
The Exon 3-Deleted Growth Hormone Receptor (d3GHR) Polymorphism—A Favorable Backdoor Mechanism for the GHR Function



Abstract
1. Introduction
2. GHR Function and Structure
3. GHR Isoforms
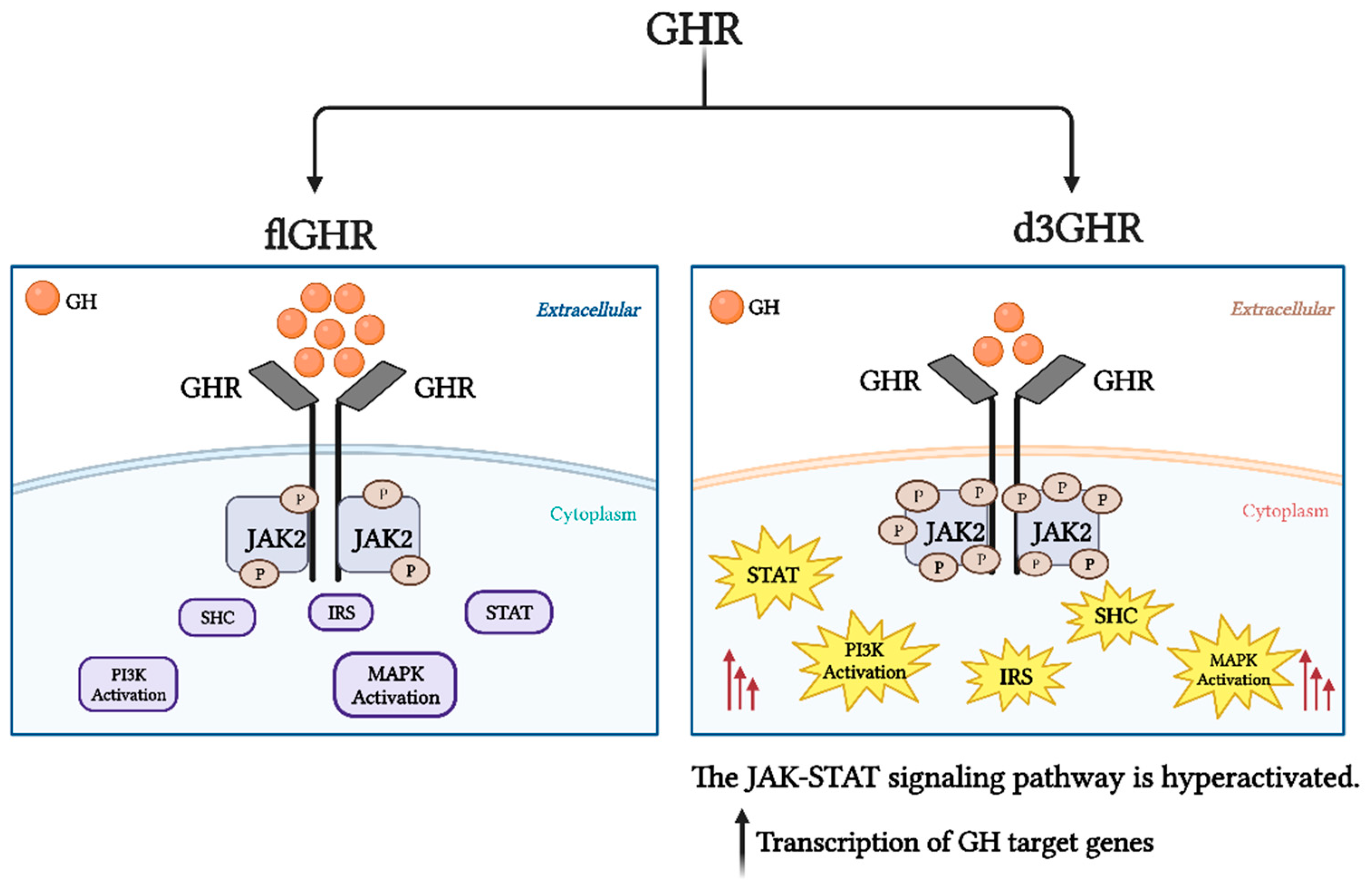
4. Deletion of Exon 3 in GHR (d3GHR)
5. flGHR and d3GHR Genotyping
6. Genetic Editing of d3GHR
7. Effects of the flGHR and d3GHR Isoforms in Healthy People
8. Effects of the flGHR and d3GHR Isoforms on Various Growth Disorders
8.1. Growth Hormone Deficiency (GHD)
8.2. Small for Gestational Age (SGA)
8.3. Turner Syndrome
8.4. Prader-Willi Syndrome (PWS)
8.5. Acromegaly

{kind=link}
{kind=link}
{kind=link}
| Authors | No. of Cases | fl/fl % | fl/d3 % | d3/d3 % | Effects |
|---|---|---|---|---|---|
| Schmid, 2007 [113] | 44 | 50 | 50 | d3GHR carriers had lower GH concentrations. | |
| Kamenicky, 2009 [123] | 105 | 51 | 30 | 19 | No differences in the baseline levels of GH and IGF-1 concentrations. |
| Pontes, 2020 [121] | 88 | 40 | 60 | No correlation between the presence of d3GHR and increasing frequency of VF, worse BMD, or bone microarchitecture. | |
| Cinar, 2015 [124] | 118 | 60.2 | 33.9 | 5.9 | No differences in the prevalence of hypertension, type 2 diabetes mellitus, or coronary artery disease. |
| Montefusco, 2010 [55] | 76 | 55.3 | 35.5 | 9.2 | d3GHR carriers had a lower BMI index. |
| Mercado, 2008 [125] | 148 | 45 | 32 | 22 | d3GHR carriers had higher post-treatment IGF-I concentrations and diabetes was more prevalent in them. |
| J E Wassenaar, 2009 [126] | 86 | 59 | 29 | 7 | d3GHR carriers had an increased prevalence of osteoarthritis, dolichocolon, and adenomatous colonic polyps. |
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| AGA | Appropriate for gestational age |
| ALS | Acid labile subunit |
| bp | Base pairs |
| CR | Caloric restriction |
| d3GHR | Exon 3-deficient growth hormone receptor |
| ERK1/2 | Extracellular signal-regulated protein kinases 1 and 2 |
| fl | Full length |
| flGHR | Full-length growth hormone receptor |
| GH | Growth hormone |
| GHD | Growth hormone deficiency |
| GHR | Growth hormone receptor |
| GHRD | Growth hormone receptor deficiency |
| GHSF | GH-stimulated binding activity |
| IGF-1 | Insulin-like growth factor 1 |
| IGFBP-3 | IGF binding protein 3 |
| IGHD | Isolated GH deficiency |
| IRS | Insulin receptor substrates |
| JAK2 | Janus kinase 2 |
| JNK | c-Jun N-terminal kinases |
| LGA | Large for gestational age |
| MAPK | Mitogen-activated protein kinase |
| P | Phosphate |
| PCR | Polymerase chain reaction |
| PI3′K | phosphatidylinositol 3-kinase |
| PLC-γ | Phospholipase C Gamma |
| PWS | Prader–Willi syndrome |
| RAP | Ras proximate |
| RAS | Rat sarcoma virus |
| rhGH | Recombinant human growth hormone |
| SDS | Standard deviation score |
| SGA | Small for gestational age |
| SNP | Single-nucleotide polymorphism |
| STAT | Signal transducers and activators of transcription |
| tagSNP | Tagging single nucleotide polymorphism |
| TS | Turner syndrome |
| VF | Vertebral fractures |
| WT | Wild type |
References
- Ashpole, N.M.; Sanders, J.E.; Hodges, E.L.; Yan, H.; Sonntag, W.E. Growth hormone, insulin-like growth factor-1 and the aging brain. Exp. Gerontol. 2015, 68, 76–81. [Google Scholar] [CrossRef]
- Dos Santos, C.; Essioux, L.; Teinturier, C.; Tauber, M.; Goffin, V.; Bougnères, P. A common polymorphism of the growth hormone receptor is associated with increased responsiveness to growth hormone. Nat. Genet. 2004, 36, 720–724. [Google Scholar] [CrossRef]
- Palizban, A.; Radmansorry, M.; Bozorgzad, M. Exon 3-deleted and full-length growth hormone receptor polymorphism frequencies in an Iranian population. Res. Pharm. Sci. 2015, 9, 489–494. [Google Scholar]
- Filopanti, M.; Giavoli, C.; Grottoli, S.; Bianchi, A.; De Marinis, L.; Ghigo, E.; Spada, A. The exon 3-deleted growth hormone receptor: Molecular and functional characterization and impact on GH/IGF-I axis in physiological and pathological conditions. J. Endocrinol. Investig. 2011, 34, 861–868. [Google Scholar] [CrossRef]
- Schreiner, F.; Stutte, S.; Bartmann, P.; Gohlke, B.; Woelfle, J. Association of the Growth Hormone Receptor d3-Variant and Catch-up Growth of Preterm Infants with Birth Weight of Less Than 1500 Grams. J. Clin. Endocrinol. Metab. 2007, 92, 4489–4493. [Google Scholar] [CrossRef][Green Version]
- Baş, F.; Darendeliler, F.; Aycan, Z.; Çetinkaya, E.; Berberoğlu, M.; Şıklar, Z.; Öcal, G.; Timirci, Ö.; Çetinkaya, S.; Darcan, S.; et al. The Exon 3-Deleted/Full-Length Growth Hormone Receptor Polymorphism and Response to Growth Hormone Therapy in Growth Hormone Deficiency and Turner Syndrome: A Multicenter Study. Horm. Res. Paediatr. 2012, 77, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, M.G.; Thankamony, A.; Roche, E.; Hoey, H.; Kirk, J.; Shaikh, G.; Ivarsson, S.A.; Söder, O.; Dunger, D.B.; Juul, A.; et al. The exon3-deleted growth hormone receptor gene polymorphism (d3-GHR) is associated with insulin and spontaneous growth in short SGA children (NESGAS). Growth Horm. IGF Res. 2017, 35, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Garrido, N.P.; Pujana, M.; Berger, M.; Ramírez, P.; Guercio, G.; Belgorosky, A.; Marino, R. Growth hormone receptor gene polymorphism. Spontaneous catch up growth in small for gestational age patients. Med. (B Aires) 2021, 81, 574–580. [Google Scholar]
- Binder, G.; Trebar, B.; Baur, F.; Schweizer, R.; Ranke, M.B. Homozygosity of the d3-growth hormone receptor polymorphism is associated with a high total effect of GH on growth and a low BMI in girls with Turner syndrome. Clin. Endocrinol. 2008, 68, 567–572. [Google Scholar] [CrossRef]
- Carrascosa, A.; Esteban, C.; Espadero, R.; Fernández-Cancio, M.; Andaluz, P.; Clemente, M.; Audí, L.; Wollmann, H.; Fryklund, L.; Parodi, L. The d3/fl-Growth Hormone (GH) Receptor Polymorphism Does Not Influence the Effect of GH Treatment (66 μg/kg per Day) or the Spontaneous Growth in Short Non-GH-Deficient Small-for-Gestational-Age Children: Results from a Two-Year Controlled Prospective Study in 170 Spanish Patients. J. Clin. Endocrinol. Metab. 2006, 91, 3281–3286. [Google Scholar] [CrossRef][Green Version]
- Bernabeu, I.; Alvarez-Escolá, C.; Quinteiro, C.; Lucas, T.; Puig-Domingo, M.; Luque-Ramírez, M.; de Miguel-Novoa, P.; Fernandez-Rodriguez, E.; Halperin, I.; Loidi, L.; et al. The Exon 3-Deleted Growth Hormone Receptor Is Associated with Better Response to Pegvisomant Therapy in Acromegaly. J. Clin. Endocrinol. Metab. 2010, 95, 222–229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blum, W.F.; Machinis, K.; Shavrikova, E.P.; Keller, A.; Stobbe, H.; Pfaeffle, R.W.; Amselem, S. The Growth Response to Growth Hormone (GH) Treatment in Children with Isolated GH Deficiency Is Independent of the Presence of the Exon 3-Minus Isoform of the GH Receptor. J. Clin. Endocrinol. Metab. 2006, 91, 4171–4174. [Google Scholar] [CrossRef] [PubMed]
- Ben-Avraham, D.; Govindaraju, D.R.; Budagov, T.; Fradin, D.; Durda, P.; Liu, B.; Ott, S.; Gutman, D.; Sharvit, L.; Kaplan, R.; et al. The GH receptor exon 3 deletion is a marker of male-specific exceptional longevity associated with increased GH sensitivity and taller stature. Sci. Adv. 2017, 3, e1602025. [Google Scholar] [CrossRef]
- Lu, M.; Flanagan, J.U.; Langley, R.J.; Hay, M.P.; Perry, J.K. Targeting growth hormone function: Strategies and therapeutic applications. Signal Transduct. Target. Ther. 2019, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. The Pituitary: Fourth Edition; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Pantel, J.; Machinis, K.; Sobrier, M.L.; Duquesnoy, P.; Goossens, M.; Amselem, S. Species-specific Alternative Splice Mimicry at the Growth Hormone Receptor Locus Revealed by the Lineage of Retroelements during Primate Evolution. J. Biol. Chem. 2000, 275, 18664–18669. [Google Scholar] [CrossRef]
- Menzies, B.R.; Shaw, G.; Fletcher, T.P.; Pask, A.J.; Renfree, M.B. Exon 3 of the growth hormone receptor (GH-R) is specific to eutherian mammals. Mol. Cell. Endocrinol. 2008, 296, 64–68. [Google Scholar] [CrossRef]
- Mohamadi, A.; Salvatori, R. Neuroendocrine Growth Disorders – Dwarfism, Gigantism. In Handbook of Neuroendocrinology; Fink, G., Pfaff, G.W., Levine, G.E., Eds.; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar] [CrossRef]
- Colosi, P.; Wong, K.; Leong, S.; Wood, W. Mutational analysis of the intracellular domain of the human growth hormone receptor. J. Biol. Chem. 1993, 268, 12617–12623. [Google Scholar] [CrossRef] [PubMed]
- Vander-Kuur, J.A.; Wang, X.; Zhang, L.; Campbell, G.S.; Allevato, G.; Billestrup, N.; Norstedt, G.; Carter-Su, C. Domains of the growth hormone receptor required for association and activation of JAK2 tyrosine kinase. J. Biol. Chem. 1994, 269, 21709–21717. [Google Scholar] [CrossRef]
- Murakami, M.; Narazaki, M.; Hibi, M.; Yawata, H.; Yasukawa, K.; Hamaguchi, M.; Taga, T.; Kishimoto, T. Critical cytoplasmic region of the interleukin 6 signal transducer gp130 is conserved in the cytokine receptor family. Proc. Natl. Acad. Sci. USA 1991, 88, 11349–11353. [Google Scholar] [CrossRef]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK–Receptor Interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Hackett, R.H.; Wang, Y.D.; Larner, A.C. Mapping of the Cytoplasmic Domain of the Human Growth Hormone Receptor Required for the Activation of Jak2 and Stat Proteins. J. Biol. Chem. 1995, 270, 21326–21330. [Google Scholar] [CrossRef]
- Postel-Vinay, M.C.; Kelly, P.A. Growth hormone receptor signalling. Baillieres Clin. Endocrinol. Metab. 1996, 10, 323–336. [Google Scholar] [CrossRef]
- Kurzer, J.H.; Carter-Su, C. Growth Hormone Induced Activation and Regulation of JAK2 and STAT Proteins. In Signal 594 Transducers and Activators of Transcription (STATs); Sehgal, P.B., Levy, D.E., Hirano, T., Eds.; Springer: Dordrecht, The Netherlands, 2003. [Google Scholar] [CrossRef]
- Carter-Su, C.; Schwartz, J.; Argetsinger, L.S. Growth hormone signaling pathways. Growth Horm. IGF Res. 2015, 28, 11–15. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Hahnel, P.S.; Thaler, S.; Antunes, E.; Huber, C.; Theobald, M.; Schuler, M. Targeting AKT Signaling Sensitizes Cancer to Cellular Immunotherapy. Cancer Res. 2008, 68, 3899–3906. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.H.; Kang, T.H.; Kim, J.H.; Pai, S.I.; Lin, K.Y.; Hung, C.F.; Wu, T.C.; Kim, T.W. Activation of Akt as a Mechanism for Tumor Immune Evasion. Mol. Ther. 2009, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Signaling via Shc family adapter proteins. Oncogene 2001, 20, 6322–6330. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.B.M.; Prigent, S.A. Insights into the Shc Family of Adaptor Proteins. J. Mol. Signal. 2017, 12, 2. [Google Scholar] [CrossRef]
- Dehkhoda, F.; Lee, C.M.M.; Medina, J.; Brooks, A.J. The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. 2018, 9, 35. [Google Scholar] [CrossRef]
- Soendergaard, C.; Young, J.A.; Kopchick, J.J. Growth Hormone Resistance—Special Focus on Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 1019. [Google Scholar] [CrossRef]
- Aguiar-Oliveira, M.H.; Bartke, A. Growth Hormone Deficiency: Health and Longevity. Endocr. Rev. 2018, 40, 575–601. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Growth hormone and aging. Rev. Endocr. Metab. Disord. 2020, 22, 71–80. [Google Scholar] [CrossRef]
- Coschigano, K.T.; Clemmons, D.; Bellush, L.L.; Kopchick, J.J. Assessment of Growth Parameters and Life Span of GHR/BP Gene-Disrupted Mice. Endocrinology 2000, 141, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Waters, M.J. The growth hormone receptor: Mechanism of activation and clinical implications. Nat. Rev. Endocrinol. 2010, 6, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef]
- Trejo, J.; Llorens-Martín, M.; Torres-Alemán, I. The effects of exercise on spatial learning and anxiety-like behavior are mediated by an IGF-I-dependent mechanism related to hippocampal neurogenesis. Mol. Cell. Neurosci. 2008, 37, 402–411. [Google Scholar] [CrossRef]
- Nashiro, K.; Guevara-Aguirre, J.; Braskie, M.N.; Hafzalla, G.W.; Velasco, R.; Balasubramanian, P.; Wei, M.; Thompson, P.M.; Mather, M.; Nelson, M.D.; et al. Brain Structure and Function Associated with Younger Adults in Growth Hormone Receptor-Deficient Humans. J. Neurosci. 2017, 37, 1696–1707. [Google Scholar] [CrossRef]
- Kranzler, J.H. Normal Intelligence with Severe Insulin-Like Growth Factor I Deficiency due to Growth Hormone Receptor Deficiency: A Controlled Study in a Genetically Homogeneous Population. J. Clin. Endocrinol. Metab. 1998, 83, 1953–1958. [Google Scholar] [CrossRef]
- Shevah, O.; Kornreich, L.; Galatzer, A.; Laron, Z. The Intellectual Capacity of Patients with Laron Syndrome (LS) Differs with Various Molecular Defects of the Growth Hormone Receptor Gene. Correlation with CNS Abnormalities. Horm. Metab. Res. 2005, 37, 757–760. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth Hormone Receptor Deficiency Is Associated with a Major Reduction in Pro-Aging Signaling, Cancer, and Diabetes in Humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef]
- Mormando, M.; Nasto, L.A.; Bianchi, A.; Mazziotti, G.; Giampietro, A.; Pola, E.; Pontecorvi, A.; Giustina, A.; De Marinis, L. GH receptor isoforms and skeletal fragility in acromegaly. Eur. J. Endocrinol. 2014, 171, 237–245. [Google Scholar] [CrossRef]
- Urbanek, M.; MacLeod, J.N.; Cooke, N.E.; Liebhaber, S.A. Expression of a human growth hormone (hGH) receptor isoform is predicted by tissue-specific alternative splicing of exon 3 of the hGH receptor gene transcript. Mol. Endocrinol. 1992, 6, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Mercado, M.; Dávila, N.; McLeod, J.F.; Baumann, G. Distribution of growth hormone receptor messenger ribonucleic acid containing and lacking exon 3 in human tissues. J. Clin. Endocrinol. Metab. 1994, 78, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Stallings-Mann, M.L.; Ludwiczak, R.L.; Klinger, K.W.; Rottman, F. Alternative splicing of exon 3 of the human growth hormone receptor is the result of an unusual genetic polymorphism. Proc. Natl. Acad. Sci. USA 1996, 93, 12394–12399. [Google Scholar] [CrossRef] [PubMed]
- Saenger, P.; Reiter, E. Genetic factors associated with small for gestational age birth and the use of human growth hormone in treating the disorder. Int. J. Pediatr. Endocrinol. 2012, 2012, 12. [Google Scholar] [CrossRef]
- Kelly, P.A.; Djiane, J.; Postel-Vinay, M.C.; Edery, M. The Prolactin/Growth Hormone Receptor Family. Endocr. Rev. 1991, 12, 235–251. [Google Scholar] [CrossRef]
- Urbanek, M.; Russell, J.; Cooke, N.; Liebhaber, S. Functional characterization of the alternatively spliced, placental human growth hormone receptor. J. Biol. Chem. 1993, 268, 19025–19032. [Google Scholar] [CrossRef]
- Sobrier, M.L.; Duquesnoy, P.; Duriez, B.; Amselem, S.; Goossens, M. Expression and binding properties of two isoforms of the human growth hormone receptor. FEBS Lett. 1993, 319, 16–20. [Google Scholar] [CrossRef]
- Seidel, B.; Glasow, A.; Schutt, M.; Kiess, W.; Wu, Z.; Strasburger, C.; Kratzsch, J. Association between the GH receptor/exon 3 genotype and the level of exon 3-positive GH-binding protein in human serum. Eur. J. Endocrinol. 2003, 148, 317–324. [Google Scholar] [CrossRef][Green Version]
- Bougnères, P. The Exon-3 Deletion of the Growth Hormone Receptor (GHR) Gene Still Has a Limited Impact in Clinical Endocrinology. J. Clin. Endocrinol. Metab. 2010, 95, 56–59. [Google Scholar] [CrossRef]
- Audí, L.; Esteban, C.; Carrascosa, A.; Espadero, R.; Pérez-Arroyo, A.; Arjona, R.; Clemente, M.; Wollmann, H.; Fryklund, L.; Parodi, L.A. Exon 3-Deleted/Full-Length Growth Hormone Receptor Polymorphism Genotype Frequencies in Spanish Short Small-for-Gestational-Age (SGA) Children and Adolescents (n = 247) and in an Adult Control Population (n = 289) Show Increasedfl/flin Short SGA. J. Clin. Endocrinol. Metab. 2006, 91, 5038–5043. [Google Scholar] [CrossRef] [PubMed]
- Montefusco, L.; Filopanti, M.; Ronchi, C.L.; Olgiati, L.; La-Porta, C.; Losa, M.; Epaminonda, P.; Coletti, F.; Beck-Peccoz, P.; Spada, A.; et al. d3-Growth hormone receptor polymorphism in acromegaly: Effects on metabolic phenotype. Clin. Endocrinol. 2010, 72, 661–667. [Google Scholar] [CrossRef]
- Tiulpakov, A.; Rubtsov, P.; Dedov, I.; Peterkova, V.; Bezlepkina, O.; Chrousos, G.P.; Hochberg, Z. A Novel C-Terminal Growth Hormone Receptor (GHR) Mutation Results in Impaired GHR-STAT5 But Normal STAT-3 Signaling. J. Clin. Endocrinol. Metab. 2005, 90, 542–547. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Butler, M.G.; Hossain, W.; Hassan, M.; Manzardo, A.M. Growth hormone receptor (GHR) gene polymorphism and scoliosis in Prader-Willi syndrome. Growth Horm. IGF Res. 2017, 39, 29–33. [Google Scholar] [CrossRef]
- Pelekanos, R.A.; Sardesai, V.S.; Nitert, M.D.; Callaway, L.K.; Fisk, N.M.; Jeffery, P.L. Rapid method for growth hormone receptor exon 3 delete (GHRd3) SNP genotyping from archival human placental samples. Endocrine 2015, 49, 643–652. [Google Scholar] [CrossRef]
- Dias, C.; Giordano, M.; Frechette, R.; Bellone, S.; Polychronakos, C.; Legault, L.; Deal, C.L.; Goodyer, C.G. Genetic variations at the human growth hormone receptor (GHR) gene locus are associated with idiopathic short stature. J. Cell. Mol. Med. 2017, 21, 2985–2999. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Resendez, S.; Pradhan, A.J.; Wu, F.; Lie, N.C.; Hall, N.J.; Zhu, Q.; Reinholdt, L.; Satta, Y.; Speidel, L.; et al. Sex-specific phenotypic effects and evolutionary history of an ancient polymorphic deletion of the human growth hormone receptor. Sci. Adv. 2021, 7, eabi4476. [Google Scholar] [CrossRef]
- Glad, C.A.; Johannsson, G.; Carlsson, L.M.; Svensson, P.A. Rapid and high throughput genotyping of the growth hormone receptor exon 3 deleted/full-length polymorphism using a tagSNP. Growth Horm. IGF Res. 2010, 20, 270–273. [Google Scholar] [CrossRef]
- Chiloiro, S.; Mirra, F.; Federico, D.; Giampietro, A.; Visconti, F.; Rossi, L.; Pontecorvi, A.; De Marinis, L.; Bianchi, A. The Role of Growth Hormone Receptor Isoforms and Their Effects in Bone Metabolism and Skeletal Fragility. Protein Pept. Lett. 2020, 27, 1260–1267. [Google Scholar] [CrossRef]
- Padidela, R.; Bryan, S.M.; Abu-Amero, S.; Hudson-Davies, R.E.; Achermann, J.C.; Moore, G.E.; Hindmarsh, P.C. The growth hormone receptor gene deleted for exon three ( GHRd3 ) polymorphism is associated with birth and placental weight. Clin. Endocrinol. 2011, 76, 236–240. [Google Scholar] [CrossRef]
- Sørensen, K.; Aksglaede, L.; Petersen, J.H.; Leffers, H.; Juul, A. The Exon 3 Deleted Growth Hormone Receptor Gene Is Associated with Small Birth Size and Early Pubertal Onset in Healthy Boys. J. Clin. Endocrinol. Metab. 2010, 95, 2819–2826. [Google Scholar] [CrossRef][Green Version]
- Jensen, R.B.; Boas, M.; Nielsen, J.E.; Maroun, L.L.; Jørgensen, A.; Larsen, T.; Main, K.M.; Juul, A. A common deletion in the growth hormone receptor gene (d3-GHR) in the offspring is related to maternal placental GH levels during pregnancy. Growth Horm. IGF Res. 2020, 55, 101360. [Google Scholar] [CrossRef]
- Martins, C.S.; Fernandes-Rosa, F.L.; Espineira, A.R.; de Souza, R.M.; de Castro, M.; Barbieri, M.A.; Bettiol, H.; Jorge, A.L.; Antonini, S.R. The growth hormone receptor exon 3 polymorphism is not associated with height or metabolic traits in healthy young adults. Growth Horm. IGF Res. 2014, 24, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, C.; Yang, S.; Yang, Y.; Chen, Y.; Zhao, X.; Zhu, W.; Zhao, Q.; Ni, C.; Huang, X.; et al. Gender Specificity and Local Socioeconomic Influence on Association of GHR fl/d3 Polymorphism With Growth and Metabolism in Children and Adolescents. Front. Pediatr. 2022, 10, 546080. [Google Scholar] [CrossRef] [PubMed]
- Kaabi, Y.A. Frequency of the exon 3-deleted/full-length growth hormone receptor polymorphism in Saudi Arabian population. Saudi Med. J. 2017, 38, 1090–1095. [Google Scholar] [CrossRef]
- Baş, F.; Keleşoğlu, F.; Timirci, Ö.; Eryılmaz, S.K.; Aydın, B.K.; Bozkurt, N.; Bundak, R.; Isbir, T.; Darendeliler, F. The Distribution of Exon 3-Deleted/Full-Length Growth Hormone Receptor Polymorphism in the Turkish Population. J. Clin. Res. Pediatr. Endocrinol. 2011, 3, 126–131. [Google Scholar] [CrossRef]
- Dauber, A.; Meng, Y.; Audi, L.; Vedantam, S.; Weaver, B.; Carrascosa, A.; Albertsson-Wikland, K.; Ranke, M.B.; Jorge, A.A.L.; Cara, J.; et al. A Genome-Wide Pharmacogenetic Study of Growth Hormone Responsiveness. J. Clin. Endocrinol. Metab. 2020, 105, 3203–3214. [Google Scholar] [CrossRef]
- Van der Klaauw, A.A.; Van der Straaten, T.; Baak-Pablo, R.; Biermasz, N.R.; Guchelaar, H.J.; Pereira, A.M.; Smit, J.W.A.; Romijn, J.A. Influence of the d3-Growth Hormone (GH) Receptor Isoform on Short-Term and Long-Term Treatment Response to GH Replacement in GH-Deficient Adults. J. Clin. Endocrinol. Metab. 2008, 93, 2828–2834. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jorge, A.A.L.; Marchisotti, F.G.; Montenegro, L.R.; Carvalho, L.R.; Mendonca, B.B.; Arnhold, I.J.P. Growth Hormone (GH) Pharmacogenetics: Influence of GH Receptor Exon 3 Retention or Deletion on First-Year Growth Response and Final Height in Patients with Severe GH Deficiency. J. Clin. Endocrinol. Metab. 2006, 91, 1076–1080. [Google Scholar] [CrossRef]
- Renehan, A.G.; Solomon, M.; Zwahlen, M.; Morjaria, R.; Whatmore, A.; Audí, L.; Binder, G.; Blum, W.; Bougnères, P.; Dos Santos, C.; et al. Growth Hormone Receptor Polymorphism and Growth Hormone Therapy Response in Children: A Bayesian Meta-Analysis. Am. J. Epidemiol. 2012, 175, 867–877. [Google Scholar] [CrossRef]
- Wassenaar, M.J.E.; Dekkers, O.M.; Pereira, A.M.; Wit, J.M.; Smit, J.W.; Biermasz, N.R.; Romijn, J.A. Impact of the Exon 3-Deleted Growth Hormone (GH) Receptor Polymorphism on Baseline Height and the Growth Response to Recombinant Human GH Therapy in GH-Deficient (GHD) and Non-GHD Children with Short Stature: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2009, 94, 3721–3730. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Leonardi, A.; Lanciotti, L.; Cofini, M.; Muzi, G.; Penta, L. Vitamin D and growth hormone in children: A review of the current scientific knowledge. J. Transl. Med. 2019, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sönksen, P. Replacement Therapy in Hypothalamo-Pituitary Insufficiency after Childhood: Management in the Adult. Horm. Res. 1990, 33, 45–51. [Google Scholar] [CrossRef]
- Yuen, K.C.J.; Biller, B.M.K.; Radovick, S.; Carmichael, J.D.; Jasim, S.; Pantalone, K.M.; Hoffman, A.R. American Association of Clinical Endocrinologists and American College of Endocrinology Guidelines for Management of Growth Hormone Deficiency in Adults and Patients Transitioning from Pediatric to Adult Care. Endocr. Pr. 2019, 25, 1191–1232. [Google Scholar] [CrossRef]
- Pilotta, A.; Mella, P.; Filisetti, M.; Felappi, B.; Prandi, E.; Parrinello, G.; Notarangelo, L.D.; Buzi, F. Common Polymorphisms of the Growth Hormone (GH) Receptor Do Not Correlate with the Growth Response to Exogenous Recombinant Human GH in GH-Deficient Children. J. Clin. Endocrinol. Metab. 2006, 91, 1178–1180. [Google Scholar] [CrossRef][Green Version]
- Raz, B.; Janner, M.; Petkovic, V.; Lochmatter, D.; Eblé, A.; Dattani, M.T.; Hindmarsh, P.C.; Fluck, C.E.; Mullis, P.E. Influence of Growth Hormone (GH) Receptor Deletion of Exon 3 and Full-Length Isoforms on GH Response and Final Height in Patients with Severe GH Deficiency. J. Clin. Endocrinol. Metab. 2008, 93, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Valsesia, A.; Chatelain, P.; Stevens, A.; Peterkova, V.A.; Belgorosky, A.; Maghnie, M.; Antoniazzi, F.; Koledova, E.; Wojcik, J.; Farmer, P.; et al. GH deficiency status combined with GH receptor polymorphism affects response to GH in children. Eur. J. Endocrinol. 2015, 173, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Adetunji, O.; Blair, J.; Javadpour, M.; Alfirevic, A.; Pirmohamed, M.; MacFarlane, I. Deletion of exon 3 in the growth hormone receptor gene in adults with growth hormone deficiency: Comparison of symptomatic and asymptomatic patients. Clin. Endocrinol. 2010, 72, 422–423. [Google Scholar] [CrossRef]
- Schlaudecker, E.P.; Munoz, F.M.; Bardají, A.; Boghossian, N.S.; Khalil, A.; Mousa, H.; Nesin, M.; Nisar, M.I.; Pool, V.; Spiegel, H.M.; et al. Small for gestational age: Case definition & guidelines for data collection, analysis, and presentation of maternal immunisation safety data. Vaccine 2017, 35, 6518–6528. [Google Scholar] [CrossRef]
- Zanelli, S.A.; Rogol, A.D. Short children born small for gestational age outcomes in the era of growth hormone therapy. Growth Horm. IGF Res. 2018, 38, 8–13. [Google Scholar] [CrossRef]
- Hwang, I.T. Efficacy and safety of growth hormone treatment for children born small for gestational age. Korean J. Pediatr. 2014, 57, 379–383. [Google Scholar] [CrossRef]
- López-Siguero, J.P.; Martínez-Aedo, M.J.; de la Vega, J.A.B.; Bosch-Muñoz, J.; Lechuga-Sancho, A.M.; Villalobos, T.; SGA Study Investigator Collaborative Group. Growth hormone treatment does not to lead to insulin resistance nor excessive rise in IGF-1 levels, while improving height in patients small for gestational age A long-term observational study. Clin. Endocrinol. 2021, 96, 558–568. [Google Scholar] [CrossRef]
- Chernausek, S.D. Update: Consequences of Abnormal Fetal Growth. J. Clin. Endocrinol. Metab. 2012, 97, 689–695. [Google Scholar] [CrossRef]
- Saenger, P.; Czernichow, P.; Hughes, I.; Reiter, E.O. Small for Gestational Age: Short Stature and Beyond. Endocr. Rev. 2007, 28, 219–251. [Google Scholar] [CrossRef] [PubMed]
- Pilling, E.; Elder, C.; Gibson, A. Growth patterns in the growth-retarded premature infant. Best Pr. Res. Clin. Endocrinol. Metab. 2008, 22, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, R.; Lee, P.A.; Ross, J.L.; Saenger, P.; Ostrow, V.; Piccoli, G. Three years of growth hormone therapy in children born small for gestational age: Results from the ANSWER Program. Endocr. Connect. 2018, 7, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.E.; Cianfarani, S.; Czernichow, P.; Johannsson, G.; Rapaport, R.; Rogol, A. Management of the Child Born Small for Gestational Age through to Adulthood: A Consensus Statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J. Clin. Endocrinol. Metab. 2007, 92, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, M.; Popa, F.I.; Corradi, M.; Gandini, A.; Teofoli, F.; Camilot, M.; Boner, A.; Cavarzere, P.; Gaudino, R.; Antoniazzi, F. Analysis of the d3-growth hormone receptor polymorphism in large cohorts of small, appropriate and large for gestational age newborns. Minerva Pediatr. 2014, 68, 157–161. [Google Scholar]
- Dörr, H.G.; Bettendorf, M.; Hauffa, B.P.; Mehls, O.; Rohrer, T.; Stahnke, N.; Pfäffle, R.; Ranke, M.B.; German KIGS Group. Different relationships between the first 2 years on growth hormone treatment and the d3-growth hormone receptor polymorphism in short small-for-gestational-age (SGA) children. Clin. Endocrinol. 2011, 75, 656–660. [Google Scholar] [CrossRef]
- Carrascosa, A.; Audí, L.; Esteban, C.; Fernández-Cancio, M.; Andaluz, P.; Gussinyé, M.; Clemente, M.; Yeste, D.; Albisu, M.A. Growth Hormone (GH) Dose, But Not Exon 3-Deleted/Full-Length GH Receptor Polymorphism Genotypes, Influences Growth Response to Two-Year GH Therapy in Short Small-for-Gestational-Age Children. J. Clin. Endocrinol. Metab. 2008, 93, 147–153. [Google Scholar] [CrossRef][Green Version]
- Binder, G.; Baur, F.; Schweizer, R.; Ranke, M.B. The d3-Growth Hormone (GH) Receptor Polymorphism Is Associated with Increased Responsiveness to GH in Turner Syndrome and Short Small-for-Gestational-Age Children. J. Clin. Endocrinol. Metab. 2006, 91, 659–664. [Google Scholar] [CrossRef]
- Turner, H.H. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology 1938, 23, 566–574. [Google Scholar] [CrossRef]
- Ullrich, O. Über typische Kombinationsbilder multipler Abartungen. Z. Kinder-Heilk. 1930, 49, 271–276. [Google Scholar] [CrossRef]
- Cui, X.; Cui, Y.; Shi, L.; Luan, J.; Zhou, X.; Han, J. A basic understanding of Turner syndrome: Incidence, complications, diagnosis, and treatment. Intractable Rare Dis. Res. 2018, 7, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, M.; Dunger, D.B.; Conway, G.S.; Wass, J.A.H. Turner’s Syndrome in Adulthood. Endocr. Rev. 2002, 23, 120–140. [Google Scholar] [CrossRef]
- Los, E.; Rosenfeld, R.G. Growth and growth hormone in Turner syndrome: Looking back, looking ahead. Am. J. Med Genet. Part C Semin. Med. Genet. 2019, 181, 74–78. [Google Scholar] [CrossRef]
- Broeck, J.V.D.; Massa, G.G.; Attanasio, A.; Matranga, A.; Chaussain, J.L.; Price, D.A.; Aarskog, D.; Wit, J.M. Final height after long-term growth hormone treatment in Turner syndrome. J. Pediatr. 1995, 127, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Cheng, F.; Xiu, L. Height outcome of the recombinant human growth hormone treatment in Turner syndrome: A meta-analysis. Endocr. Connect. 2018, 7, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.M.; Kim, J.M.; Cheon, C.K.; Kim, D.H.; Lee, D.Y.; Cheong, W.Y.; Kim, E.Y.; Park, M.J.; Yoo, H.W. The common exon 3 polymorphism of the growth hormone receptor gene and the effect of growth hormone therapy on growth in Korean patients with Turner syndrome. Clin. Endocrinol. 2010, 72, 196–202. [Google Scholar] [CrossRef]
- Chernausek, S.D.; Attie, K.M.; Cara, J.F.; Rosenfeld, R.G.; Frane, J. Growth Hormone Therapy of Turner Syndrome: The Impact of Age of Estrogen Replacement on Final Height. J. Clin. Endocrinol. Metab. 2000, 85, 2439–2445. [Google Scholar] [CrossRef]
- Chung, M.S.; Langouët, M.; Chamberlain, S.J.; Carmichael, G.G. Prader-Willi syndrome: Reflections on seminal studies and future therapies. Open Biol. 2020, 10, 200195. [Google Scholar] [CrossRef]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef]
- McQuillan, D.J.; Handley, C.J.; Campbell, M.A.; Bolis, S.; Milway, V.E.; Herington, A.C. Stimulation of proteoglycan biosynthesis by serum and insulin-like growth factor-I in cultured bovine articular cartilage. Biochem. J. 1986, 240, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.E.; Carrel, A.L.; Whitman, B.Y.; Allen, D.B. Sustained benefit after 2 years of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome. J. Pediatr. 2000, 137, 42–49. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, S.T.; Sohn, Y.B.; Kim, S.H.; Cho, S.Y.; Ko, A.R.; Ji, S.T.; Kwon, J.Y.; Yeau, S.; Paik, K.H.; et al. A polymorphism in the growth hormone receptor is associated with height in children with Prader-Willi syndrome. Am. J. Med. Genet. Part A 2011, 155, 2970–2973. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Roberts, J.; Hayes, J.; Tan, X.; Manzardo, A.M. Growth hormone receptor (GHR) gene polymorphism and prader-willi syndrome. Am. J. Med. Genet. Part A 2013, 161, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Acromegaly: Medical Progress. N. Engl. J. Med. 2006, 24, 355. [Google Scholar]
- Melmed, S. Acromegaly pathogenesis and treatment. J. Clin. Investig. 2009, 119, 3189–3202. [Google Scholar] [CrossRef] [PubMed]
- Dobrashian, R.D.; O’Halloran, D.J.; Hunt, A.; Beardwell, C.G.; Shalet, S.M. Relationships between insulin-like growth factor-1 levels and growth hormone concentrations during diurnal profiles and following oral glucose in acromegaly. Clin. Endocrinol. 1993, 38, 589–593. [Google Scholar] [CrossRef]
- Schmid, C.; Krayenbuehl, P.A.; Bernays, R.L.; Zwimpfer, C.; E Maly, F.; Wiesli, P. Growth Hormone (GH) Receptor Isoform in Acromegaly: Lower Concentrations of GH but Not Insulin-Like Growth Factor-1 in Patients with a Genomic Deletion of Exon 3 in the GH Receptor Gene. Clin. Chem. 2007, 53, 1484–1488. [Google Scholar] [CrossRef][Green Version]
- Parkinson, C.; Ryder, W.D.J.; Trainer, P.J. The Relationship between Serum GH and Serum IGF-I in Acromegaly Is Gender-Specific. J. Clin. Endocrinol. Metab. 2001, 86, 5240–5244. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van der Lely, A.J.; Hutson, R.K.; Trainer, P.J.; Besser, G.M.; Barkan, A.L.; Katznelson, L.; Klibanski, A.; Herman-Bonert, V.; Melmed, S.; Vance, M.L.; et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 2001, 358, 1754–1759. [Google Scholar] [CrossRef]
- Bianchi, A.; Mazziotti, G.; Tilaro, L.; Cimino, V.; Veltri, F.; Gaetani, E.; Pecorini, G.; Pontecorvi, A.; Giustina, A.; De Marinis, L. Growth hormone receptor polymorphism and the effects of pegvisomant in acromegaly. Pituitary 2008, 12, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Franck, S.E.; Broer, L.; van der Lely, A.J.; Kamenicky, P.; Bernabéu, I.; Malchiodi, E.; Delhanty, P.J.; Rivadeneira, F.; Neggers, S.J. The Effect of the Exon-3-Deleted Growth Hormone Receptor on Pegvisomant-Treated Acromegaly: A Systematic Review and Meta-Analysis. Neuroendocrinology 2016, 105, 131–140. [Google Scholar] [CrossRef]
- Colao, A.; Ferone, D.; Marzullo, P.; Lombardi, G. Systemic Complications of Acromegaly: Epidemiology, Pathogenesis, and Management. Endocr. Rev. 2004, 25, 102–152. [Google Scholar] [CrossRef]
- Colao, A.; Pivonello, R.; Auriemma, R.S.; Galdiero, M.; Ferone, D.; Minuto, F.; Marzullo, P.; Lombardi, G. The Association of Fasting Insulin Concentrations and Colonic Neoplasms in Acromegaly: A Colonoscopy-Based Study in 210 Patients. J. Clin. Endocrinol. Metab. 2007, 92, 3854–3860. [Google Scholar] [CrossRef]
- Colao, A.; Spinelli, L.; Marzullo, P.; Pivonello, R.; Petretta, M.; Di Somma, C.; Vitale, G.; Bonaduce, D.; Lombardi, G. High Prevalence of Cardiac Valve Disease in Acromegaly: An Observational, Analytical, Case-Control Study. J. Clin. Endocrinol. Metab. 2003, 88, 3196–3201. [Google Scholar] [CrossRef] [PubMed]
- Pontes, J.; Madeira, M.; Lima, C.H.A.; Ogino, L.L.; Neto, F.d.P.P.; de Mendonça, L.M.C.; Farias, M.L.F.; Kasuki, L.; Gadelha, M.R. Exon 3-deleted growth hormone receptor isoform is not related to worse bone mineral density or microarchitecture or to increased fracture risk in acromegaly. J. Endocrinol. Investig. 2019, 43, 163–171. [Google Scholar] [CrossRef]
- Park, H.Y.; Hwang, I.R.; Seo, J.B.; Kim, S.W.; Seo, H.A.; Lee, I.K.; Kim, J.G. Association between the Growth Hormone Receptor Exon 3 Polymorphism and Metabolic Factors in Korean Patients with Acromegaly. Endocrinol. Metab. 2015, 30, 312–317. [Google Scholar] [CrossRef]
- Kamenicky, P.; Dos Santos, C.; Espinosa, C.; Salenave, S.; Galland, F.; Le Bouc, Y.; Maison, P.; Bougnères, P.; Chanson, P. D3 GH receptor polymorphism is not associated with IGF1 levels in untreated acromegaly. Eur. J. Endocrinol. 2009, 161, 231–235. [Google Scholar] [CrossRef]
- Cinar, N.; Dagdelen, S.; Yorgun, H.; Canpolat, U.; Kabakçı, G.; Erbas, T. The clinical and cardiometabolic effects of d3-growth hormone receptor polymorphism in acromegaly. Pituitary 2014, 18, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Mercado, M.; González, B.; Sandoval, C.; Esquenazi, Y.; Mier, F.; Vargas, G.; Monteros, A.L.E.d.L.; Sosa, E. Clinical and Biochemical Impact of the d3 Growth Hormone Receptor Genotype in Acromegaly. J. Clin. Endocrinol. Metab. 2008, 93, 3411–3415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wassenaar, M.J.E.; Biermasz, N.R.; Pereira, A.M.; van der Klaauw, A.A.; Smit, J.W.A.; Roelfsema, F.; van der Straaten, T.; Cazemier, M.; Hommes, D.W.; Kroon, H.M.; et al. The Exon-3 Deleted Growth Hormone Receptor Polymorphism Predisposes to Long-Term Complications of Acromegaly. J. Clin. Endocrinol. Metab. 2009, 94, 4671–4678. [Google Scholar] [CrossRef] [PubMed]



| Authors | No. of Cases | Effects |
|---|---|---|
| Wegmann, 2017 [7] | 96 | d3GHR carriers had increased spontaneous growth, lower IS, and a compensatory increase in glucose, C-peptide, and insulin at baseline. |
| Garrido, 2021 [8] | 65 | d3GHR carriers have increased spontaneous growth. |
| Dos Santos, 2004 [2] | 76 | d3GHR carriers responded to hGH therapy 1.7 to 2 times better than those who had the flGHR isoform. |
| Dörr, 2011 [92] | 142 | d3GHR carriers exhibited a better growth response to growth hormone only in the first year but not in the second year. |
| Binder, 2006 [94] | 60 | d3GHR carriers grew significantly more quickly than anticipated. During the first year of rhGH treatment, the average height gain linked to d3GHR was roughly 0.75 cm. |
| Carrascosa, 2006 [10] | 170 | Spontaneous growth rate and responsiveness to 66 micro/k·d GH therapy are comparable in flGHR and d3GHR carriers. |
| Carrascosa, 2008 [93] | 60 | Both genotypes responded similarly to GH therapy after two years of response (32.1 ± 3.8 microg/kg·d). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falah, G.; Sharvit, L.; Atzmon, G. The Exon 3-Deleted Growth Hormone Receptor (d3GHR) Polymorphism—A Favorable Backdoor Mechanism for the GHR Function. Int. J. Mol. Sci. 2023, 24, 13908. https://doi.org/10.3390/ijms241813908
Falah G, Sharvit L, Atzmon G. The Exon 3-Deleted Growth Hormone Receptor (d3GHR) Polymorphism—A Favorable Backdoor Mechanism for the GHR Function. International Journal of Molecular Sciences. 2023; 24(18):13908. https://doi.org/10.3390/ijms241813908
Chicago/Turabian StyleFalah, Ghadeer, Lital Sharvit, and Gil Atzmon. 2023. "The Exon 3-Deleted Growth Hormone Receptor (d3GHR) Polymorphism—A Favorable Backdoor Mechanism for the GHR Function" International Journal of Molecular Sciences 24, no. 18: 13908. https://doi.org/10.3390/ijms241813908
APA StyleFalah, G., Sharvit, L., & Atzmon, G. (2023). The Exon 3-Deleted Growth Hormone Receptor (d3GHR) Polymorphism—A Favorable Backdoor Mechanism for the GHR Function. International Journal of Molecular Sciences, 24(18), 13908. https://doi.org/10.3390/ijms241813908