
A Robust Drug–Target Interaction Prediction Framework with Capsule Network and Transfer Learning

 , , ,
, , ,
Abstract
:1. Introduction
- To overcome the limitation of other studies in which negative DTI data are randomly selected from unknown drug–target pairs, we established two experimentally validated datasets.
- Protein sequences are treated as natural language and are vectorized by the state-of-the-art ProtBert model through transfer learning, while drug molecules are transformed by MPNN. Both encoding approaches represent protein targets and drug molecules more precisely.
- The proposed capsule network-based DTI prediction model describes the internal hierarchical representation of features. It outperforms other existing established SOTA DTI prediction tools on seven experimentally validated DTI datasets with varying amounts of data, including human (Homo sapiens) and worm (Caenorhabditis elegans) species datasets.
2. Results
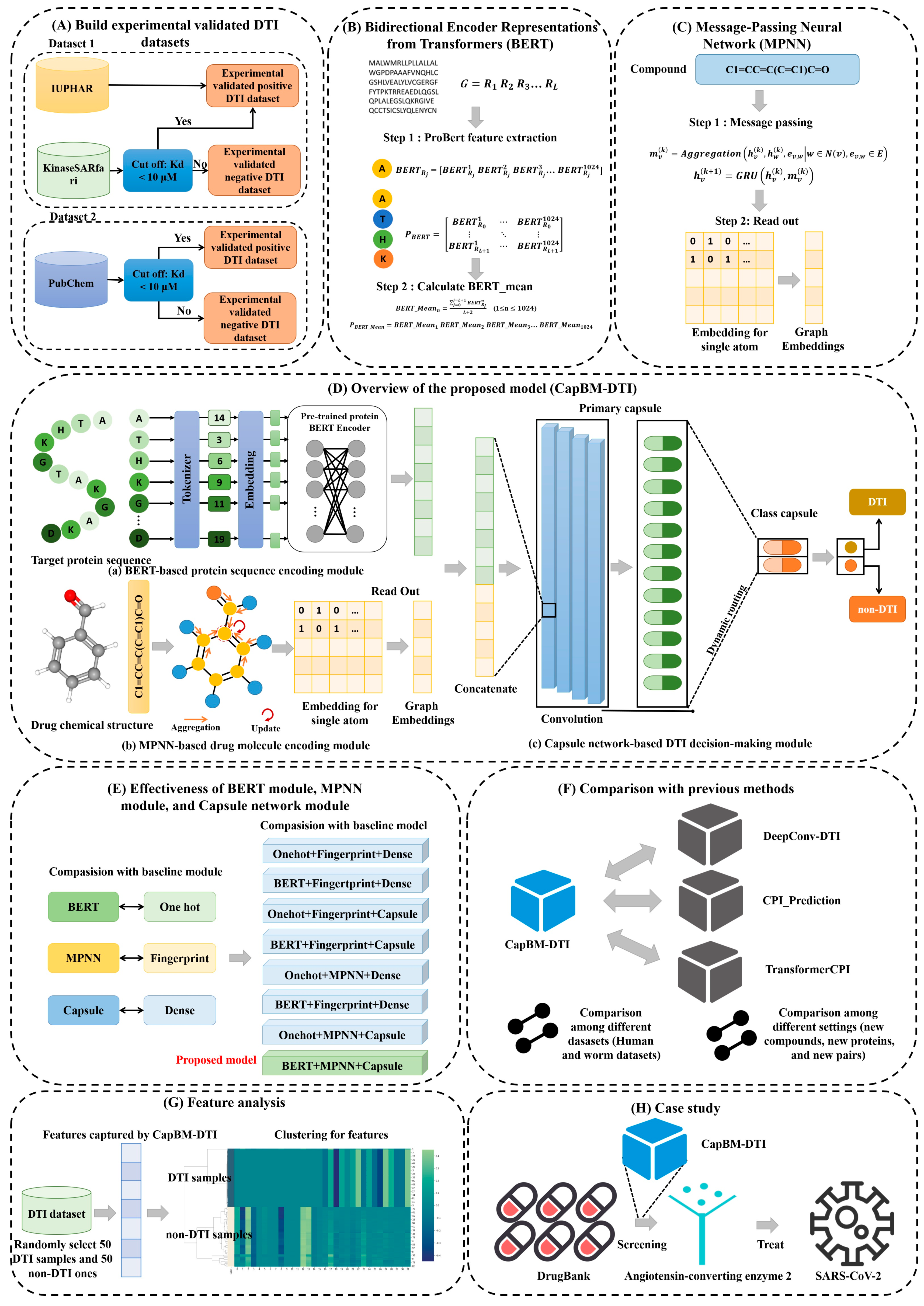
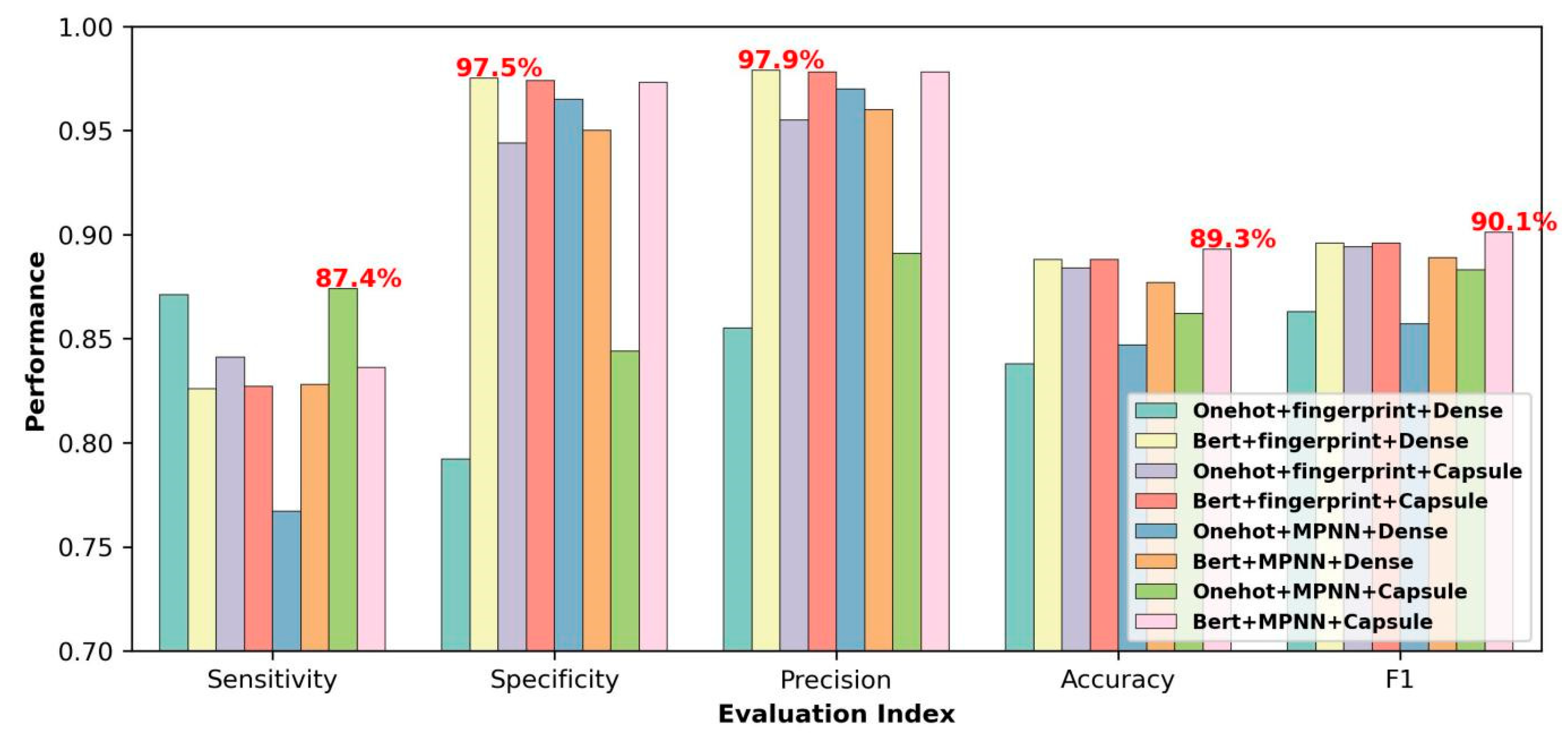
2.1. Effectiveness of BERT Module, MPNN Module, and Capsule Network Module
2.2. Performance and Generalization Comparison with Existing SOTA Predictors
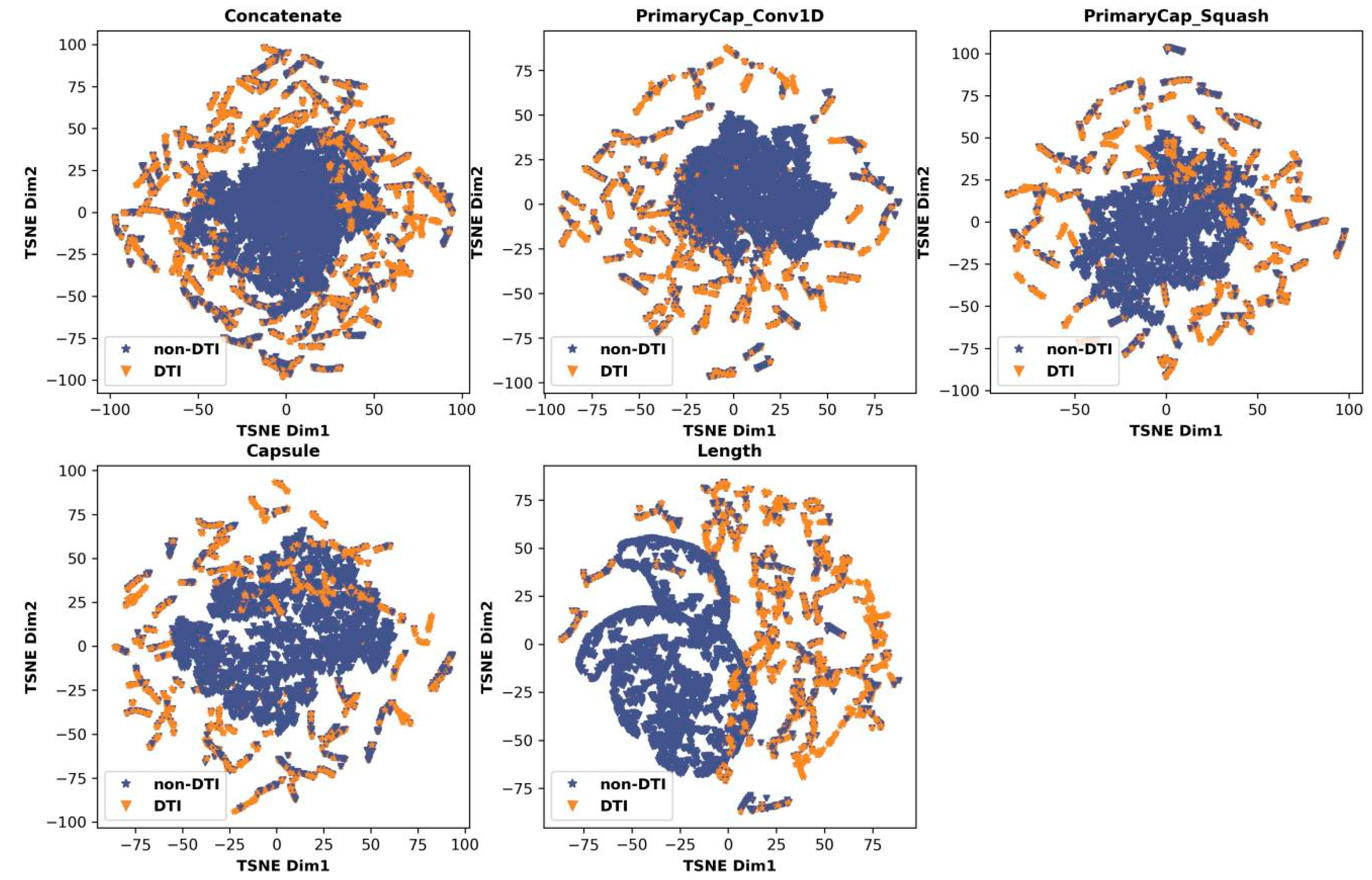
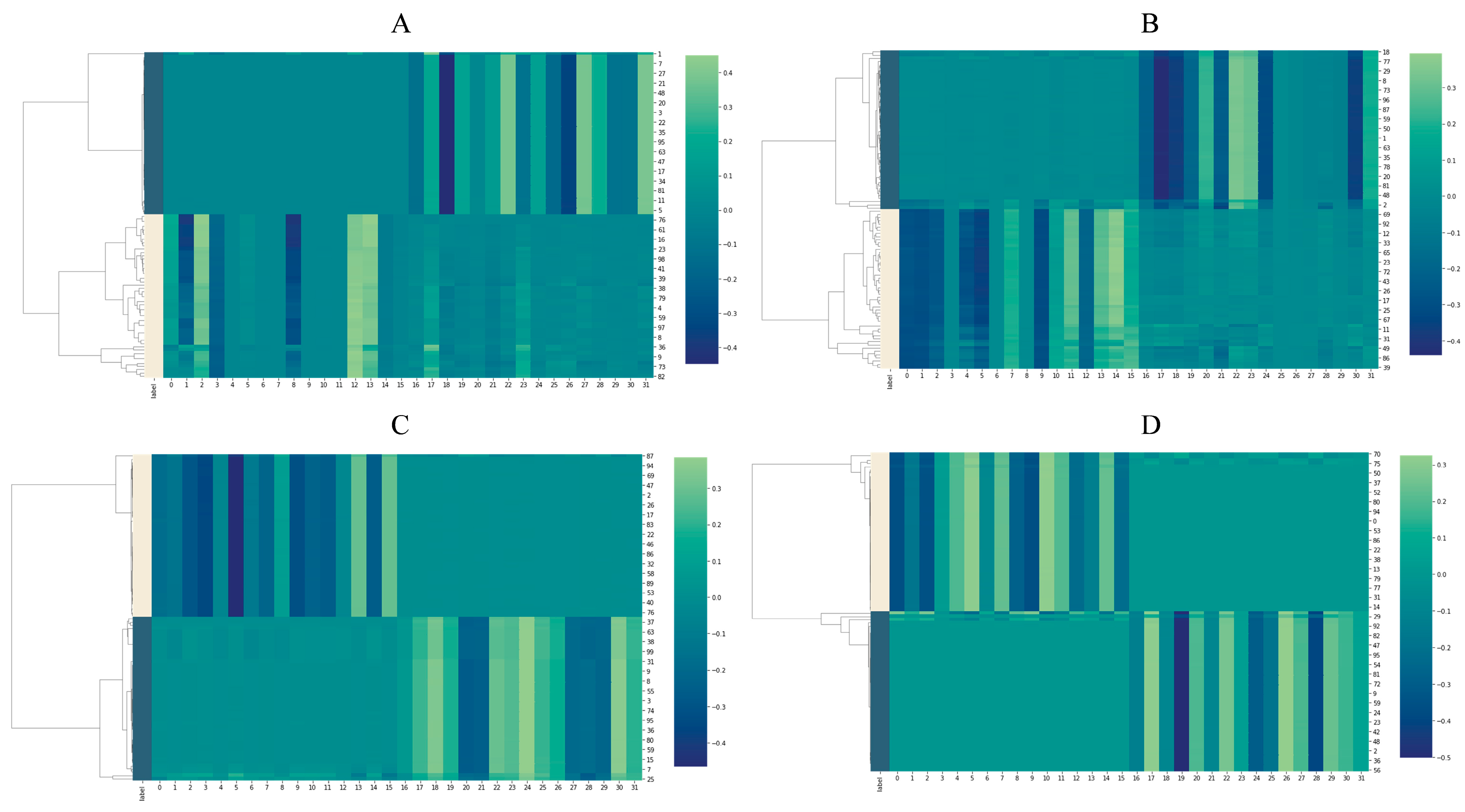
2.3. Feature Analysis
2.4. Case Study of Drug Repurposing to Treat COVID-19
3. Discussion
4. Materials and Methods
4.1. Experimentally Validated Datasets
4.2. Framework of the Constructed Model
4.2.1. Feature Extraction from Protein
4.2.2. Feature Extraction from Drug Molecule
Message Passing
Readout
- The k-step-aggregated node states will be partitioned into the subgraphs (corresponding to each molecule in the batch);
- Each subgraph will then be padded to match the subgraph with the greatest number of nodes;
- The (stacked padded) tensor, encoding subgraphs (each subgraph containing a set of node states), are masked to make sure the paddings do not interfere with training;
- Finally, the tensor is passed to the transformer followed by average pooling.
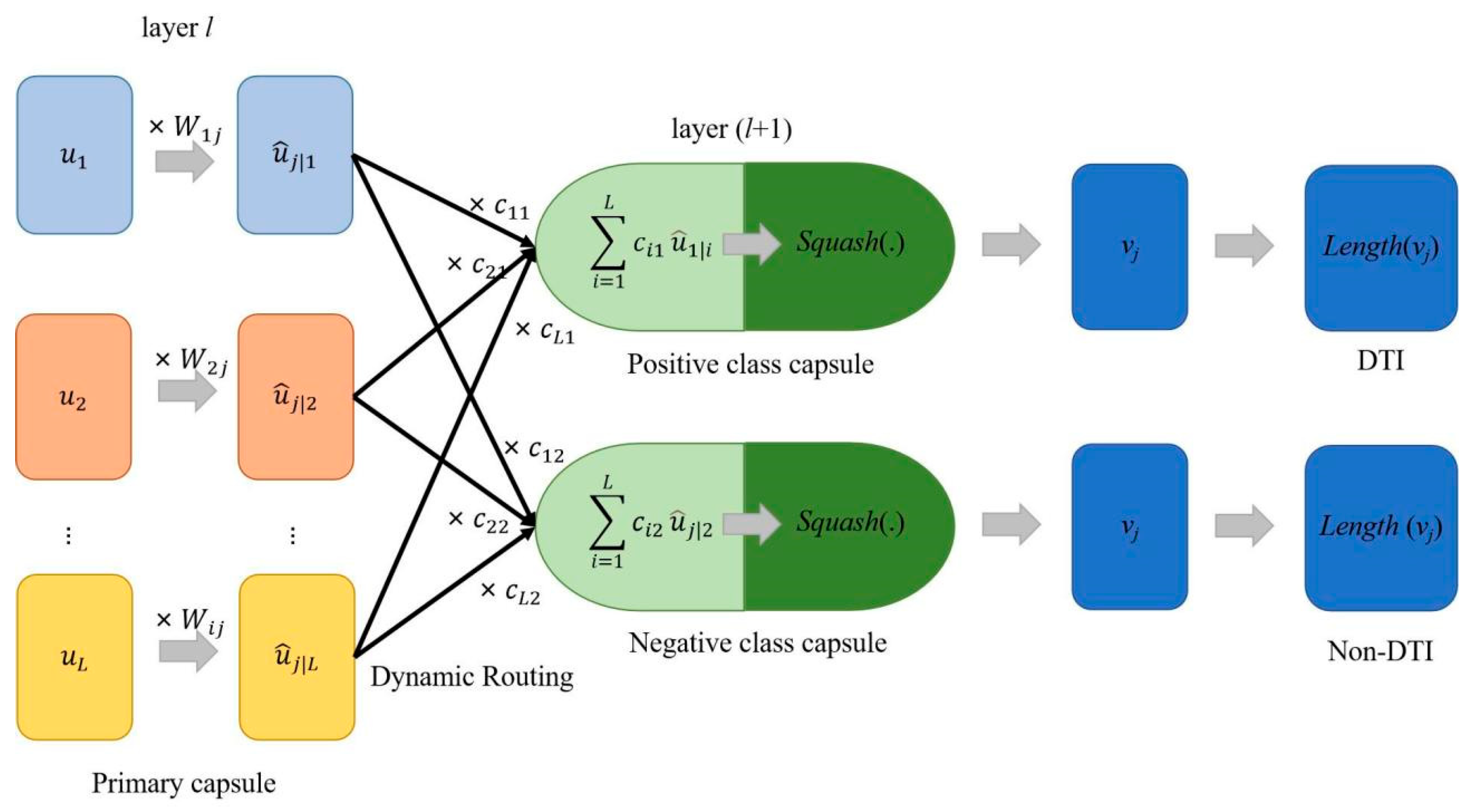
4.2.3. Capsule Network
| Algorithm 1: Dynamic Routing |
| Input: , r and l Output: vj
|
4.3. Performance Metrics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sachdev, K.; Gupta, M.K. A comprehensive review of feature based methods for drug target interaction prediction. J. Biomed. Inform. 2019, 93, 103159. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Kotera, M.; Moriya, Y.; Sawada, R.; Kanehisa, M.; Goto, S. DINIES: Drug-target interaction network inference engine based on supervised analysis. Nucleic Acids Res. 2014, 42, W39–W45. [Google Scholar] [CrossRef] [PubMed]
- Bagherian, M.; Sabeti, E.; Wang, K.; Sartor, M.A.; Nikolovska-Coleska, Z.; Najarian, K. Machine learning approaches and databases for prediction of drug–target interaction: A survey paper. Brief. Bioinform. 2021, 22, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xiao, X.; Qiu, W.-R. DTI-BERT: Identifying drug-target interactions in cellular networking based on BERT and deep learning method. Front. Genet. 2022, 13, 859188. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Sethi, A.; Joshi, K.; Sasikala, K.; Alvala, M. Molecular docking in modern drug discovery: Principles and recent applications. Drug Discov. Dev.-New Adv. 2019, 2, 27–47. [Google Scholar]
- He, Z.; Zhang, J.; Shi, X.H.; Hu, L.L.; Kong, X.; Cai, Y.D.; Chou, K.C. Predicting drug-target interaction networks based on functional groups and biological features. PLoS ONE 2010, 5, e9603. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Yang, Z.-X.; Wang, Y.; Deng, N.-Y. Computationally probing drug-protein interactions via support vector machine. Lett. Drug Des. Discov. 2010, 7, 370–378. [Google Scholar] [CrossRef]
- Tabei, Y.; Yamanishi, Y. Scalable prediction of compound-protein interactions using minwise hashing. BMC Syst. Biol. 2013, 7 (Suppl. S6), S3. [Google Scholar] [CrossRef]
- Yu, H.; Chen, J.; Xu, X.; Li, Y.; Zhao, H.; Fang, Y.; Li, X.; Zhou, W.; Wang, W.; Wang, Y. A systematic prediction of multiple drug-target interactions from chemical, genomic, and pharmacological data. PLoS ONE 2012, 7, e37608. [Google Scholar] [CrossRef]
- Sawada, R.; Kotera, M.; Yamanishi, Y. Benchmarking a Wide Range of Chemical Descriptors for Drug-Target Interaction Prediction Using a Chemogenomic Approach. Mol. Inform. 2014, 33, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, H.; Ozgur, A.; Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Xiao, C.; Glass, L.M.; Sun, J. MolTrans: Molecular Interaction Transformer for drug-target interaction prediction. Bioinformatics 2021, 37, 830–836. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhao, Q.; Li, Y.; Wang, J. IIFDTI: Predicting drug-target interactions through interactive and independent features based on attention mechanism. Bioinformatics 2022, 38, 4153–4161. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Walters, R.; Shafi, Z.; Ahmed, O.S.; Sebek, M.; Gysi, D.; Yu, R.; Eliassi-Rad, T.; Barabási, A.-L.; Menichetti, G. Improving the generalizability of protein-ligand binding predictions with AI-Bind. Nat. Commun. 2023, 14, 1989. [Google Scholar] [CrossRef]
- You, J.; McLeod, R.D.; Hu, P. Predicting drug-target interaction network using deep learning model. Comput. Biol. Chem. 2019, 80, 90–101. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rihawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M. ProtTrans: Towards cracking the language of Life’s code through self-supervised deep learning and high performance computing. arXiv 2020, arXiv:2007.06225. [Google Scholar]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 2019, 35, 309–318. [Google Scholar] [CrossRef]
- Chen, L.; Tan, X.; Wang, D.; Zhong, F.; Liu, X.; Yang, T.; Luo, X.; Chen, K.; Jiang, H.; Zheng, M. TransformerCPI: Improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 2020, 36, 4406–4414. [Google Scholar] [CrossRef]
- Liu, H.; Sun, J.; Guan, J.; Zheng, J.; Zhou, S. Improving compound–protein interaction prediction by building up highly credible negative samples. Bioinformatics 2015, 31, i221–i229. [Google Scholar] [CrossRef]
- Mulling, N.; Rohn, H. Angiotensin-converting enzyme 2 (ACE2): Role in the pathogenesis of diseases outside of COVID-19. Der nephrologe 2021, 16, 185–188. [Google Scholar] [CrossRef]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef]
- Touret, F.; Gilles, M.; Barral, K.; Nougairède, A.; Van Helden, J.; Decroly, E.; De Lamballerie, X.; Coutard, B. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Sci. Rep. 2020, 10, 13093. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; Zhang, X.; Dai, F.; Yin, J.; Zhang, Y. Drug–target interaction prediction: Databases, web servers and computational models. Brief. Bioinform. 2016, 17, 696–712. [Google Scholar] [CrossRef] [PubMed]
- Gönen, M. Predicting drug–target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics 2012, 28, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhu, W.; Liao, B.; Duan, Y.; Chen, M.; Chen, Y.; Yang, J. Screening drug-target interactions with positive-unlabeled learning. Sci. Rep. 2017, 7, 8087. [Google Scholar] [CrossRef]
- Ezzat, A.; Wu, M.; Li, X.-L.; Kwoh, C.-K. Computational prediction of drug–target interactions using chemogenomic approaches: An empirical survey. Brief. Bioinform. 2019, 20, 1337–1357. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, X.; Jin, S.; Lin, J.; Liu, J. Machine learning for drug-target interaction prediction. Molecules 2018, 23, 2208. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Harmar, A.J.; Hills, R.A.; Rosser, E.M.; Jones, M.; Buneman, O.P.; Dunbar, D.R.; Greenhill, S.D.; Hale, V.A.; Sharman, J.L.; Bonner, T.I. IUPHAR-DB: The IUPHAR database of G protein-coupled receptors and ion channels. Nucleic Acids Res. 2009, 37 (Suppl. S1), D680–D685. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Niijima, S.; Shiraishi, A.; Okuno, Y. Dissecting kinase profiling data to predict activity and understand cross-reactivity of kinase inhibitors. J. Chem. Inf. Model. 2012, 52, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xiao, X.; Qiu, W.-R. iCDI-W2vCom: Identifying the Ion channel–Drug interaction in cellular networking based on word2vec and node2vec. Front. Genet. 2021, 12, 738274. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Bianchi, F.; Terragni, S.; Hovy, D. Pre-training is a hot topic: Contextualized document embeddings improve topic coherence. arXiv 2020, arXiv:2004.03974. [Google Scholar]
- Hu, B.; Xia, J.; Zheng, J.; Tan, C.; Huang, Y.; Xu, Y.; Li, S.Z. Protein language models and structure prediction: Connection and progression. arXiv 2022, arXiv:2211.16742. [Google Scholar]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Dumortier, B.; Liutkus, A.; Carré, C.; Krouk, G. PeTriBERT: Augmenting BERT with tridimensional encoding for inverse protein folding and design. bioRxiv 2022, 2022, 503344. [Google Scholar]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Chandra, A.; Tünnermann, L.; Löfstedt, T.; Gratz, R. Transformer-based deep learning for predicting protein properties in the life sciences. eLife 2023, 12, e82819. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural Message Passing for Quantum Chemistry. In Proceedings of the International Conference on Machine Learning 2017, Sydney, Australia, 6–11 August 2017; PMLR: Cambridge, UK, 2017; pp. 1263–1272. [Google Scholar]
- Withnall, M.; Lindelöf, E.; Engkvist, O.; Chen, H. Building attention and edge message passing neural networks for bioactivity and physical–chemical property prediction. J. Cheminform. 2020, 12, 1. [Google Scholar] [CrossRef]
- Jo, J.; Kwak, B.; Choi, H.-S.; Yoon, S. The message passing neural networks for chemical property prediction on SMILES. Methods 2020, 179, 65–72. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, M.; Luo, Y.; Xu, Z.; Xie, Y.; Wang, L.; Cai, L.; Qi, Q.; Yuan, Z.; Yang, T. Advanced graph and sequence neural networks for molecular property prediction and drug discovery. Bioinformatics 2022, 38, 2579–2586. [Google Scholar] [CrossRef]
- Datta, R.; Das, D.; Das, S. Efficient lipophilicity prediction of molecules employing deep-learning models. Chemom. Intell. Lab. Syst. 2021, 213, 104309. [Google Scholar] [CrossRef]
- Lu, L.; Yi, Y.; Huang, F.; Wang, K.; Wang, Q. Integrating local CNN and global CNN for script identification in natural scene images. IEEE Access 2019, 7, 52669–52679. [Google Scholar] [CrossRef]
- Cao, X.; He, W.; Chen, Z.; Li, Y.; Wang, K.; Zhang, H.; Wei, L.; Cui, L.; Su, R.; Wei, L. PSSP-MVIRT: Peptide secondary structure prediction based on a multi-view deep learning architecture. Brief. Bioinform. 2021, 22, bbab203. [Google Scholar] [CrossRef]
- Khanal, J.; Nazari, I.; Tayara, H.; Chong, K.T. 4mCCNN: Identification of N4-methylcytosine sites in prokaryotes using convolutional neural network. IEEE Access 2019, 7, 145455–145461. [Google Scholar] [CrossRef]
- Khanal, J.; Tayara, H.; Chong, K.T. Identifying enhancers and their strength by the integration of word embedding and convolution neural network. IEEE Access 2020, 8, 58369–58376. [Google Scholar] [CrossRef]
- Luo, X.; Kang, X.; Schönhuth, A. Predicting the prevalence of complex genetic diseases from individual genotype profiles using capsule networks. Nat. Mach. Intell. 2023, 5, 114–125. [Google Scholar] [CrossRef]
- Ali, S.D.; Kim, J.H.; Tayara, H.; to Chong, K. Prediction of RNA 5-hydroxymethylcytosine modifications using deep learning. IEEE Access 2021, 9, 8491–8496. [Google Scholar] [CrossRef]
- LaLonde, R.; Bagci, U. Capsules for object segmentation. arXiv 2018, arXiv:1804.04241. [Google Scholar]
- Sabour, S.; Frosst, N.; Hinton, G.E. Dynamic routing between capsules. Adv. Neural Inf. Process. Syst. 2017, 30, 3859–3869. [Google Scholar]
- Hinton, G.E.; Sabour, S.; Frosst, N. Matrix capsules with EM routing. In Proceedings of the International Conference on Learning Representations 2018, Vancouver, BC, Canada, 30 April–3 May 2018. [Google Scholar]
- Baell, J.B. Feeling nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- Rimassa, L. Drugs in development for hepatocellular carcinoma. Gastroenterol. Hepatol. 2018, 14, 542. [Google Scholar]
- Lee, I.; Nam, H. Sequence-based prediction of protein binding regions and drug–target interactions. J. Cheminform. 2022, 14, 5. [Google Scholar] [CrossRef]
- Verhasselt, S.; Roman, B.I.; Bracke, M.E.; Stevens, C.V. Improved synthesis and comparative analysis of the tool properties of new and existing D-ring modified (S)-blebbistatin analogs. Eur. J. Med. Chem. 2017, 136, 85–103. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Dataset 1 | Dataset 2 | Dataset 3 [21] | Dataset 4 [21] |
|---|---|---|---|---|
| Species | H. sapiens | H. sapiens | H. sapiens | C. elegans |
| The number of compounds | 6602 | 14,737 | 2726 | 1767 |
| The number of proteins | 1900 | 2709 | 2001 | 1876 |
| The number of positive interactions a | 16,627 | 32,013 | 3364 | 3893 |
| The number of negative interactions b | 11,768 | 32,013 | 3364 | 3893 |
| Density (%) | 0.226 | 0.16 | 0.123 | 0.235 |
| Model | Dataset 1 | Dataset 2 | Dataset 3 | Dataset 4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy | F1 | AUC | AUPR | Accuracy | F1 | AUC | AUPR | Accuracy | F1 | AUC | AUPR | Accuracy | F1 | AUC | AUPR | |
| DeepConv-DTI | 0.877 | 0.894 | 0.941 | 0.964 | 0.825 | 0.843 | 0.933 | 0.932 | 0.611 | 0.662 | 0.636 | 0.780 | 0.943 | 0.936 | 0.978 | 0.975 |
| CPI_prediction | 0.885 | 0.894 | 0.943 | 0.967 | 0.864 | 0.852 | 0.935 | 0.938 | 0.891 | 0.901 | 0.936 | 0.945 | 0.926 | 0.931 | 0.965 | 0.972 |
| TransformerCPI | 0.872 | 0.883 | 0.940 | 0.965 | 0.855 | 0.851 | 0.938 | 0.935 | 0.892 | 0.893 | 0.954 | 0.958 | 0.914 | 0.911 | 0.977 | 0.977 |
| IIFDTI | 0.857 | 0.879 | 0.946 | 0.968 | 0.736 | 0.631 | 0.952 | 0.943 | 0.880 | 0.890 | 0.951 | 0.963 | 0.938 | 0.942 | 0.980 | 0.983 |
| CapBM-DTI | 0.893 | 0.901 | 0.946 | 0.970 | 0.87 | 0.862 | 0.935 | 0.944 | 0.915 | 0.915 | 0.958 | 0.961 | 0.941 | 0.938 | 0.982 | 0.983 |
| Dataset | New Compounds Dataset c | New Proteins Dataset d | New Pairs Dataset e | |||
|---|---|---|---|---|---|---|
| Training Set | Test Set | Training Set | Test Set | Training Set | Test Set | |
| The number of compounds | 2000 | 726 | 2406 | 824 | 1770 | 227 |
| The number of proteins | 1797 | 915 | 1500 | 501 | 1351 | 125 |
| The number of positive interactions a | 2569 | 795 | 2888 | 476 | 2218 | 125 |
| The number of negative interactions b | 2445 | 919 | 2532 | 832 | 1834 | 221 |
| Density (%) | 0.140 | 0.258 | 0.150 | 0.317 | 0.169 | 0.674 |
| Model | New Compounds | New Proteins | New Pairs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy | F1 | AUC | AUPR | Accuracy | F1 | AUC | AUPR | Accuracy | F1 | AUC | AUPR | |
| DeepConv-DTI | 0.744 | 0.714 | 0.808 | 0.699 | 0.792 | 0.719 | 0.849 | 0.668 | 0.668 | 0.590 | 0.729 | 0.645 |
| CPI_prediction | 0.571 | 0.613 | 0.599 | 0.669 | 0.399 | 0.462 | 0.443 | 0.53 | 0.455 | 0.567 | 0.527 | 0.645 |
| TransformerCPI | 0.779 | 0.743 | 0.847 | 0.832 | 0.829 | 0.744 | 0.892 | 0.841 | 0.723 | 0.575 | 0.753 | 0.661 |
| IIFDTI | 0.750 | 0.765 | 0.834 | 0.818 | 0.851 | 0.814 | 0.922 | 0.912 | 0.705 | 0.608 | 0.765 | 0.668 |
| CapBM-DTI | 0.788 | 0.766 | 0.832 | 0.848 | 0.906 | 0.874 | 0.945 | 0.908 | 0.725 | 0.615 | 0.751 | 0.704 |
| DrugBank ID | Name | Interact Status a | Non-DTI Possibility | DTI Possibility | Drug Mechanism of Action | Ref |
|---|---|---|---|---|---|---|
| DB00691 | Moexipril | 1 | 0.03831145 | 0.98414844 | An angiotensin-converting enzyme inhibitor (ACE inhibitor) used for the treatment of hypertension and congestive heart failure | [23] |
| DB00477 | Chlorpromazine | 1 | 0.01079374 | 0.995042 | Facilitating ACE2 endocytosis, reducing virus-receptor binding capacity in vitro | [23] |
| DB13609 | Umifenovir | 1 | 0.12442836 | 0.84877896 | Decreased viral endocytosis | [24] |
| DB12466 | Favipiravir | 1 | 0.03333556 | 0.98836476 | Decreased viral endocytosis | [24] |
| DB13729 | Camostat | 1 | 0.0929359 | 0.83505136 | TMPRSS2 hydrolyzes ACE2 and thus degrades ACE2 | [25] |
| DB12598 | Nafamostat | 1 | 0.04878383 | 0.9957441 | TMPRSS2 hydrolyzes ACE2 and thus degrades ACE2 | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Huang, H.-Y.; Chen, Y.; Lin, Y.-C.-D.; Yao, L.; Lin, T.; Leng, J.; Chang, Y.; Zhang, Y.; Zhu, Z.; et al. A Robust Drug–Target Interaction Prediction Framework with Capsule Network and Transfer Learning. Int. J. Mol. Sci. 2023, 24, 14061. https://doi.org/10.3390/ijms241814061
Huang Y, Huang H-Y, Chen Y, Lin Y-C-D, Yao L, Lin T, Leng J, Chang Y, Zhang Y, Zhu Z, et al. A Robust Drug–Target Interaction Prediction Framework with Capsule Network and Transfer Learning. International Journal of Molecular Sciences. 2023; 24(18):14061. https://doi.org/10.3390/ijms241814061
Chicago/Turabian StyleHuang, Yixian, Hsi-Yuan Huang, Yigang Chen, Yang-Chi-Dung Lin, Lantian Yao, Tianxiu Lin, Junlin Leng, Yuan Chang, Yuntian Zhang, Zihao Zhu, and et al. 2023. "A Robust Drug–Target Interaction Prediction Framework with Capsule Network and Transfer Learning" International Journal of Molecular Sciences 24, no. 18: 14061. https://doi.org/10.3390/ijms241814061
APA StyleHuang, Y., Huang, H.-Y., Chen, Y., Lin, Y.-C.-D., Yao, L., Lin, T., Leng, J., Chang, Y., Zhang, Y., Zhu, Z., Ma, K., Cheng, Y.-N., Lee, T.-Y., & Huang, H.-D. (2023). A Robust Drug–Target Interaction Prediction Framework with Capsule Network and Transfer Learning. International Journal of Molecular Sciences, 24(18), 14061. https://doi.org/10.3390/ijms241814061