

The Role of SNAP-25 in Autism Spectrum Disorders Onset Patterns
 , ,
, ,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Results
2.1. SNAP-25 rs363050, rs363039, rs363043, and rs1051312 Genotype Distribution in Children with ASD and Healthy Controls
2.2. SNAP-25 rs363050, rs363039, rs363043, and rs1051312 Allele and Genotype Distributions in Children with ASD with Early and Regressive Onsets
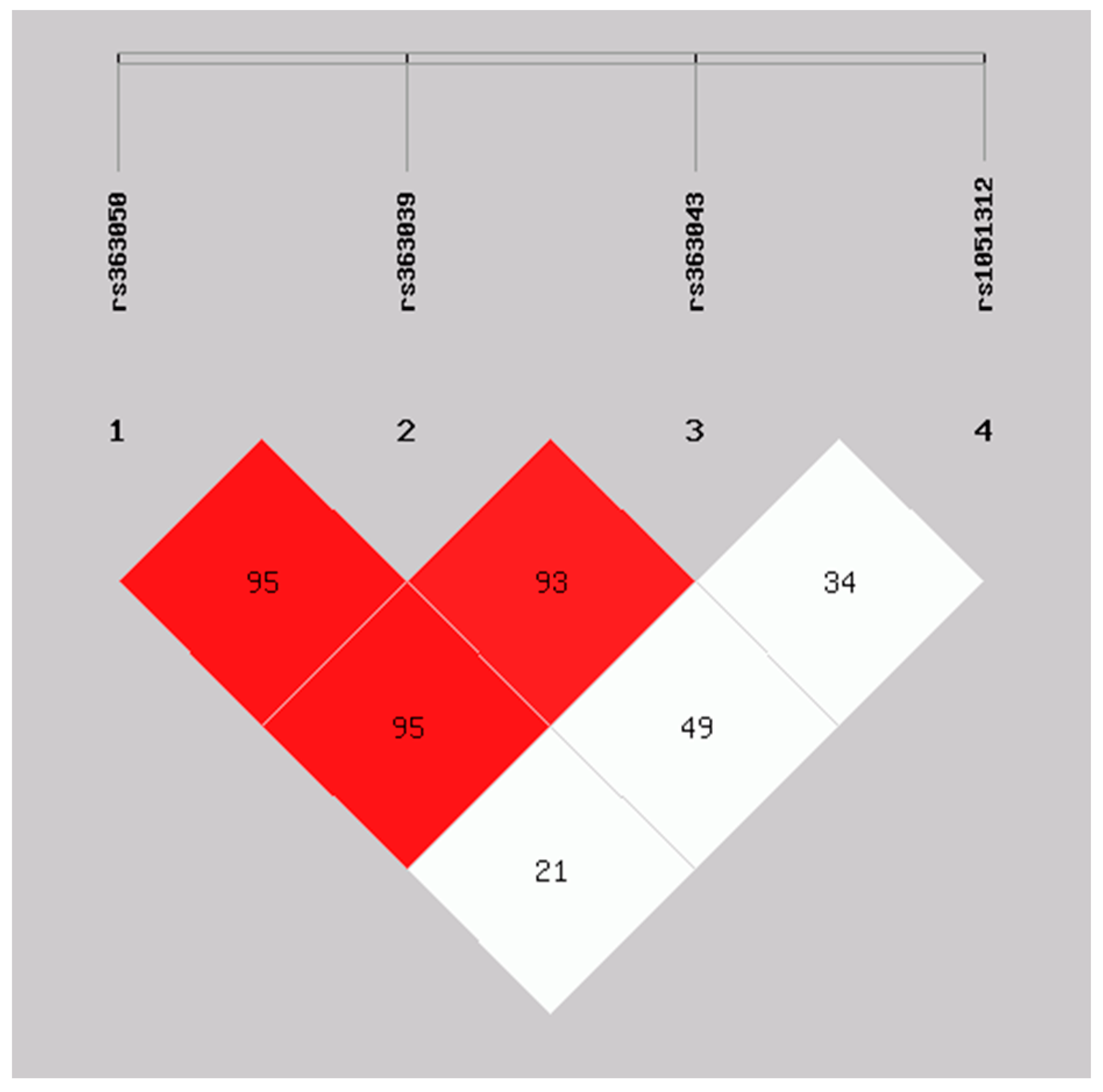
2.3. Haplotype Analysis of SNAP-25 rs363050, rs363039, rs363043, and rs1051312 in the ASD Early and Regressive Onsets and Healthy Controls Groups
2.4. Correlation of Clinical and Functioning Scales with the ASD Early Onset, Regressive Onset, and SNAP-25 Genotypes
3. Discussion
4. Material and Methods
4.1. Patients and Controls
4.2. SNPs Genotyping
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association [APA]. Diagnostic and Statistical Manual of Mental Disorders (DSM-V-TR), 5th ed.; American Psychiatry Association Publishing: Washington, DC, USA, 2022. [Google Scholar] [CrossRef]
- Maenner, M.J.; Warren, Z.; Williams, A.R.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Fitzgerald, R.T.; Furnier, S.M.; Hughes, M.M.; et al. Prevalence and characteristics of Autism Spectrum Disorder Among Children Aged 8 Years-Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2020. MMWR. Surveill. Summ. 2023, 72, 1–14. [Google Scholar] [CrossRef]
- OssNA. Available online: https://osservatorionazionaleautismo.iss.it/ (accessed on 1 June 2023).
- Trost, B.; Thiruvahindrapuram, B.; Chan, A.J.S.; Engchuan, W.; Higginbotham, E.J.; Howe, J.L.; Loureiro, L.O.; Reuter, M.S.; Roshandel, D.; Whitney, J.; et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 2022, 185, 4409–4427.e18. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; Wamsley, B.; Klei, L.; Wang, L.; Hao, S.P.; et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 2022, 54, 1320–1331. [Google Scholar] [CrossRef]
- Zhou, X.; Feliciano, P.; Shu, C.; Wang, T.; Astrovskaya, I.; Hall, J.B.; Obiajulu, J.U.; Wright, J.R.; Murali, S.C.; Xu, S.X.; et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat. Genet. 2022, 54, 1305–1319. [Google Scholar] [CrossRef]
- Barger, B.D.; Campbell, J.M.; McDonough, J.D. Prevalence and onset of regression within autism spectrum disorders: A meta-analytic review. J. Autism Dev. Disord. 2013, 43, 817–828. [Google Scholar] [CrossRef]
- Ozonoff, S.; Iosif, A.M. Changing conceptualizations of regression: What prospective studies reveal about the onset of autism spectrum disorder. Neurosci. Biobehav. Rev. 2019, 100, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Lainhart, J.E.; Ozonoff, S.; Coon, H.; Krasny, L.; Dinh, E.; Nice, J.; McMahon, W. Autism, regression, and the broader autism phenotype. Am. J. Med. Genet. 2002, 113, 231–237. [Google Scholar] [CrossRef]
- Barger, B.D.; Campbell, J.; Simmons, C. The relationship between regression in autism spectrum disorder, epilepsy, and atypical epileptiform EEGs: A meta-analytic review. J. Intellect. Dev. Disabil. 2017, 42, 45–60. [Google Scholar] [CrossRef]
- Nordahl, C.W.; Lange, N.; Li, D.D.; Barnett, L.A.; Lee, A.; Buonocore, M.H.; Simon, T.J.; Rogers, S.; Ozonoff, S.; Amaral, D.G. Brain enlargement is associated with regression in preschool-age boys with autism spectrum disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 20195–20200. [Google Scholar] [CrossRef]
- Valvo, G.; Baldini, S.; Retico, A.; Rossi, G.; Tancredi, R.; Ferrari, A.R.; Calderoni, S.; Apicella, F.; Muratori, F.; Santorelli, F.M.; et al. Temporal lobe connects regression and macrocephaly to autism spectrum disorders. Eur. Child. Adolesc. Psychiatry 2016, 25, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Scott, O.; Shi, D.; Andriashek, D.; Clark, B.; Goez, H.R. Clinical clues for autoimmunity and neuroinflammation in patients with autistic regression. Dev. Med. Child. Neurol. 2017, 59, 947–951. [Google Scholar] [CrossRef]
- Prosperi, M.; Guiducci, L.; Peroni, D.G.; Narducci, C.; Gaggini, M.; Calderoni, S.; Tancredi, R.; Morales, M.A.; Gastaldelli, A.; Muratori, F.; et al. Inflammatory Biomarkers are Correlated with Some Forms of Regressive Autism Spectrum Disorders. Brain Sci. 2019, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.M.; Soke, G.N.; Sabourin, K.R.; Croen, L.A.; Daniels, J.L.; Fallin, M.D.; Kral, T.V.E.; Lee, L.C.; Newschaffer, C.J.; Pinto-Martin, J.A.; et al. Gastrointestinal Symptoms in 2- to 5-Year-Old Children in the Study to Explore Early Development. J. Autism Dev. Disord. 2021, 51, 3806–3817. [Google Scholar] [CrossRef] [PubMed]
- Goin-Kochel, R.P.; Trinh, S.; Barber, S.; Bernier, R. Gene Disrupting Mutations Associated with Regression in Autism Spectrum Disorder. J. Autism Dev. Disord. 2017, 47, 3600–3607. [Google Scholar] [CrossRef]
- Gupta, A.R.; Westphal, A.; Yang, D.Y.J.; Sullivan, C.A.W.; Eilbott, J.; Zaidi, S.; Voos, A.; Vander Wyk, B.C.; Ventola, P.; Waqar, Z.; et al. Neurogenetic analysis of childhood disintegrative disorder. Mol. Autism 2017, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chun, C.A.; Zavadenko, N.N.; Pechatnikova, N.L.; Naumova, O.Y.; Doddapaneni, H.V.; Hu, J.; Muzny, D.M.; Schaaf, C.P.; Grigorenko, E.L. Next Generation Sequencing of 134 Children with Autism Spectrum Disorder and Regression. Genes 2020, 11, 853. [Google Scholar] [CrossRef]
- Singh, K.; Singh, I.N.; Diggins, E.; Connors, S.L.; Karim, M.A.; Lee, D.; Zimmerman, A.W.; Frye, R.E. Developmental regression and mitochondrial function in children with autism. Ann. Clin. Transl. Neurol. 2020, 7, 683–694. [Google Scholar] [CrossRef]
- Veeraragavan, S.; Wan, Y.W.; Connolly, D.R.; Hamilton, S.M.; Ward, C.S.; Soriano, S.; Pitcher, M.R.; McGraw, C.M.; Huang, S.G.; Green, J.R.; et al. Loss of MeCP2 in the rat models regression, impaired sociability and transcriptional deficits of Rett syndrome. Hum. Mol. Genet. 2016, 25, 3284–3302. [Google Scholar] [CrossRef]
- Xi, C.Y.; Ma, H.W.; Lu, Y.; Zhao, Y.J.; Hua, T.Y.; Zhao, Y.; Ji, Y.H. MeCP2 gene mutation analysis in autistic boys with developmental regression. Psychiatr. Genet. 2007, 17, 113–116. [Google Scholar] [CrossRef]
- Tammimies, K. Genetic mechanisms of regression in autism spectrum disorder. Neurosci. Biobehav. Rev. 2019, 102, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, B.; Fang, D.; Zeng, H.; Chen, X.; Peng, G.; Cheng, Q.; Liang, G. The impact of SNAP-25 on brain functional connectivity density and working memory in ADHD. Biol. Psychol. 2018, 138, 35–40. [Google Scholar] [CrossRef]
- Liu, Y.S.; Dai, X.; Wu, W.; Yuan, F.F.; Gu, X.; Chen, J.G.; Zhu, L.Q.; Wu, J. The Association of SNAP25 Gene Polymorphisms in Attention Deficit/Hyperactivity Disorder: A Systematic Review and Meta-Analysis. Mol. Neurobiol. 2017, 54, 2189–2200. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Lau, E.W.; Liang, A.; de Dios, C.; Suchting, R.; Östlundh, L.; Masdeu, J.C.; Fujita, M.; Sanches, M.; Soares, J.C.; et al. Alterations in brain synaptic proteins and mRNAs in mood disorders: A systematic review and meta-analysis of postmortem brain studies. Mol. Psychiatry 2022, 27, 1362–1372. [Google Scholar] [CrossRef]
- Guerini, F.R.; Bolognesi, E.; Chiappedi, M.; Manca, S.; Ghezzo, A.; Agliardi, C.; Sotgiu, S.; Usai, S.; Matteoli, M.; Clerici, M. SNAP-25 single nucleotide polymorphisms are associated with hyperactivity in autism spectrum disorders. Pharmacol. Res. 2011, 64, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Braida, D.; Guerini, F.R.; Ponzoni, L.; Corradini, I.; De Astis, S.; Pattini, L.; Bolognesi, E.; Benfante, R.; Fornasari, D.; Chiappedi, M.; et al. Association between SNAP-25 gene polymorphisms and cognition in autism: Functional consequences and potential therapeutic strategies. Transl. Psychiatry 2015, 5, e500. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, J.; Zhang, T.; Lu, T.; Wang, H.; Jia, M.; Liu, J.; Xiong, J.; Zhang, D.; Wang, L. Family-based association study identifies SNAP-25 as a susceptibility gene for autism in the Han Chinese population. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 105, 109985. [Google Scholar] [CrossRef]
- Safari, M.R.; Omrani, M.D.; Noroozi, R.; Sayad, A.; Sarrafzadeh, S.; Komaki, A.; Manjili, F.A.; Mazdeh, M.; Ghaleiha, A.; Taheri, M. Synaptosome-Associated Protein 25 (SNAP25) Gene Association Analysis Revealed Risk Variants for ASD, in Iranian Population. J. Mol. Neurosci. 2017, 61, 305–311. [Google Scholar] [CrossRef]
- Guerini, F.R.; Farina, E.; Costa, A.S.; Baglio, F.; Saibene, F.L.; Margaritella, N.; Calabrese, E.; Zanzottera, M.; Bolognesi, E.; Nemni, R.; et al. ApoE and SNAP-25 Polymorphisms Predict the Outcome of Multidimensional Stimulation Therapy Rehabilitation in Alzheimer’s Disease. Neurorehabilit. Neural Repair 2016, 30, 883–893. [Google Scholar] [CrossRef]
- Agostini, S.; Mancuso, R.; Costa, A.S.; Guerini, F.R.; Trecate, F.; Miglioli, R.; Menna, E.; Arosio, B.; Clerici, M.; SA. M. B. A. Project. Sarcopenia associates with SNAP-25 SNPs and a miRNAs profile which is modulated by structured rehabilitation treatment. J. Transl. Med. 2021, 19, 315. [Google Scholar] [CrossRef]
- Yalın, O.Ö.; Gökdoğan Edgünlü, T.; Karakaş Çelik, S.; Emre, U.; Güneş, T.; Erdal, Y.; Eroğlu Ünal, A. Novel SNARE Complex Polymorphisms Associated with Multiple Sclerosis: Signs of Synaptopathy in Multiple Sclerosis. Balkan Med. J. 2019, 36, 174–178. [Google Scholar] [CrossRef]
- Palfreyman, M.T.; West, S.E.; Jorgensen, E.M. SNARE Proteins in Synaptic Vesicle Fusion. Adv. Neurobiol. 2023, 33, 63–118. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Corradini, I.; Fossati, G.; Tomasoni, R.; Menna, E.; Matteoli, M. SNAP-25, a Known Presynaptic Protein with Emerging Postsynaptic Functions. Front. Synaptic Neurosci. 2016, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Tomasoni, R.; Repetto, D.; Morini, R.; Elia, C.; Gardoni, F.; Di Luca, M.; Turco, E.; Defilippi, P.; Matteoli, M. SNAP-25 regulates spine formation through postsynaptic binding to p140Cap. Nat. Commun. 2013, 4, 2136. [Google Scholar] [CrossRef]
- Gopaul, K.R.; Irfan, M.; Miry, O.; Vose, L.R.; Moghadam, A.; Subah, G.; Hökfelt, T.; Bark, C.; Stanton, P.K. Developmental Time Course of SNAP-25 Isoforms Regulate Hippocampal Long-Term Synaptic Plasticity and Hippocampus-Dependent Learning. Int. J. Mol. Sci. 2020, 21, 1448. [Google Scholar] [CrossRef] [PubMed]
- Fossati, G.; Morini, R.; Corradini, I.; Antonucci, F.; Trepte, P.; Edry, E.; Sharma, V.; Papale, A.; Pozzi, D.; Defilippi, P.; et al. Reduced SNAP-25 increases PSD-95 mobility and impairs spine morphogenesis. Cell Death Differ. 2015, 22, 1425–1436. [Google Scholar] [CrossRef]
- Hussain, S.; Ringsevjen, H.; Schupp, M.; Hvalby, Ø.; Sørensen, J.B.; Jensen, V.; Davanger, S. A possible postsynaptic role for SNAP-25 in hippocampal synapses. Brain Struct. Funct. 2019, 224, 521–532. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Association: Washington, DC, USA, 1994. [Google Scholar]
- Barbeau, W.E. Neonatal and regressive forms of autism: Diseases with similar symptoms but a different etiology. Med. Hypotheses 2017, 109, 46–52. [Google Scholar] [CrossRef]
- Kovacs-Nagy, R.; Sarkozy, P.; Hu, J.; Guttman, A.; Sasvari-Szekely, M.; Ronai, Z. Haplotyping of putative microRNA-binding sites in the SNAP-25 gene. Electrophoresis 2011, 32, 2013–2020. [Google Scholar] [CrossRef]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Wei, C.; Thatcher, E.J.; Olena, A.F.; Cha, D.J.; Perdigoto, A.L.; Marshall, A.F.; Carter, B.D.; Broadie, K.; Patton, J.G. miR-153 regulates SNAP-25, synaptic transmission, and neuronal development. PLoS ONE 2013, 8, e57080. [Google Scholar] [CrossRef] [PubMed]
- Machitani, M.; Sakurai, F.; Wakabayashi, K.; Nakatani, K.; Tachibana, M.; Mizuguchi, H. MicroRNA miR-27 Inhibits Adenovirus Infection by Suppressing the Expression of SNAP-25 and TXN2. J. Virol. 2017, 91, e00159-17. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Bolognesi, E.; Mancuso, R.; Marventano, I.; Citterio, L.A.; Guerini, F.R.; Clerici, M. miR-23a-3p and miR-181a-5p modulate SNAP-25 expression. PLoS ONE 2023, 18, e0279961. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.; Munson, J.; Webb, S.J.; Nalty, T.; Abbott, R.; Toth, K. Rate of head growth decelerates and symptoms worsen in the second year of life in autism. Biol. Psychiatry 2007, 61, 458–464. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Enstrom, A.; Onore, C.; Tarver, A.; Hertz-Picciotto, I.; Hansen, R.; Croen, L.; Van de Water, J.; Ashwood, P. Peripheral Blood Leukocyte Production of BDNF following Mitogen Stimulation in Early Onset and Regressive Autism. Am. J. Biochem. Biotechnol. 2008, 4, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, P.; Li, Q.; Peng, B.; Cun, Y.; Dai, Y.; Wei, H.; Liu, X.; Yu, Y.; Jiang, Z.; et al. Regressive Autism Spectrum Disorder: High Levels of Total Secreted Amyloid Precursor Protein and Secreted Amyloid Precursor Protein-α in Plasma. Front. Psychiatry 2022, 13, 809543. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143, Erratum in Neuron 2014, 83, 1482. [Google Scholar] [CrossRef]
- Vogel-Ciernia, A.; Wood, M.A. Neuron-specific chromatin remodeling: A missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology 2014, 80, 18–27. [Google Scholar] [CrossRef]
- Ebert, D.H.; Greenberg, M.E. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013, 493, 327–337. [Google Scholar] [CrossRef]
- Tamouza, R.; Fernell, E.; Eriksson, M.A.; Anderlid, B.M.; Manier, C.; Mariaselvam, C.M.; Boukouaci, W.; Leboyer, M.; Gillberg, C. HLA Polymorphism in Regressive and Non-Regressive Autism: A Preliminary Study. Autism Res. 2020, 13, 182–186. [Google Scholar] [CrossRef]
- Frye, R.E.; Cakir, J.; McCarty, P.J.; Rose, S.; Delhey, L.M.; Palmer, R.F.; Austin, C.; Curtin, P.; Yitshak-Sade, M.; Arora, M. Pollution and Maximum Temperature Are Associated with Neurodevelopmental Regressive Events in Autism Spectrum Disorder. J. Pers. Med. 2022, 12, 1809. [Google Scholar] [CrossRef] [PubMed]
- Martin-Borreguero, P.; Gómez-Fernández, A.R.; De La Torre-Aguilar, M.J.; Gil-Campos, M.; Flores-Rojas, K.; Perez-Navero, J.L. Children with Autism Spectrum Disorder and Neurodevelopmental Regression Present a Severe Pattern after a Follow-Up at 24 Months. Front. Psychiatry 2021, 12, 644324. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yang, F.; Yang, T.; Chen, J.; Dai, Y.; Jia, F.; Wu, L.; Hao, Y.; Li, L.; Zhang, J.; et al. A Multi-Center Study on the Relationship Between Developmental Regression and Disease Severity in Children with Autism Spectrum Disorders. Front. Psychiatry 2022, 13, 796554. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.; Gillberg, C.; Landberg, S.; Kantzer, A.K.; Miniscalco, C.; Barnevik Olsson, M.; Eriksson, M.A.; Fernell, E. Autism with and Without Regression: A Two-Year Prospective Longitudinal Study in Two Population-Derived Swedish Cohorts. J. Autism Dev. Disord. 2019, 49, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Damiani, S.; Leali, P.; Nosari, G.; Caviglia, M.; Puci, M.V.; Monti, M.C.; Brondino, N.; Politi, P. Association of Autism Onset, Epilepsy, and Behavior in a Community of Adults with Autism and Severe Intellectual Disability. Brain Sci. 2020, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Fernandez, A.; de la Torre-Aguilar, M.J.; Gil-Campos, M.; Flores-Rojas, K.; Cruz-Rico, M.D.; Martin-Borreguero, P.; Perez-Navero, J.L. Children with Autism Spectrum Disorder With Regression Exhibit a Different Profile in Plasma Cytokines and Adhesion Molecules Compared to Children Without Such Regression. Front. Pediatr. 2018, 6, 264. [Google Scholar] [CrossRef]
- Heuer, L.S.; Croen, L.A.; Jones, K.L.; Yoshida, C.K.; Hansen, R.L.; Yolken, R.; Zerbo, O.; DeLorenze, G.; Kharrazi, M.; Ashwood, P.; et al. An Exploratory Examination of Neonatal Cytokines and Chemokines as Predictors of Autism Risk: The Early Markers for Autism Study. Biol. Psychiatry 2019, 86, 255–264. [Google Scholar] [CrossRef]
- Lord, C.; Rutter, M.; DiLavore, P.C.; Risi, S.; Gotham, K.; Bishop, S. Autism Diagnostic Observation Schedule–Second Edition (ADOS-2); Los Angeles: Western Psychological Services Italian Edition; Colombi, C., Tancredi, R., Persico, A., Faggioli, A., Eds.; Hogrefe Editor: Firenze, Italy, 2012. [Google Scholar]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef]
- Schopler, E.; Reichler, R.J.; Renner, B.R. The Childhood Autism Rating Scale (CARS), for Diagnostic Screening and Classification in Autism; Irvington: New York, NY, USA, 1986. [Google Scholar]
- Roid, G.H.; Miller, L.J.; Pomplun, M.; Koch, C. Leiter International Performance Scale (Leiter-3), 3rd ed.; Los Angeles: Western Psychological Services Italian Edition; Cornoldi, C., Giofrè, D., Belacchi, C., Eds.; Giunti Organizzazioni Speciali: Firenze, Italy, 2013. [Google Scholar]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; Harcourt Assessment: San Antonio, TX, USA, 2003. [Google Scholar]
- Raven, J.C.; Court, J.H.; Raven, J. Manual for Raven’s Progressive Matrices and Vocabulary Scales; Section 2: Coloured Progressive Matrices. London: H. K. Lewis Italian Edition; Belacchi, C., Scalisi, T.G., Cannoni, E., Cornoldi, C., Eds.; Giunti Organizzazioni Speciali: Firenze, Italy, 1984. [Google Scholar]
- Shaffer, D.; Gould, M.S.; Brasic, J.; Ambrosini, P.; Fisher, P.; Bird, H.; Aluwahlia, S. A children’s global assessment scale (CGAS). Arch. Gen. Psychiatry 1983, 40, 1228–1231. [Google Scholar] [CrossRef]
- Shi, Y.Y.; He, L. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. 2005, 15, 97–98. [Google Scholar] [CrossRef]
- Panisi, C.; Guerini, F.R.; Abruzzo, P.M.; Balzola, F.; Biava, P.M.; Bolotta, A.; Brunero, M.; Burgio, E.; Chiara, A.; Clerici, M.; et al. Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift. J. Pers. Med. 2021, 11, 70. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Allele Frequency | Continental ASD N (%) | Sardinian ASD N (%) | p * | Total ASD N (%) | HC N (%) | p ° |
|---|---|---|---|---|---|---|
| Rs363050 | ||||||
| A | 79 (61.0) | 75 (61.0) | 154 (61.0) | 244 (60.0) | ||
| G | 51 (39.0) | 49 (39.0) | 0.9 | 100 (39.0) | 166 (40.0) | 0.8 |
| Rs363039 | ||||||
| G | 97 (75.0) | 81 (65.0) | 178 (70.0) | 257 (63.0) | ||
| A | 33 (25.0) | 43 (35.0) | 0.1 | 76 (30.0) | 153 (37.0) | 0.05 |
| Rs363043 | ||||||
| C | 94 (72.0) | 84 (68.0) | 178 (70.0) | 298 (73.0) | ||
| T | 36 (28.0) | 40 (32.0) | 0.4 | 76 (30.0) | 112 (27.0) | 0.4 |
| Rs1051312 | ||||||
| T | 97 (75.0) | 103 (83.0) | 200 (79.0) | 301 (73.0) | ||
| C | 33 (25.0) | 21 (17.0) | 0.1 | 54 (21.0) | 109 (27.0) | 0.1 |
| Total | 130 | 124 | 254 | 410 | ||
| Genotype frequency | ||||||
| Rs363050 | ||||||
| AA | 24 (37.0) | 26 (42.0) | 50 (39.0) | 70 (34.0) | ||
| AG | 31(48.0) | 23(37.0) | 54(43.0) | 104 (43.0) | ||
| GG | 10 (15.0) | 13 (21.0) | 0.4 | 23 (18.0) | 31 (19.0) | 0.3 |
| Rs363039 | ||||||
| GG | 35 (54.0) | 28 (45.0) | 63 (50.0) | 78 (38.0) | ||
| GA | 27 (42.0) | 25 (40.0) | 52 (41.0) | 101 (49.0) | ||
| AA | 3 (5.0) | 9 (14.0) | 0.1 | 12 (9.0) | 26 (13.0) | 0.1 |
| Rs363043 | ||||||
| CC | 35 (54.0) | 32 (54.0) | 67 (53.0) | 111 (54.0) | ||
| CT | 24 (37.0) | 20 (32.0) | 44 (35.0) | 76 (37.0) | ||
| TT | 6 (9.0) | 10 (16.0) | 0.5 | 16 (13.0) | 18 (9.0) | 0.5 |
| Rs1051312 | ||||||
| TT | 36 (55.0) | 42 (68.0) | 78 (61.0) | 108 (53.0) | ||
| TC | 25 (38.0) | 19 (31.0) | 44 (35.0) | 85 (41.0) | ||
| CC | 4 (6.0) | 1 (2.0) | 0.2 | 5 (4.0) | 12 (6.0) | 0.2 |
| Total | 127 | 127 | 127 | 205 |
| Allele Frequency | ASD Early Onset N (%) | ASD Regressive Onset N (%) | p | HC N (%) | p |
|---|---|---|---|---|---|
| Rs363050 | |||||
| A | 86 (62.0) * | 68 (59.0)° | 244 (60.0) | ||
| G | 52 (38.0) | 48 (41.0) | 0.5 | 166 (40.0) | * 0.5. ° 0.8 |
| Rs363039 | |||||
| G | 103 (75.0) * | 75 °(65.0) | 257 (63.0) | ||
| A | 35 (25.0) | 41 (35.0) | 0.08 | 153 (37.0) | * 0.009, ° 0.7 |
| Rs363043 | |||||
| C | 91 (66.0) * | 87 (75.0)° | 298 (73.0) | ||
| T | 47 (34.0) | 29 (25.0) | 0.12 | 112 (27.0) | * 0.1. ° 0.6 |
| Rs1051312 | |||||
| T | 99 (72.0) * | 101 (87.0)° | 301 (73.0) | ||
| C | 39 (28.0) | 15 (13.0) | 0.003 | 109 (27.0) | * 0.7, ° 0.002 |
| Total | 148 | 116 | |||
| Genotype frequency | |||||
| Rs363050 | |||||
| AA | 30 (43.0) * | 20(34.0)° | 70 (34.0) | ||
| AG | 26(38.0) | 28(48.0) | 104 (43.0) | ||
| GG | 13 (19.0) | 10 (17.0) | 0.47 | 31 (19.0) | * 0.2. ° 0.5 |
| Rs363039 | |||||
| GG | 40 (58.0) * | 23° (40.0) | 78 (38.0) | ||
| GA | 23 (33.0) | 29 (50.0) | 101 (49.0) | ||
| AA | 6 (9.0) | 6 (10.0) | 0.11 | 26 (13.0) | * 0.015, ° 0.9 |
| Rs363043 | |||||
| CC | 32 (46.0) * | 35 (60.0)° | 111 (54.0) | ||
| CT | 27 (39.0) | 17 (29.0) | 76 (37.0) | ||
| TT | 10 (14.0) | 6 (10.0) | 0.29 | 18 (9.0) | * 0.3. ° 0.5 |
| Rs1051312 | |||||
| TT | 34 * (49.0) | 44 (76.0)° | 108 (53.0) | ||
| TC | 31 (45.0) | 13 (22.0) | 85 (41.0) | ||
| CC | 4 (6.0) | 1 (2.0) | 0.008 | 12 (6.0) | * 0.9, ° 0.006 |
| Total | 69 | 58 | 205 |
| Genotype Frequency | ASD Early Onset N (%) | ASD Regressive Onset N (%) | p | OR (95%IC) | HC N (%) | p | OR (95%IC) |
|---|---|---|---|---|---|---|---|
| Rs363039 | |||||||
| GG | 40 (58.0) * | 23 (40.0) | 78 (38.0) | ||||
| GA/AA | 29 (42.0) | 35 (60.0) | 0.04 | 2.09 (1.02–4.3) | 127 (62.0) | * 0.004 | 2.2 (1.28–3.9) |
| rs1051312 | |||||||
| TT | 34 (49.0) | 44 (76.0)° | 108 (53.0) | ||||
| TC/CC | 35 (51.0) | 14 (24.0) | 0.002 | 3.2 (1.5–6.9) | 97 (47.0) | ° 0.001 | 2.8 (1.47–5.6) |
| N | 69 | 58 | 205 |
| ASD Early Onset | ASD Regressive Onset | HC | |||||||
|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | p | OR (95%IC) | N | % | p | |
| 4 SNPs HAPLOTYPE | |||||||||
| A-G-T-C | 24 * | 17.5 | 5 | 4.2 | 0.0008 | 4.7 (1.8–14.1) | 30 | 7.2 | * 0.0006 |
| G-A-C-T | 28 | 20.3 | 36° | 31.0 | 0.05 | 1.7 (1.9–3.1) | 101 | 24.7 | ° 0.17 |
| 2 SNPs HAPLOTYPE | |||||||||
| G-C | 33 * | 23.8 | 12 | 10.5 | 0.004 | 2.7 (1.3–5.7) | 72 | 17.5 | * 0.1 |
| A-T | 29 | 20.9 | 39° | 33.3 | 0.02 | 1.9 (1.1–3.3) | 116 | 28.3 | ° 0.3 |
| Total N | 138 | 116 | 410 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolognesi, E.; Guerini, F.R.; Carta, A.; Chiappedi, M.; Sotgiu, S.; Mensi, M.M.; Agliardi, C.; Zanzottera, M.; Clerici, M. The Role of SNAP-25 in Autism Spectrum Disorders Onset Patterns. Int. J. Mol. Sci. 2023, 24, 14042. https://doi.org/10.3390/ijms241814042
Bolognesi E, Guerini FR, Carta A, Chiappedi M, Sotgiu S, Mensi MM, Agliardi C, Zanzottera M, Clerici M. The Role of SNAP-25 in Autism Spectrum Disorders Onset Patterns. International Journal of Molecular Sciences. 2023; 24(18):14042. https://doi.org/10.3390/ijms241814042
Chicago/Turabian StyleBolognesi, Elisabetta, Franca Rosa Guerini, Alessandra Carta, Matteo Chiappedi, Stefano Sotgiu, Martina Maria Mensi, Cristina Agliardi, Milena Zanzottera, and Mario Clerici. 2023. "The Role of SNAP-25 in Autism Spectrum Disorders Onset Patterns" International Journal of Molecular Sciences 24, no. 18: 14042. https://doi.org/10.3390/ijms241814042
APA StyleBolognesi, E., Guerini, F. R., Carta, A., Chiappedi, M., Sotgiu, S., Mensi, M. M., Agliardi, C., Zanzottera, M., & Clerici, M. (2023). The Role of SNAP-25 in Autism Spectrum Disorders Onset Patterns. International Journal of Molecular Sciences, 24(18), 14042. https://doi.org/10.3390/ijms241814042