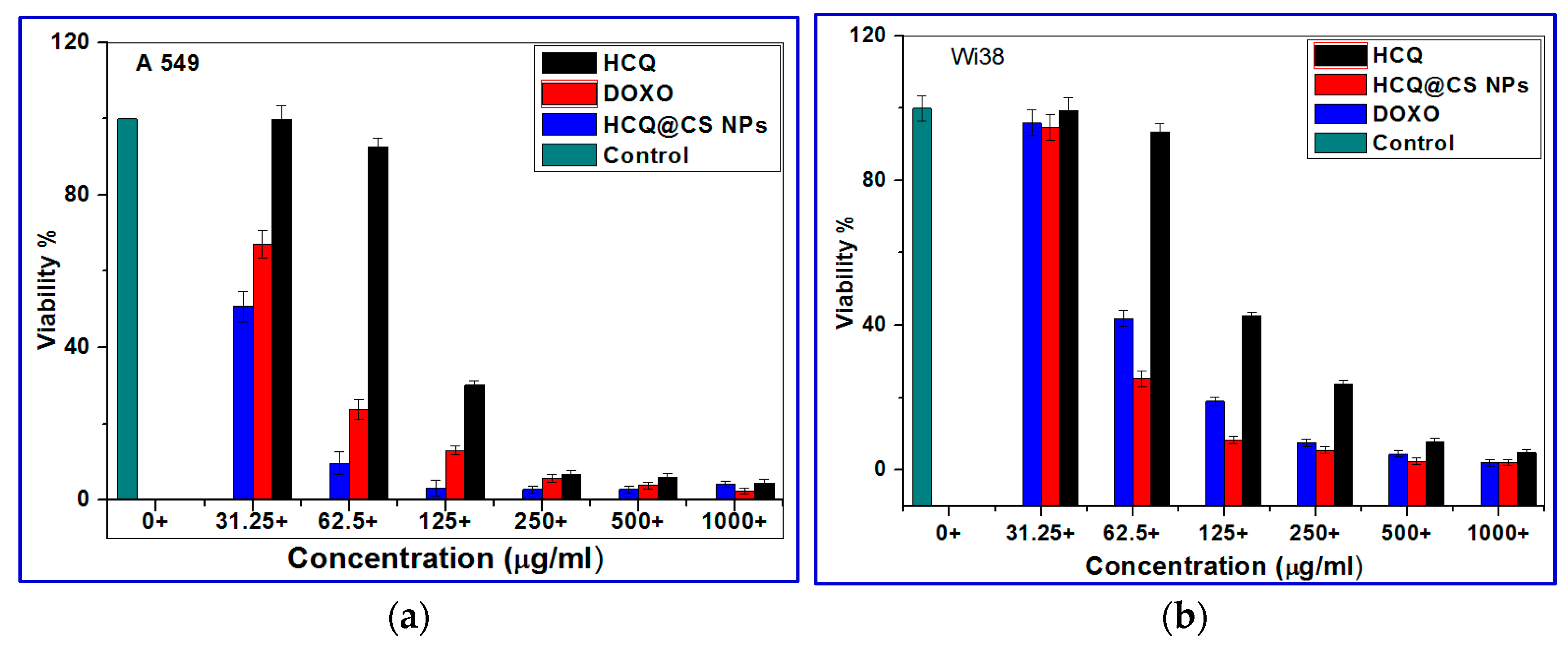
2.4. In Vitro Cytotoxicity Studies
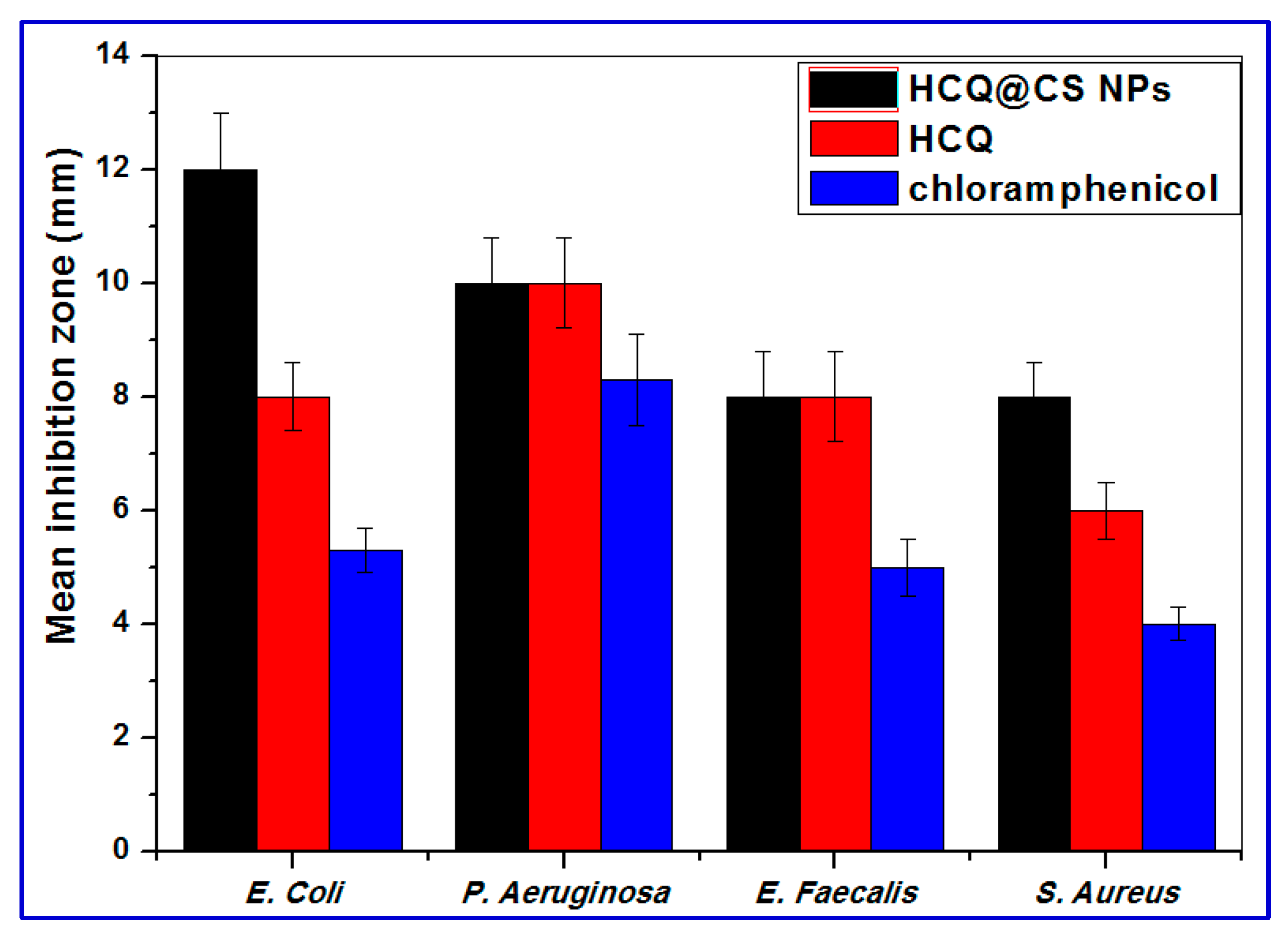
2.5. Antibacterial Activity
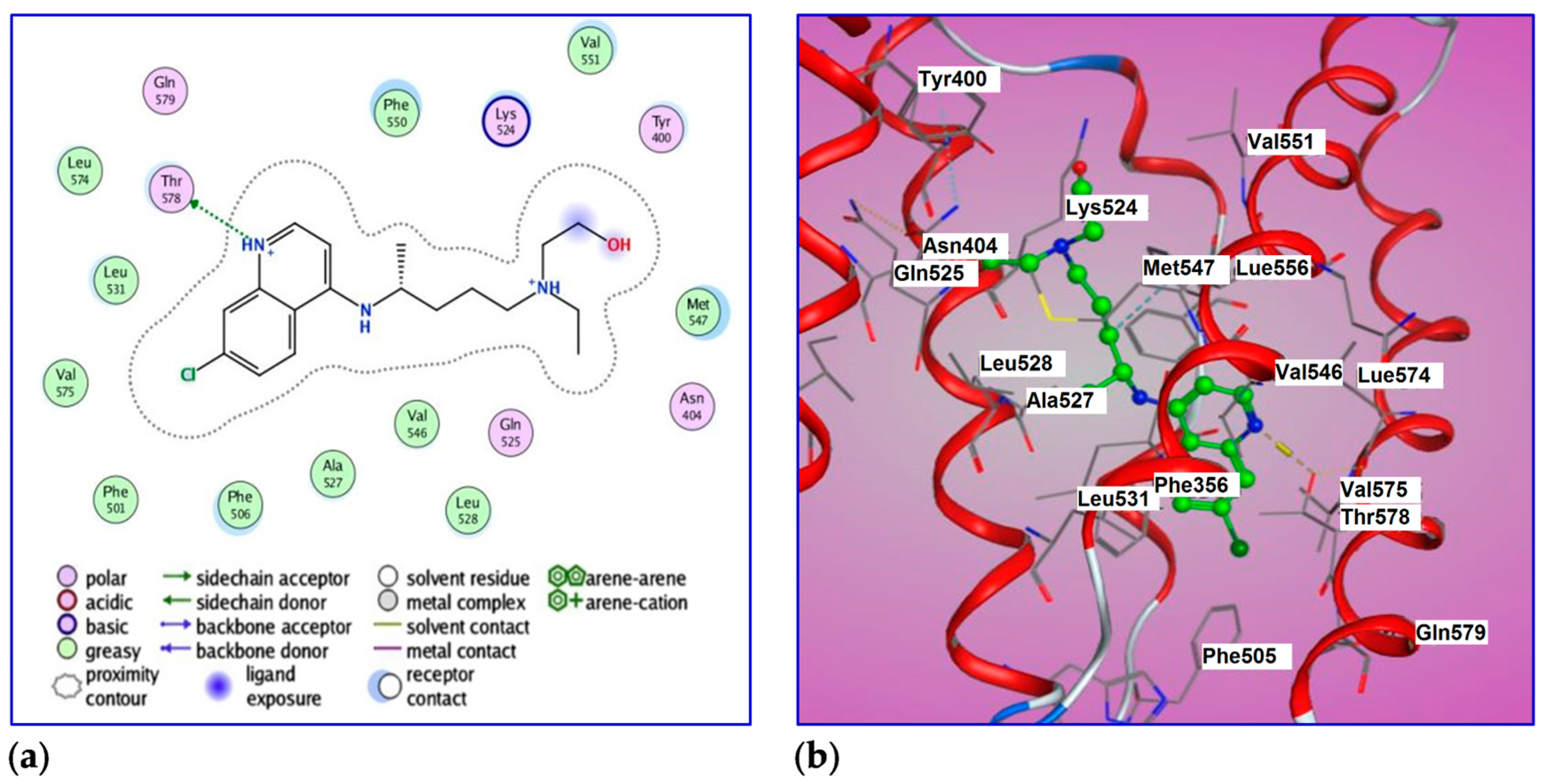
2.6. Molecular Docking of BSA with HCQ
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Preparation of HCQ@CS NPs
4.3. Characterization
4.4. BSA Binding Study Procedures
4.5. Stopped-Flow Fluorescence Kinetic Studies
4.6. Cell Cytotoxicity Assay
4.7. Antibacterial Activity
4.8. Computational Studies
4.9. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Casaluce, F.; Sgambato, A.; Maione, P. The potential role of new targeted therapies in the treatment of advanced non-small-cell lung cancer. Clin. Investig. 2013, 3, 369–383. [Google Scholar] [CrossRef]
- Ahmad, S. Platinum–DNA interactions and subsequent cellular processes controlling sensitivity to anticancer platinum complexes. Chem. Biodivers. 2010, 7, 543–566. [Google Scholar] [CrossRef]
- Sterlacci, W.; Fiegl, M.; Tzankov, A. Prognostic and predictive value of cell cycle deregulation in non-small-cell lung cancer. Pathobiology 2012, 79, 175–194. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Burgess, J.T.; Croft, L.V.; Wallace, N.C.; Stephenson, S.-A.; Adams, M.N.; Ashton, N.W.; Solomon, B.; O’Byrne, K.; Richard, D.J. DNA repair pathways and their therapeutic potential in lung cancer. Lung Cancer Manag. 2014, 3, 159–173. [Google Scholar] [CrossRef]
- Mirkin, C.A.; Nel, A.; Thaxton, C.S. Applications: Nano biosystems, medicine and health. Sci. Policy Rep. 2011, 1, 305–374. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, Y.S.; Park, K.; Lee, S.; Nam, H.Y.; Min, K.H.; Jo, H.G.; Park, J.H.; Choi, K.S.Y.; Jeong, R.W.; et al. Antitumor efficacy of cisplatin-loaded glycol chitosan nanoparticles in tumor-bearing mice. J. Control. Release 2008, 127, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, X.; Liu, L.; Zhang, Q. Thiolated chitosan-modified PLA-PCL-TPGS nanoparticles for oral chemotherapy of lung cancer. Nanoscale Res. Lett. 2013, 8, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Lonso, M.J.; Sanchez, A. The potential of chitosan in ocular drug delivery. J. Pharm. Pharmacol. 2003, 55, 451–1463. [Google Scholar] [CrossRef]
- Yee Kuen, C.; Masarudin, M.J. Chitosan Nanoparticle-Based System: A New Insight into the Promising Controlled Release System for Lung Cancer Treatment. Molecules 2022, 27, 473. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Victor, R.; Marcelo da Cunha Santos, A.; Viana de Sousa, B.; de Araújo Neves, G.; Navarro de Lima Santana, L.; Rodrigues Menezes, R. A Review on Chitosan’s Uses as Biomaterial: Tissue Engineering, Drug Delivery Systems and Cancer Treatment. Materials 2020, 13, 4995. [Google Scholar] [CrossRef] [PubMed]
- Aranaz, I.; Alcántara, A.R.; Civera, M.C.; Arias, C.; Elorza, B.; Heras Caballero, A.; Acosta, N. Chitosan: An Overview of Its Properties and Applications. Polymers 2021, 13, 3256. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wan, A.J. Preparation of nanoparticles composed of chitosan and its derivatives as delivery systems for macromolecules. J. Appl. Polym. Sci. 2007, 105, 552–561. [Google Scholar] [CrossRef]
- Taşkonak, B.; Aylaz, G.; Andac, M.; Güven, E.; Ozkahraman, B.; Süloğlu, A.K. Hypericin-Loaded Chitosan Nanoparticles for Enhanced Photodynamic Therapy in A549 Lung Cancer Cells. BioNanoScience 2023, 13, 352–364. [Google Scholar] [CrossRef]
- Pandya, A.D.; Øverbye, A.; Sahariah, P.; Gaware, V.S.; Høgset, H.; Masson, M.; Høgset, A.; Mælandsmo, G.M.; Skotland, T.; Sandvig, K.; et al. Drug-Loaded Photosensitizer-Chitosan Nanoparticles for Combinatorial Chemo- and Photodynamic-Therapy of Cancer. Biomacromolecules 2020, 21, 1489–1498. [Google Scholar] [CrossRef]
- Swarnalatha, Y.; Gunna, G.K.; Jacob, C.M. Synthesis of alkaloid loaded chitosan nanoparticles for enhancing the anticancer activity in A549 lung cancer cell line. Der Pharm. Lett. 2015, 7, 378–390. [Google Scholar]
- Zhou, S.H.; Hong, Y.; Fang, G.J. Preparation, characterization and anticancer effect of chitosan nanoparticles. CRTER 2007, 11, 9688–9691. [Google Scholar]
- Kumar, S.; Kurkjian, C. Advances in the treatment of metastatic colorectal cancer. Am. J. Ther. 2009, 16, 412–420. [Google Scholar] [CrossRef]
- Johnston, S.R.D. Acquired tamoxifen resistance in human breast cancer-potential mechanisms and clinical implications. Anticancer Drugs 1997, 8, 911–930. [Google Scholar] [CrossRef]
- Brown, K. Breast cancer chemoprevention: Risk-benefit effects of the antioestrogen tamoxifen. Expert Opin. Drug Saf. 2002, 1, 253–267. [Google Scholar] [CrossRef]
- Balunas, M.J.; Su, B.; Brueggemeier, R.W.; Kinghorn, A.D. Natural products as aromatase inhibitors. Anticancer Agents Med. Chem. 2008, 8, 646–682. [Google Scholar] [CrossRef] [PubMed]
- Anbu, S.A.; Velmuruganb, P.; Leec, J.H.; Ohb, B.T.; Venkatachalama, P. Biomolecule-loaded chitosan nanoparticles induce apoptosis and molecular changes in cancer cell line (SiHa). Int. J. Biol. Macromol. 2016, 88, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Cyr, M. The history of lupus erythematosus. From Hippocrates to Osler. Rheum. Dis. Clin. N. Am. 1988, 14, 1–14. [Google Scholar] [CrossRef]
- Ben-Zvi, I.; Kivity, S.; Langevitz, P.; Shoenfeld, Y. Hydroxychloroquine: From malaria to autoimmunity. Clin. Rev. Allergy Immunol. 2012, 42, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Safer, A.-M.; Leporatti, S. Chitosan Nanoparticles for Antiviral Drug Delivery: A Novel Route for COVID-19 Treatment. Int. J. Nanomed. 2021, 16, 8141–8158. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Patrizia Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and hydroxychloroquine as anti-cancer agents. Ecancer 2017, 11, 781. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.-H.; Ko, H.-L.; Yang, K.-L.; Lee, C.-Y.; Chi, M.-S.; Kao, S.-J. Addition of rapamycin and hydroxychloroquine to metronomic chemotherapy as a second line treatment results in high salvage rates for refractory metastatic solid tumors: A pilot safety and effectiveness analysis in a small patient cohort. Oncotarget 2015, 6, 16735–16745. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.B.; Supko, J.G.; Neal, J.W.; Muzikansky, A.; Digumarthy, S.; Fidias, P.; Temel, J.S.; Heist, R.S.; Shaw, A.T.; McCarthy, P.O.; et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.-S.B.; Neal, J.W.; Wakelee, H.A.; Sequist, L.V.; Marmor, M.F. Rapid onset of retinal toxicity from high-dose hydroxychloroquine given for cancer therapy American. Am. J. Ophthalmol. 2015, 160, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, Q.; Zhou, Z.; He, N.; Li, S.; Zhao, C. Co-delivery of doxorubicin and hydroxychloroquine via chitosan/alginate nanoparticles for blocking autophagy and enhancing chemotherapy in breast cancer therapy. Front. Pharmacol. 2023, 14, 1176232. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Pfab, C.; Schnobrich, L.; Eldnasoury, S.; Gessner, A.; El-Najjar, N. Repurposing of Antimicrobial Agents for Cancer Therapy: What Do We Know? Cancers 2021, 13, 3193. [Google Scholar] [CrossRef]
- Simović, A.R.; Masnikosa, R.; Bratsos, I.; Alessio, E. Chemistry and reactivity of ruthenium(II) complexes: DNA/protein binding mode and anticancer activity are related to the complex structure. Coord. Chem. Rev. 2019, 398, 113011–113037. [Google Scholar] [CrossRef]
- Wen, P.; Ke, W.; Dirisala, A.; Toh, K.; Tanaka, M.; Li, J. Stealth and pseudo-stealth nanocarriers. Adv. Drug Deliv. Rev. 2023, 198, 114895. [Google Scholar] [CrossRef]
- Shereef, H.A.; Moemen, Y.S.; Elshami, F.I.; El-Nahas, A.M.; Shaban, S.Y.; van Eldik, R. DNA Binding and Cleavage, Stopped-Flow Kinetic, Mechanistic, and Molecular Docking Studies of Cationic Ruthenium(II) Nitrosyl Complexes Containing “NS4” Core. Molecules 2023, 28, 3028. [Google Scholar] [CrossRef] [PubMed]
- Shereef, H.A.; Shaban, S.Y.; Moemen, Y.S.; El-Khouly, M.E.; El-Nahas, A.M. Biophysicochemical studies of a ruthenium(II) nitrosyl thioether-thiolate complex binding to BSA: Mechanistic information, molecular docking, and relationship to antibacterial and cytotoxic activities. Appl. Organomet. Chem. 2022, 34, 6583. [Google Scholar] [CrossRef]
- Shaban, N.Z.; Aboelsaad, A.M.; Shoueir, K.R.; Abdulmalek, S.A.; Awad, D.; Shaban, S.Y.; Mansour, H. Chitosan-based dithi-ophenolato nanoparticles: Preparation, mechanistic information of DNA binding, antibacterial and cytotoxic activities. J. Mol. Liq. 2020, 318, 114252. [Google Scholar] [CrossRef]
- Elshami, F.I.; Ramadan, A.E.-M.M.; Ibrahim, M.M.; El-Mehasseb, I.M.; Al-Juaid, S.; Shaban, S.Y. Metformin Containing Nickel(II) Complexes: Synthesis, Structural Characterization, Binding and Kinetic Interactions with BSA, Antibacterial and in-vitro Cytotoxicity Studies. Appl. Organomet. Chem. 2020, 34, 5437. [Google Scholar] [CrossRef]
- Shaban, N.Z.; Yehia, S.A.; Shoueir, K.R.; Saleh, S.R.; Awad, D.; Shaban, S.Y. Design, DNA binding and kinetic studies, anti-bacterial and cytotoxic activities of stable dithiophenolato titanium(IV)-chitosan Nanocomposite. J. Mol. Liq. 2019, 287, 111002. [Google Scholar] [CrossRef]
- Ramzy, E.; Ibrahim, M.M.; El-Mehasseb, I.M.; Ramadan, A.E.-M.M.; Elshami, F.I.; Shaban, Y.S.; van Eldik, R. Synthesis, Bio-physical Interaction of DNA/BSA, Equilibrium and Stopped-Flow Kinetic Studies, and Biological Evaluation of bis(2-Picolyl)amine-Based Nickel(II) Complex. Biomimetics 2022, 7, 172. [Google Scholar] [CrossRef]
- Domard, A. pH and c.d. measurements on a fully deacetylated chitosan: Application to CuII-polymer interactions. Int. J. Biol. Macromol. 1987, 9, 98–104. [Google Scholar] [CrossRef]
- Rinaudo, M.; Pavlov, G.; Desbrières, J. Solubilization of Chitosan in Strong Acid Medium. Int. J. Polym. Anal. Charact. 1999, 5, 267–276. [Google Scholar] [CrossRef]
- Ejeromedoghene, O.; Oderinde, O.; Egejuru, G.; Adewuyi, S. Chitosan-drug encapsulation as a potential candidate for COVID-19 drug delivery systems: A review. J. Turk. Chem. Soc. A Chem. 2020, 7, 851–864. [Google Scholar] [CrossRef]
- Sun, L.; Legood, R.; Sadique, Z.; Dos-Santos-Silva, I.; Yang, L. Cost effectiveness of risk-based breast cancer screening programme. China Bull. World Health Organ. 2019, 96, 568–577. [Google Scholar] [CrossRef]
- Du, J.Z.; Du, X.J.; Mao, C.Q.; Wang, J. Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 2011, 133, 17560–17563. [Google Scholar] [CrossRef]
- Tripathy, S.; Das, S.; Chakrabortya, S.P.; Sahu, S.K.; Pramanik, P.; Roya, S. Synthesis, characterization of chitosan–tripolyphosphate conjugated chloroquine nanoparticle and its in vivo anti-malarial efficacy against rodent parasite: A dose and duration dependent approach. Int. J. Pharm. 2012, 434, 292–305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Parr, R.G.; Yang, W. Density-functional theory of atoms and molecules. Int. J. Quantum Chem. 1993, 47, 101. [Google Scholar]
- Ziegler, T. Approximate density functional theory as a practical tool in molecular energetics and dynamics. Chem. Rev. 1991, 91, 651–667. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Onoka, I.; Pogrebnoi, A.; Pogrebnaya, T. Geometrical structure, vibrational spectra and thermodynamic properties of chitosan constituents by DFT method. Int. J. Mater. Sci. Appl. 2014, 311, 121–128. [Google Scholar] [CrossRef]
- Zhurko, G.A.; Zhurko, D.A. ChemCraft Version 1.7 (build132). Available online: https://www.chemcraftprog.com (accessed on 21 April 2013).
- Barmpa, A.; Frousiou, O.; Kalogiannis, S.; Perdih, F.; Turel, I.; Psomas, G. Manganese(II) complexes of the quinolone family member flumequine: Structure, antimicrobial activity and affinity for albumins and calf-thymus DNA. Polyhedron 2018, 145, 166–175. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Plenum Press: New York, NY, USA, 2006. [Google Scholar]
- Tan, C.; Liu, J.; Li, H.; Zheng, W.; Shi, S.; Chen, L.; Ji, L. Synthesis, structural characteristics, DNA binding properties and cytotoxicity studies of a series of Ru(III) complexes. J. Inorg. Biochem. 2008, 102, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Rajendiran, V.; Karthik, R.; Palaniandavar, M.; Stoeckli-Evans, H.; Periasamy, V.S.; Akbarsha, M.A.; Srinag, B.S.; Krishnamurthy, H. Mixed-ligand copper(II)-phenolate complexes: Effect of coligand on enhanced DNA and protein binding, DNA cleavage, and anticancer activity. Inorg. Chem. 2007, 46, 8208–8221. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Zhang, G.; Tao, W.; Tang, S. Interaction of the flavonoid hesperidin with bovine serum albumin: A fluorescence quenching study. J. Lumin. 2007, 126, 211–218. [Google Scholar] [CrossRef]
- Sahoo, B.K.; Ghosh, K.S.; Dasgupta, S. Molecular interactions of isoxazolcurcumin with human serum albumin: Spectroscopic and molecular modeling studies. Biopolymers 2009, 91, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Wani, T.A.; Alsaif, N.A.; Alanazi, M.M.; Bakheit, A.H.; Khan, A.A.; Zargar, S. Binding of colchicine and ascorbic acid (vitamin C) to bovine serum albumin: An invitro interaction study using multispectroscopic, molecular docking and molecular dynamics simulation study. J. Mol. Liq. 2021, 342, 117542. [Google Scholar] [CrossRef]
- Hashemnia, S.; Fard, F.K.; Mokhtari, Z.A. study of the interactions between ephedrine and human serum albumin based on spectroscopic, electrochemical and docking assessments. J. Mol. Liq. 2022, 348, 118058. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Y.; Liang, H. Interactive association of drugs binding to human serum albumin. Int. J. Mol. Sci. 2014, 15, 3580–3595. [Google Scholar] [CrossRef]
- Abd Halim, A.A.; Ridzwan, F.W.; Bin Mohamad, S.; Tayyab, S. Targeting the Nalidixic Acid Binding Site on Human Serum Albumin Through Computational Approach: A Re-Investigation. Biointerface Res. Appl. Chem. 2022, 12, 1520–1525. [Google Scholar] [CrossRef]
- Abdollahpour, N.; Soheili, V.; Saberi, M.R.; Chamani, J. Investigation of the interaction between human serum albumin and two drugs as binary and ternary systems. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 705–721. [Google Scholar] [CrossRef]
- Basak, P.; Debnath, T.; Banerjee, R.; Bhattacharyya, M. Selective binding of divalent cations toward heme proteins. Front. Biol. 2016, 11, 32–42. [Google Scholar] [CrossRef]
- Linciano, S.; Moro, G.; Zorzi, A.; Angelini, A. Molecular analysis and therapeutic applications of human serum albumin-fatty acid interactions. J. Control. Release 2022, 348, 15–26. [Google Scholar] [CrossRef]
- Bodapati, A.T.; Reddy, R.S.; Lavanya, K.; Madku, S.R.; Sahoo, B.K. A comprehensive biophysical and theoretical study on the binding of dexlansoprazole with human serum albumin. J. Mol. Liq. 2023, 380, 121777. [Google Scholar] [CrossRef]
- Parsekar, S.U.; Paliwal, K.; Haldar, P.; Antharjanam, P.K.S.; Kumar, M. Synthesis, Characterization, Crystal Structure, DNA and HSA Interactions, and Anticancer Activity of a Mononuclear Cu(II) Complex with a Schiff Base Ligand Containing a Thiadiazoline Moiety. ACS Omega 2022, 7, 2881–2896. [Google Scholar] [CrossRef]
- Shen, F.; Liu, Y.X.; Li, S.M.; Jiang, C.K.; Wang, B.F.; Xiong, Y.H.; Mao, Z.W.; Le, X.Y. Synthesis, crystal structures, molecular docking and in vitro cytotoxicity studies of two new copper(II) complexes: Special emphasis on their binding to HSA. New J. Chem. 2017, 41, 12429–12441. [Google Scholar] [CrossRef]
- Aseman, M.D.; Aryamanesh, S.; Shojaeifard, Z.; Hemmateenejad, B.; Nabavizadeh, S.M. Cycloplatinated(II) derivatives of mercaptopurine capable of binding interactions with HSA/DNA. Inorg. Chem. 2019, 58, 16154–16170. [Google Scholar] [CrossRef]
- Hussain, A.; AlAjmi, M.F.; Rehman, M.T.; Amir, S.; Husain, F.M.; Alsalme, A.; Siddiqui, M.A.; AlKhedhairy, A.A.; Khan, R.A. Copper(II) complexes as potential anticancer and Nonsteroidal anti-inflammatory agents: In vitro and in vivo studies. Sci. Rep. 2019, 9, 5237. [Google Scholar] [CrossRef] [PubMed]
- Bodapati, A.T.S.; Sahoo, B.K.; Ragaiahgari, S.R.; Kandikonda, L.; Madku, S.R. Deciphering the nature of binding of dexlansoprazole with DNA: Biophysical and docking approaches. Int. J. Biol. Macromol. 2022, 217, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Ovung, A.; Bhattacharyya, J. Binding effects of antibiotic drug sulfamethazine on the serum albumins: Multi-spectroscopic and computation approach. Chem. Phys. 2022, 5, 100087. [Google Scholar] [CrossRef]
- Huang, Z.Y.; Li, X.Y.; Hu, L.Y.; Bai, A.M.; Hu, Y.J. Comparative study of two antipsychotic drugs binding to human serum albumin: By multispectroscopic and molecular docking methods. J. Mol. Liq. 2022, 365, 120084. [Google Scholar] [CrossRef]
- Denny, W.A.; Wakelin, L.P. Kinetic and equilibrium studies of the interaction of amsacrine and anilino ring-substituted analogues with DNA. Cancer Res. 1986, 46 Pt 1, 1717–1721. [Google Scholar] [PubMed]
- Daviter, T.; Johnson, C.M.; McLaughlin, S.H.; Williams, M.A. Protein-Ligand Interactions Methods and Applications, 3rd ed.; Springer Science Business Media, LLC: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics; Portland Press: London, UK, 1995; 343p, ISBN 1-85578-072-0. [Google Scholar]
- Gutfreund, H. Kinetics for the Life Sciences: Receptors, Transmitters and Catalysts; Cambridge University Press: Cambridge, UK, 1995; 360p. [Google Scholar]
- Williams, M.A.; Daviter, T. Protein-Ligand Interactions. Methods Mol. Biol. 2013, 13, 85. [Google Scholar]
- Li, J.; Kataoka, K. Chemo-physical Strategies to Advance the in Vivo Functionality of Targeted Nanomedicine: The Next Generation. J. Am. Chem. Soc. 2021, 143, 538–559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qin, X.; Kong, F.; Chen, P.; Pan, G. Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Deliv. 2019, 26, 328–342. [Google Scholar] [CrossRef]
- Yan, D.; Li, Y.; Liu, Y.; Li, N.; Xue Zhang, X.; Yan, C. Antimicrobial Properties of Chitosan and Chitosan Derivatives in the Treatment of Enteric Infections. Molecules 2021, 26, 7136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, Y.; Ding, Y.; Povey, M.; York, D. Investigation into the antibacterial behavior of suspensions of ZnO nanoparticles (ZnO nanofluids). J. Nanopart. Res. 2007, 9, 479–489. [Google Scholar] [CrossRef]
- Dey, D.; Maiti, C.; Maiti, S.; Dhara, D. Interaction between Calf Thymus DNA and Cationic Bottle-Brush Copolymers: Equilibrium and Stopped-Flow Kinetic Studies. Phys. Chem. Chem. Phys. 2015, 17, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- The Molecular Operating Environment (MOE2019.01) Software Available from Chemical Computing Group Inc. 1010 Sherbrooke Street West, Suite 910, Montreal, Canda H3A 2R7. Available online: http://www.chemcomp.com (accessed on 5 June 2010).
- Owuama, C. Determination of minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) using a novel dilution tube method. Afr. J. Microbiol. Res. 2017, 11, 977–980. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270. [Google Scholar] [CrossRef]
- Roy, U.; Luck, L.A. Molecular modeling of estrogen receptor using molecular operating environment. Biochem. Mol. Biol. Educ. 2007, 35, 238–243. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size (nm) | PDI | Zeta Potential (mV) | |
|---|---|---|---|
| HCQ | 255.7 ± 18.5 | 0.224 ± 0.083 | −16.4 ± 0.4 |
| HCQ@CS NPs | 159.3 ± 7.1 | 0.337 ± 0.101 | +46.6 ± 0.8 |
| Parameter | EHOMO (eV) | ELUMO (eV) | IP | EA | ELUMO–EHOMO (eV) | Χ (eV) | Pi (eV) | η (eV) | σ (eV−1) | S (eV−1) | ω (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HCQ | +1.46 | +3.42 | −1.46 | −3.42 | 1.96 | −2.44 | 2.44 | 0.982 | 1.018 | 0.509 | 3.037 |
| M | −10.27 | −3.07 | +10.27 | +3.07 | 7.2 | 6.67 | −6.67 | 3.601 | 0.277 | 0.138 | 6.177 |
| MM | −10.15 | −1.78 | +10.15 | +1.78 | 8.36 | 5.97 | −5.97 | 4.183 | 0.239 | 0.119 | 4.261 |
| KSV [M−1 × 104] | Kq [M−1s−1 × 1012] | n | fa | K [M−1 × 104] | Ka [M−1 × 104] | ΔG [kJ Mol−1] | Kb [M−1 × 104] | |
|---|---|---|---|---|---|---|---|---|
| HCQ | 0.20 | 0.20 | 0.8 | 1.10 | 0.41 | 0.15 | −7.8 | 0.10 |
| HCQ@CS NPs | 0.70 | 0.70 | 1.0 | 1.21 | 0.62 | 0.80 | −9.7 | 0.55 |
| First Step | Second Step | Overall Reaction | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| k1 [M−1s−1] | k-1 [s−1] | Ka1 [M−1] | Kd1 [M] | ΔG1 [kJ mol−1] | K2 [M−1s−1] | k-2 [s−1] | Ka2 [M−1] | Kd2 [M] | ΔG [kJ mol−1] | Kd [10−2M] | Ka [M−1] | ΔG [kJmol−1] | |
| HCQ@CS NPs | 175 ± 10 | 7.9 ± 0.8 | 22.2 | 0.045 | −7.7 | 4.1 ± 0.2 | 0.4 ± 0.0 | 10 | 0.1 | −5.7 | 0.4 | 244.5 | −13.6 |
| HCQ | 5.9± 0.4 | 3.5 ± 0.6 | 1.67 | 0.6 | −12.7 | Only one step has been detected | |||||||
| IC50 Values 1 (μg/mL) | ||
|---|---|---|
| Wi38 | A549 | |
| HCQ | 115 ± 1.4 | 102.21 ± 0.67 |
| HCQ@CS NPs | 66.37 ± 0.51 | 28.57 ± 1.72 |
| Doxo | 57.81 ± 0.79 | 50.68 ± 3.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elshami, F.I.; Shereef, H.A.; El-Mehasseb, I.M.; Shaban, S.Y.; van Eldik, R. Hydroxychloroquine-Loaded Chitosan Nanoparticles Induce Anticancer Activity in A549 Lung Cancer Cells: Design, BSA Binding, Molecular Docking, Mechanistic, and Biological Evaluation. Int. J. Mol. Sci. 2023, 24, 14103. https://doi.org/10.3390/ijms241814103
Elshami FI, Shereef HA, El-Mehasseb IM, Shaban SY, van Eldik R. Hydroxychloroquine-Loaded Chitosan Nanoparticles Induce Anticancer Activity in A549 Lung Cancer Cells: Design, BSA Binding, Molecular Docking, Mechanistic, and Biological Evaluation. International Journal of Molecular Sciences. 2023; 24(18):14103. https://doi.org/10.3390/ijms241814103
Chicago/Turabian StyleElshami, Fawzia I., Hadeer A. Shereef, Ibrahim M. El-Mehasseb, Shaban Y. Shaban, and Rudi van Eldik. 2023. "Hydroxychloroquine-Loaded Chitosan Nanoparticles Induce Anticancer Activity in A549 Lung Cancer Cells: Design, BSA Binding, Molecular Docking, Mechanistic, and Biological Evaluation" International Journal of Molecular Sciences 24, no. 18: 14103. https://doi.org/10.3390/ijms241814103
APA StyleElshami, F. I., Shereef, H. A., El-Mehasseb, I. M., Shaban, S. Y., & van Eldik, R. (2023). Hydroxychloroquine-Loaded Chitosan Nanoparticles Induce Anticancer Activity in A549 Lung Cancer Cells: Design, BSA Binding, Molecular Docking, Mechanistic, and Biological Evaluation. International Journal of Molecular Sciences, 24(18), 14103. https://doi.org/10.3390/ijms241814103