
Chemical Diversity of Mo5S5 Clusters with Pyrazole: Synthesis, Redox and UV-vis-NIR Absorption Properties

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results and Discussion
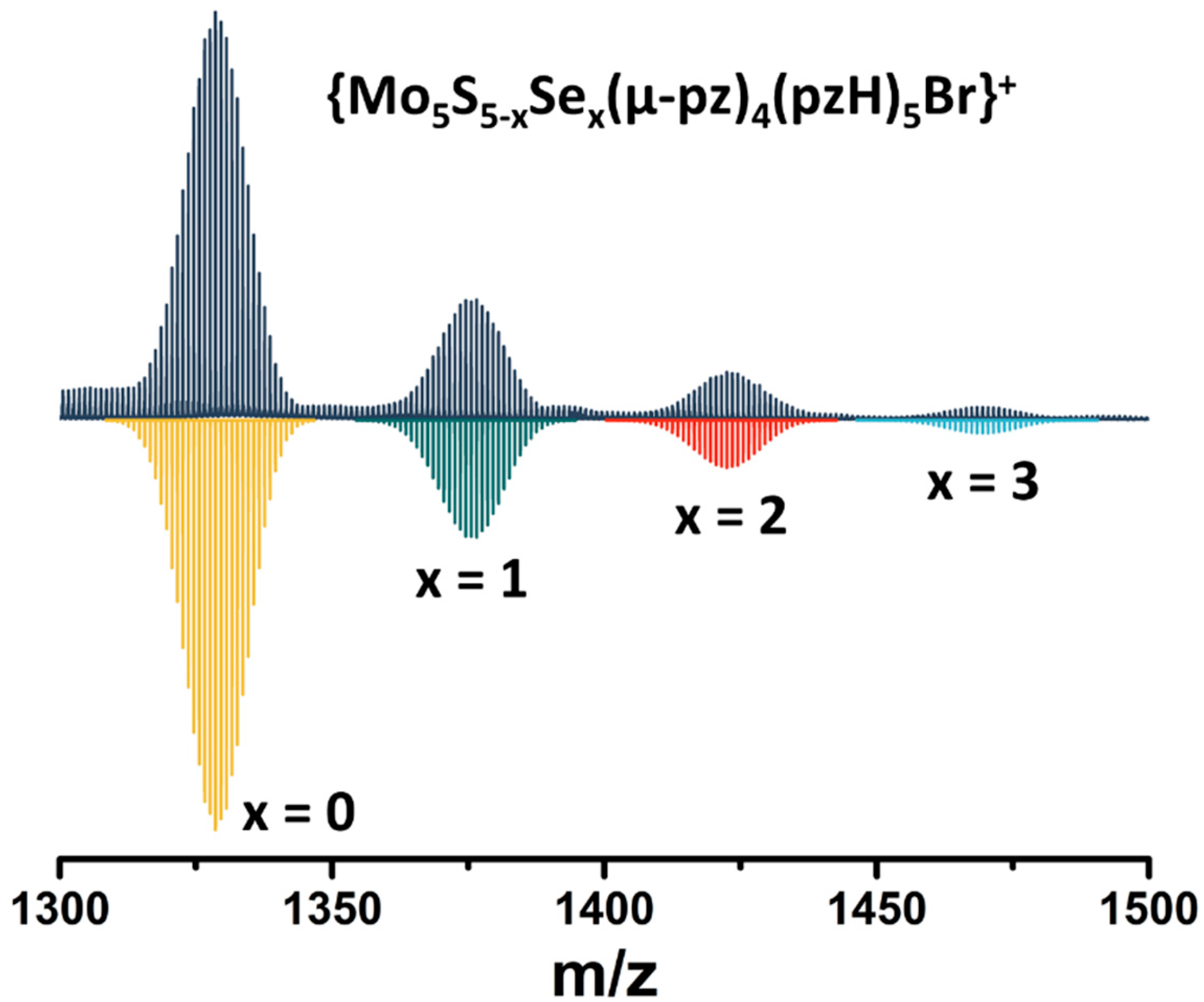
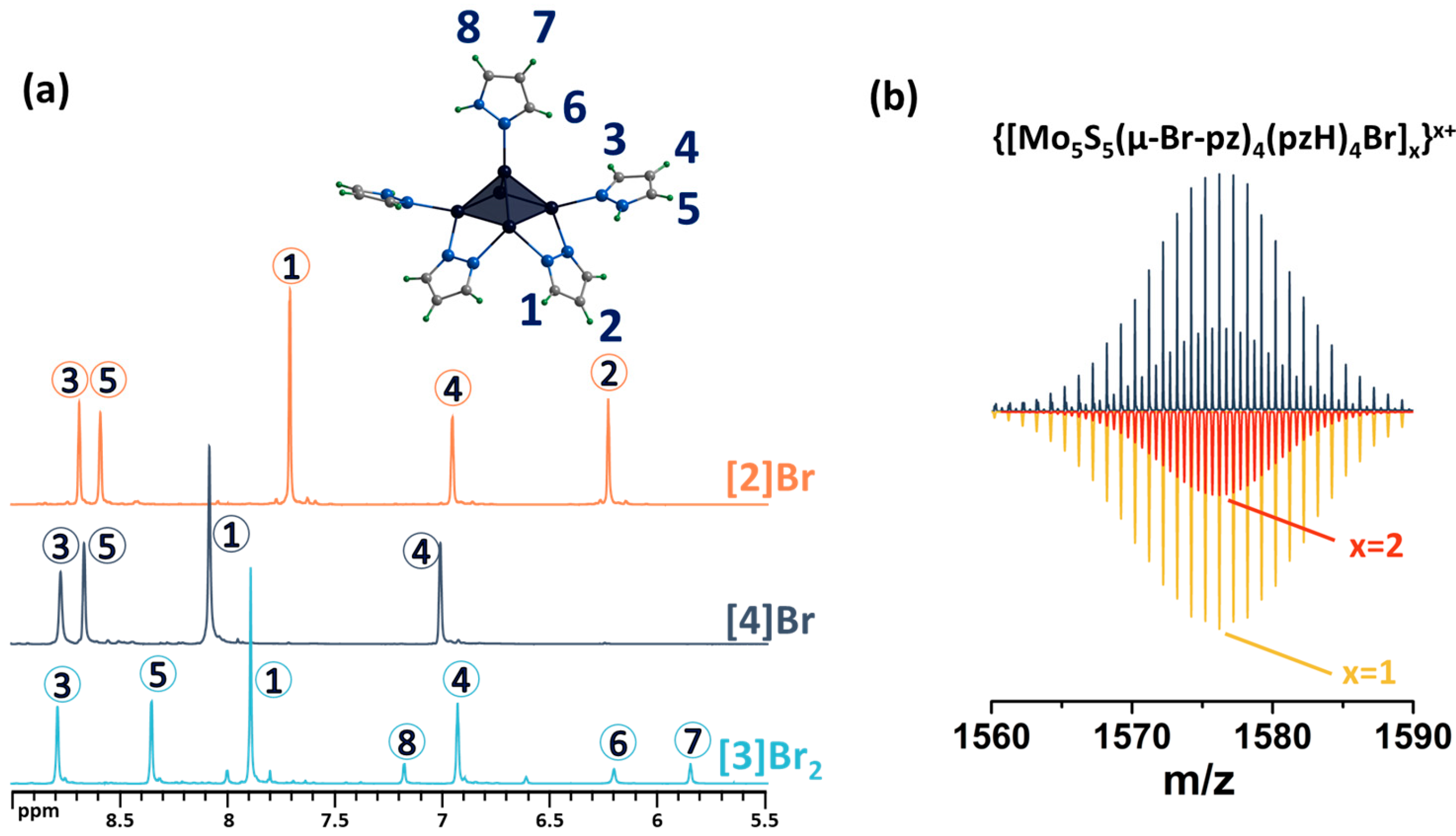
2.1. Synthesis and General Characterization of Square Pyramidal Clusters
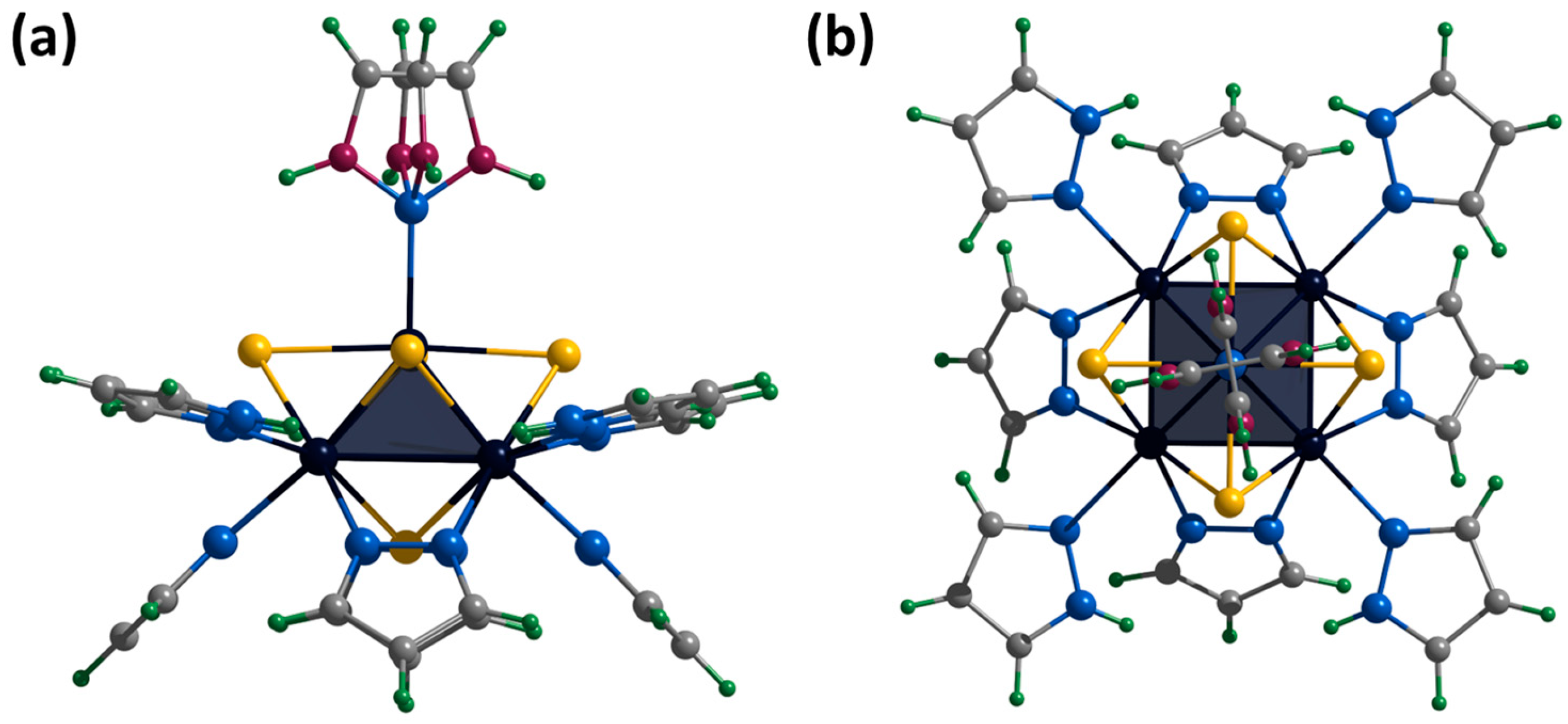
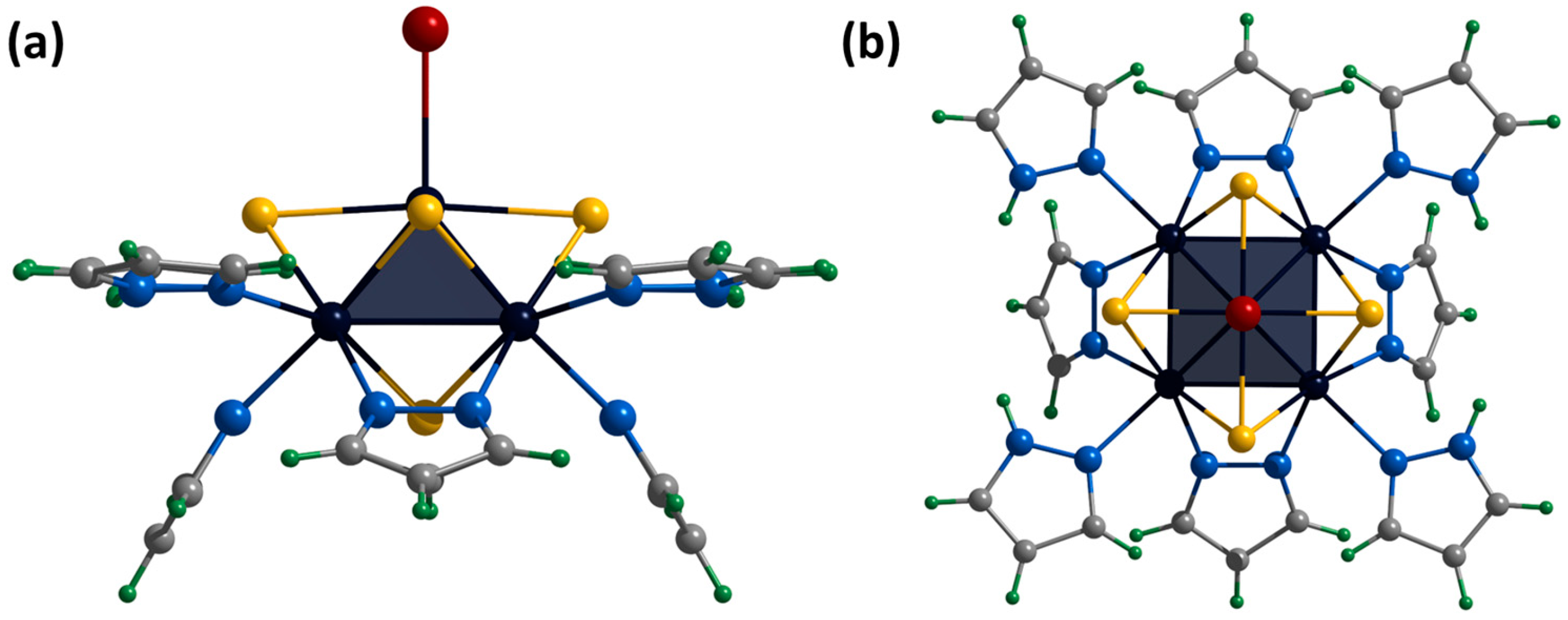
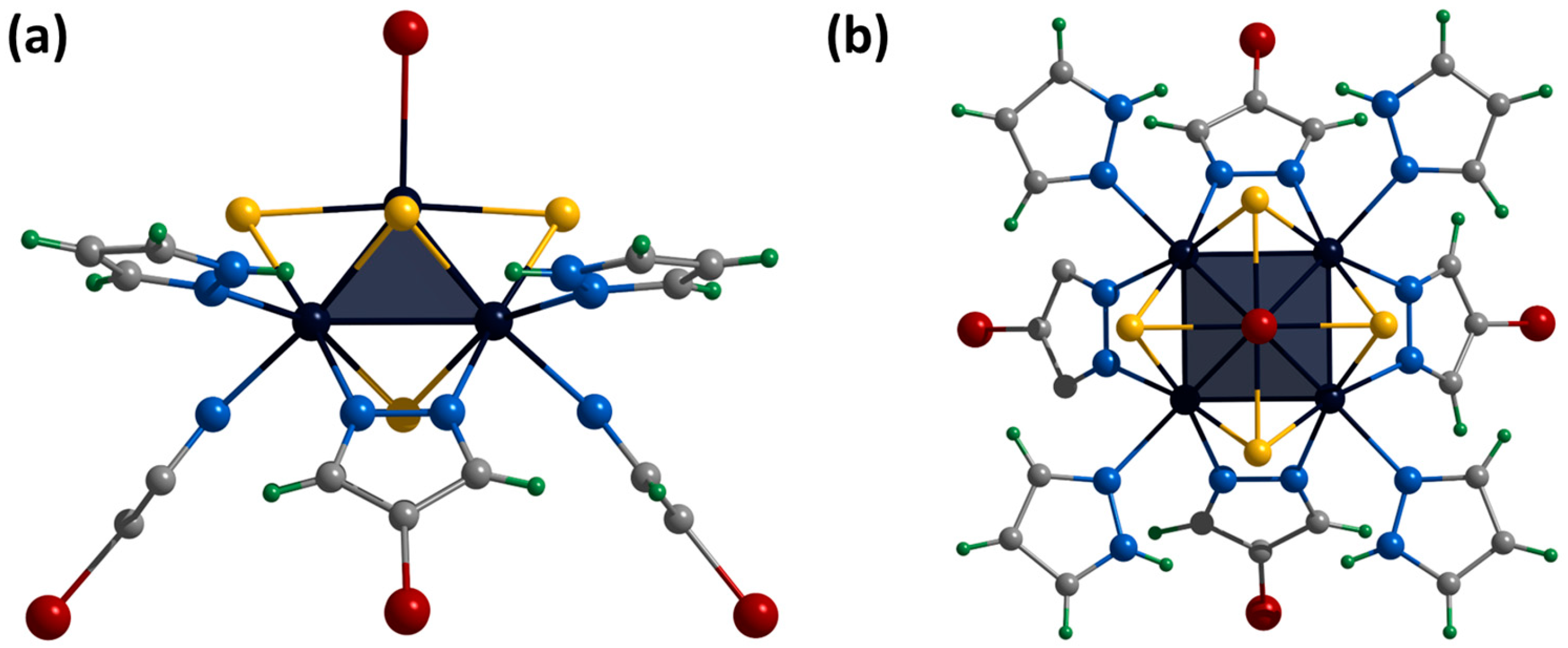
2.2. Crystal Structure of Compounds
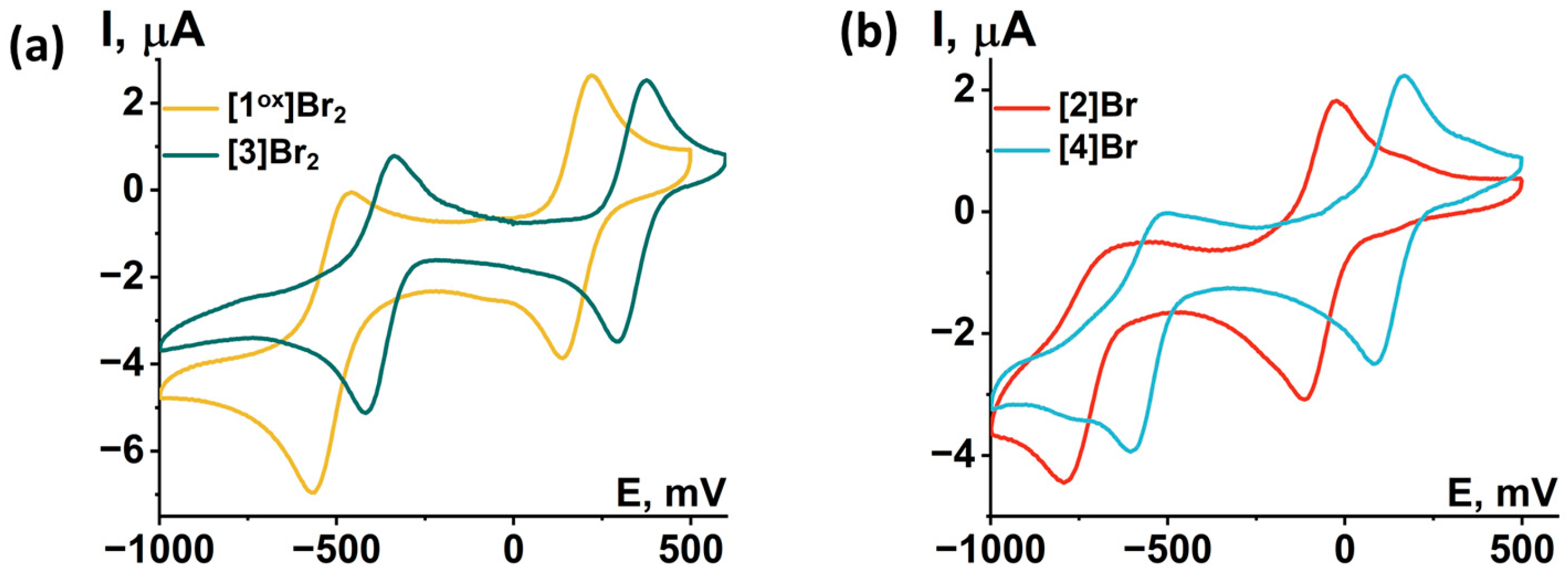
2.3. Physicochemical Properties
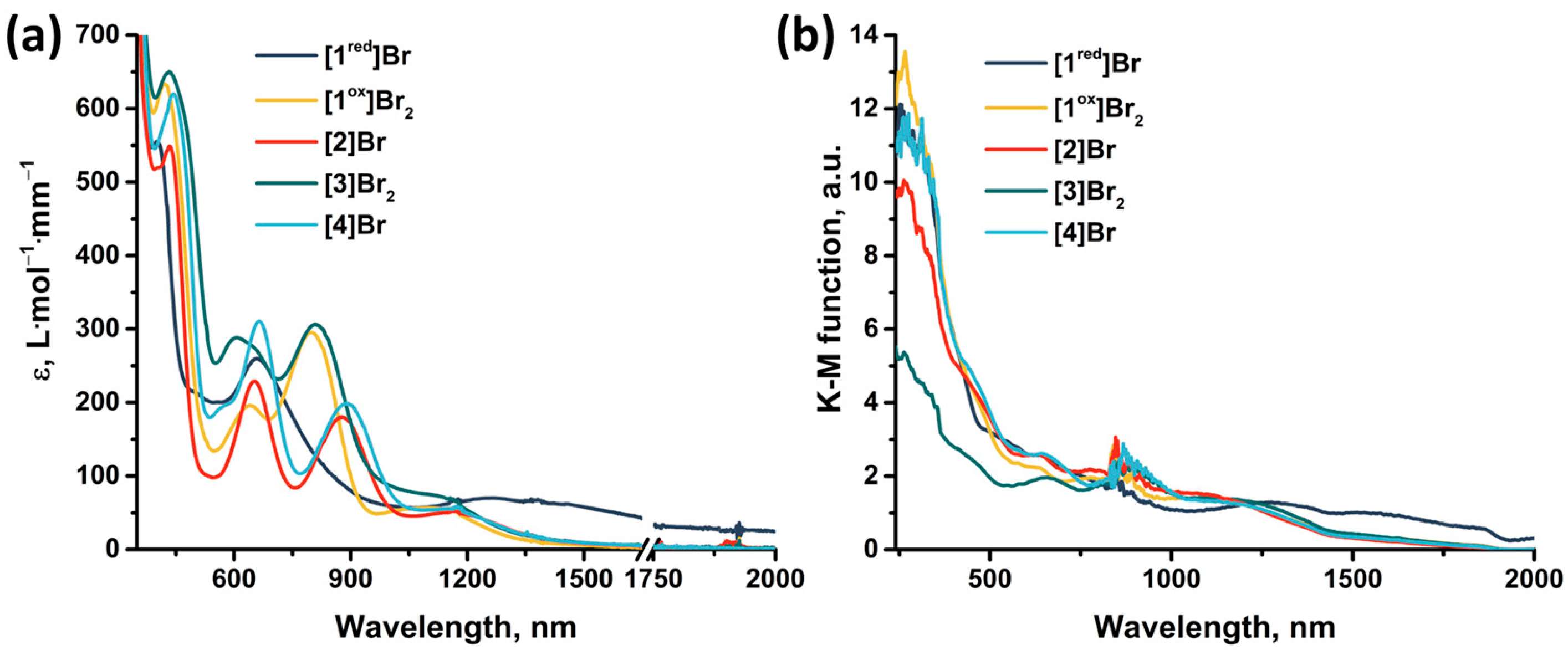
2.4. UV-Vis-NIR Absorption
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Syntheses
3.2.1. [{Mo5(μ3-S)i4(μ4-S)i(μ-pz)i4}(pzH)t5]Br pzH·H2O (Denoted as [1red]Br)
3.2.2. [{Mo5(μ3-S)i4(μ4-S)i(μ-pz)i4}(pzH)t5]Br2 2H2O (Denoted as [1ox]Br2)
3.2.3. [{Mo5(μ3-S)i4(μ4-S)i(μ-pz)i4}(pzH)bs4Bra]Br (Denoted as [2]Br)
3.2.4. [{Mo5(μ3-S)i4(μ4-S)i(μ-4-Br-pz)i4}(pzH)t5]Br2 (Denoted as [3]Br2)
3.2.5. [{Mo5(μ3-S)i4(μ4-S)i(μ-4-Br-pz)i4}(pzH)bs4Bra]Br (Denoted as [4]Br)
3.3. Physical Methods
3.4. Single-Crystal X-ray Diffraction Analysis (XRD)
3.5. Cyclic Voltammetry
3.6. EPR
3.7. Magnetic Susceptibility
3.8. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braunstein, P.; Raithby, P.R. Metal Clusters in Chemistry; Wiley-VCH: Weinheim, Germany, 1999; pp. 2–7. [Google Scholar]
- Fedorov, V.E. Metal clusters. As they were born in Siberia. J. Clust. Sci. 2015, 26, 3–15. [Google Scholar] [CrossRef]
- Mikhaylov, M.A.; Sokolov, M.N. Molybdenum iodides—From obscurity to bright luminescence. Eur. J. Inorg. Chem. 2019, 39–40, 4181–4197. [Google Scholar] [CrossRef]
- Fedorov, V.E.; Mironov, Y.V.; Naumov, N.G.; Sokolov, M.N.; Fedin, V.P. Chalcogenide clusters of group 5–7 metals. Russ. Chem. Rev. 2007, 76, 529–552. [Google Scholar] [CrossRef]
- Gayfulin, Y.M.; Mironov, Y.V.; Naumov, N.G. High-valence cluster compounds of transition metals containing interstitial heteroatoms: Geometry, electronic structure, and physicochemical properties. J. Struct. Chem. 2021, 62, 331–355. [Google Scholar] [CrossRef]
- Kirakci, K.; Shestopalov, M.A.; Lang, K. Recent developments on luminescent octahedral transition metal cluster complexes towards biological applications. Coord. Chem. Rev. 2023, 481, 215048. [Google Scholar] [CrossRef]
- Khanna, S.N.; Reber, A.C.; Bista, D.; Sengupta, T.; Lambert, R. The superatomic state beyond conventional magic numbers: Ligated metal chalcogenide superatoms. J. Chem. Phys. 2021, 155, 120901. [Google Scholar] [CrossRef]
- Nguyen, N.T.K.; Lebastard, C.; Wilmet, M.; Dumait, N.; Renaud, A.; Cordier, S.; Ohashi, N.; Uchikoshi, T.; Grasset, F. A review on functional nanoarchitectonics nanocomposites based on octahedral metal atom clusters (Nb6, Mo6, Ta6, W6, Re6): Inorganic 0D and 2D powders and films. Sci. Technol. Adv. Mater. 2022, 23, 547–578. [Google Scholar] [CrossRef]
- Molard, Y.; Dorson, F.; Cîrcu, V.; Roisnel, T.; Artzner, F.; Cordier, S. Clustomesogens: Liquid crystal materials containing transition-metal clusters. Angew. Chem. Int. Ed. 2010, 49, 3351–3355. [Google Scholar] [CrossRef]
- Khlifi, S.; Fournier Le Ray, N.; Paofai, S.; Amela-Cortes, M.; Akdas-Kiliç, H.; Taupier, G.; Derien, S.; Cordier, S.; Achard, M.; Molard, Y. Self-erasable inkless imprinting using a dual emitting hybrid organic-inorganic material. Mater. Today 2020, in press. [Google Scholar] [CrossRef]
- Gushchin, A.L.; Laricheva, Y.A.; Sokolov, M.N.; Llusar, R. Tri- and tetranuclear molybdenum and tungsten chalcogenide clusters: On the way to new materials and catalysts. Russ. Chem. Rev. 2018, 87, 670–706. [Google Scholar] [CrossRef]
- Szczepura, L.F.; Soto, E. Exploring the breadth of terminal ligands coordinated in [Mo6X8]4+ and [Re6Q8]2+ based cluster complexes. In Ligated Transition Metal Clusters in Solid-State Chemistry: The Legacy of Marcel Sergent; Halet, J.-F., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 75–108. [Google Scholar]
- Vorotnikova, N.A.; Bardin, V.A.; Vorotnikov, Y.A.; Kirakci, K.; Adamenko, L.S.; Alekseev, A.Y.; Meyer, E.E.; Kubát, P.; Lang, K.; Shestopalov, M.A. Heterogeneous photoactive antimicrobial coatings based on a fluoroplastic doped with an octahedral molybdenum cluster compound. Dalton Trans. 2021, 50, 8467–8475. [Google Scholar] [CrossRef] [PubMed]
- Efremova, O.A.; Shestopalov, M.A.; Chirtsova, N.A.; Smolentsev, A.I.; Mironov, Y.V.; Kitamura, N.; Brylev, K.A.; Sutherland, A.J. A highly emissive inorganic hexamolybdenum cluster complex as a handy precursor for the preparation of new luminescent materials. Dalton Trans. 2014, 43, 6021–6025. [Google Scholar] [CrossRef] [PubMed]
- Kirakci, K.; Kubáňová, M.; Přibyl, T.; Rumlová, M.; Zelenka, J.; Ruml, T.; Lang, K. A cell membrane targeting molybdenum-iodine nanocluster: Rational ligand design toward enhanced photodynamic activity. Inorg. Chem. 2022, 61, 5076–5083. [Google Scholar] [CrossRef] [PubMed]
- Takei, I.; Wakebe, Y.; Suzuki, K.; Enta, Y.; Suzuki, T.; Mizobe, Y.; Hidai, M. Synthesis of cubane-type Mo3NiS4 clusters and their catalytic activity for the cyclization of alkynoic acids to enol lactones. Organometallics 2003, 22, 4639–4641. [Google Scholar] [CrossRef]
- Tourneur, J.; Fabre, B.; Loget, G.; Vacher, A.; Mériadec, C.; Ababou-Girard, S.; Gouttefangeas, F.; Joanny, L.; Cadot, E.; Haouas, M.; et al. Molecular and material engineering of photocathodes derivatized with polyoxometalate-supported {Mo3S4} HER catalysts. J. Am. Chem. Soc. 2019, 141, 11954–11962. [Google Scholar] [CrossRef]
- Feliz, M.; Garriga, J.M.; Llusar, R.; Uriel, S.; Humphrey, M.G.; Lucas, N.T.; Samoc, M.; Luther-Davies, B. Synthesis, Structure, and optical-limiting properties of heterobimetallic [M3CuS4] cuboidal clusters (M = Mo or W) with terminal phosphine ligands. Inorg. Chem. 2001, 40, 6132–6138. [Google Scholar] [CrossRef]
- Cebotari, D.; Calancea, S.; Marrot, J.; Guillot, R.; Falaise, C.; Guérineau, V.; Touboul, D.; Haouas, M.; Gulea, A.; Floquet, S. Tuning the nuclearity of [Mo2O2S2]2+-based assemblies by playing with the degree of flexibility of bis-thiosemicarbazone ligands. Dalton Trans. 2023, 52, 3059–3071. [Google Scholar] [CrossRef]
- Gougeon, P.; Padiou, J.; Le Marouille, J.Y.; Potel, M.; Sergent, M. Ag3.6Mo9Se11: Premier compose a clusters Mo9 dans des motifs Mo9Se11. J. Solid State Chem. 1984, 51, 218–226. [Google Scholar] [CrossRef]
- Picard, S.; Gougeon, P.; Potel, M. Synthesis, structural evolution, and electrical properties of the novel Mo12 cluster compounds K1+xMo12S14 (x = 0, 1.1, 1.3, and 1.6) with a Tunnel Structure. Inorg. Chem. 2006, 45, 1611–1616. [Google Scholar] [CrossRef]
- Stewart, D.F.; O’Donnell, T.A. Metal clusters in a new mixed halide of molybdenum (IV) and (V). Nature 1966, 210, 836. [Google Scholar] [CrossRef]
- Zietlow, T.C.; Gray, H.B. Preparation and characterization of pentanuclear molybdenum halide clusters. Inorg. Chem. 1986, 25, 631–634. [Google Scholar] [CrossRef]
- Savina, I.V.; Ivanov, A.A.; Evtushok, D.V.; Gayfulin, Y.M.; Komarovskikh, A.Y.; Syrokvashin, M.M.; Ivanova, M.N.; Asanov, I.P.; Eltsov, I.V.; Kuratieva, N.V.; et al. Unusual square pyramidal chalcogenide Mo5 cluster with bridging pyrazolate-ligands. Int. J. Mol. Sci. 2023, 24, 3440. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Popp, F.; Boettcher, S.; Yuan, M.; Oertel, C.M.; DiSalvo, F.J. Synthesis, characterization and properties of Mo6S8(4-tert-butylpyridine)6 and related M6S8L6 cluster complexes (M = Mo, W). J. Chem. Soc. Dalton Trans. 2002, 2002, 3096–3100. [Google Scholar] [CrossRef]
- Xie, X.; McCarley, R.E. Synthesis, characterization, and structure of neutral and anionic complexes containing octahedral W6Te8 cluster units. Inorg. Chem. 1997, 36, 4665–4675. [Google Scholar] [CrossRef] [PubMed]
- Novikova, E.D.; Gassan, A.D.; Ivanov, A.A.; Vorotnikov, Y.A.; Shestopalov, M.A. Neutral Mo6Q8-clusters with terminal phosphane ligands—A route to water-soluble molecular units of Chevrel phases. New J. Chem. 2022, 46, 2218–2223. [Google Scholar] [CrossRef]
- Jödden, K.; von Schnering, H.G.; Schäfer, H. [(n-C4H9)4N]2Mo5Cl13— a compound with the cluster group [Mo5Cli8]. Angew. Chem. 1975, 14, 570–571. [Google Scholar] [CrossRef]
- Winder, W. Synthesis and Characterization of Several Molybdenum Chloride Cluster Compounds. Ph.D. Thesis, Iowa State University, Ames, IA, USA, 1983. [Google Scholar]
- Saito, T.; Yamamoto, N.; Nagase, T.; Tsudoi, T.; Kobayashi, K.; Yamagata, T.; Imoto, H.; Unoura, K. Molecular models of the superconducting chevrel phases: Syntheses and structures of [Mo6X8(PEt3)6] and [PPN][Mo6X8(PEt3)6] (X = S, Se; PPN = (Ph3P)2N). Inorg. Chem. 1990, 29, 764–770. [Google Scholar] [CrossRef]
- Gassan, A.D.; Ivanov, A.A.; Pozmogova, T.N.; Eltsov, I.V.; Kuratieva, N.V.; Mironov, Y.V.; Shestopalov, M.A. Water-soluble chalcogenide W6-clusters: On the way to biomedical applications. Int. J. Mol. Sci. 2022, 23, 8734. [Google Scholar] [CrossRef]
- Nguyen, T.K.N.; Dierre, B.; Grasset, F.; Dumait, N.; Cordier, S.; Lemoine, P.; Renaud, A.; Fudouzi, H.; Ohashi, N. Electrophoretic coating of octahedral molybdenum metal clusters for UV/NIR light screening. Coatings 2017, 7, 114. [Google Scholar] [CrossRef]
- Nguyen, T.K.N.; Renaud, A.; Wilmet, M.; Dumait, N.; Paofai, S.; Dierre, B.; Chen, W.; Ohashi, N.; Cordier, S.; Grasset, F.; et al. New ultra-violet and near-infrared blocking filters for energy saving applications: Fabrication of tantalum metal atom cluster-based nanocomposite thin films by electrophoretic deposition. J. Mater. Chem. C 2017, 5, 10477–10484. [Google Scholar] [CrossRef]
- Chen, W.; Nguyen, T.K.N.; Wilmet, M.; Dumait, N.; Makrygenni, O.; Matsui, Y.; Takei, T.; Cordier, S.; Ohashi, N.; Uchikoshi, T.; et al. ITO@SiO2 and ITO@{M6Br12}@SiO2 (M = Nb, Ta) nanocomposite films for ultraviolet-near infrared shielding. Nanoscale Adv. 2019, 1, 3693–3698. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Dou, J.-H.; Zhao, N.; Zhang, S.; Zheng, Y.-Q.; Zhang, J.-P.; Wang, J.Y.; Pei, J.; Wang, Y. Highly efficient NIR-II photothermal conversion based on an organic conjugated polymer. Chem. Mater. 2017, 29, 718–725. [Google Scholar] [CrossRef]
- Khutornoi, V.A.; Naumov, N.G.; Mironov, Y.V.; Oeckler, O.; Simon, A.; Fedorov, V.E. Novel complexes [M(DMF)6][Mo6Br8(NCS)6] (M = Mn2+, Co2+, Ni2+, Cu2+, and Cd2+): Synthesis, structure determination, and properties. Russ. J. Coord. Chem. 2002, 28, 183–190. [Google Scholar] [CrossRef]
- Kirakci, K.; Cordier, S.; Perrin, C. Synthesis and characterization of Cs2Mo6X14 (X = Br or I) hexamolybdenum cluster halides: Efficient Mo6 cluster precursors for solution chemistry syntheses. Z. Anorg. Allg. Chem. 2005, 631, 411–416. [Google Scholar] [CrossRef]
- Bruker. APEX2 (Version 1.08), SAINT (Version 07.03), SADABS (Version 02.11), SHELXTL (Version 06.12); Bruker AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Autschbach, J.; Pritchard, B. Calculation of molecular g-tensors using the zeroth-order regular approximation and density functional theory: Expectation value versus linear response approaches. Theor. Chem. Acc. 2011, 129, 453–466. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF 2023.1, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 16 May 2023).
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mobs–Mobs (Average) | Mobs–Moa (Average) | Mo–μ3-S (Average) | Mo–μ4-S (Average) |
|---|---|---|---|---|
| [1red]Br | 2.7980(3)–2.8010(4) (2.8000) | 2.6384(4)–2.6459(4) (2.6422) | 2.3982(8)–2.4395(8) (2.4187) | 2.4235(6)–2.4318(6) (2.4277) |
| [1ox][Mo6I14] | 2.8136(6)–2.8249(6) (2.8193) | 2.6288(6)–2.6322(6) (2.6305) | 2.3781(14)–2.4523(12) (2.4168) | 2.4296(12)–2.4390(12) (2.4343) |
| [2]Br·2.5H2O·0.5DMF | 2.7951(17)–2.8217(17) (2.8080) | 2.6285(18)–2.6345(18) (2.6310) | 2.368(4)–2.479(4) (2.423) | 2.443(4)–2.447(4) (2.446) |
| [3]Br2·CH3CN·Et2O | 2.8073(10)–2.8244(10) (2.8168) | 2.6185(10)–2.6341(10) (2.6269) | 2.363(2)–2.465(2) (2.402) | 2.436(2)–2.449(2) (2.441) |
| [4]Br·1.25H2O·2DMSO | 2.7816(9)–2.7985(17) (2.790) | 2.6468(11)–2.6526(10) (2.6497) | 2.387(2)–2.441(3) (2.411) | 2.432(2)–2.436(2) (2.434) |
| Mo5Se5red | 2.8398(2) | 2.6823(3) | 2.5572(2) | 2.5329(3) |
| Mo5Se5ox | 2.865(1) | 2.660(1)–2.674(1) (2.667) | 2.499(1)–2.5712(8) (2.5326) | 2.547(1)–2.550(1) (2.548) |
| Mo–Nμ-pz (average) | Mobs–NpzH (average) | Moa–NpzH (average) | Moa–Br (average) | |
| [1red]Br | 2.183(3)–2.194(2) (2.186) | 2.207(3)–2.210(3) (2.209) | 2.199(4) | – |
| [1ox][Mo6I14] | 2.155(4)–2.177(4) (2.165) | 2.189(4)–2.198(4) (2.194) | 2.197(6) | – |
| [2]Br·2.5H2O·0.5DMF | 2.145(14)–2.180(15) (2.167) | 2.212(12)–2.225(13) (2.216) | – | 2.632(3)–2.640(3) (2.636) |
| [3]Br2·CH3CN·Et2O | 2.149(7)–2.188(7) (2.175) | 2.190(7)–2.209(7) (2.200) | 2.236(8) | – |
| [4]Br·1.25H2O·2DMSO | 2.223(7) | 2.189(6)–2.193(6) (2.191) | – | 2.6327(15) |
| Mo5Se5red (a) | 2.184(2)–2.194(2) (2.189) | 2.217(2) | 2.239(4) | – |
| Mo5Se5ox (a) | 2.164(8)–2.209(7) (2.179) | 2.214(7)–2.230(7) (2.222) | 2.25(1) | – |
| Compound | Solvent | Process [a] | Ea | Ec | E1/2 | Ref. |
|---|---|---|---|---|---|---|
| Mo5Se5red | ACN | 15 VEC to 16 VEC, qrev | –0.54 | –0.60 | –0.57 | [24] |
| 15 VEC to 14 VEC, rev | 0.14 | 0.07 | 0.11 | |||
| [1ox]Br2 | ACN | 14 VEC to 15 VEC, rev | 0.17 | 0.10 | 0.14 | This work |
| 15 VEC to 16 VEC, qrev | –0.53 | –0.61 | –0.57 | |||
| DCM | 14 VEC to 15 VEC, rev | 0.22 | 0.14 | 0.18 | ||
| 15 VEC to 16 VEC, qrev | –0.46 | –0.57 | –0.52 | |||
| [2]Br | DCM | 14 VEC to 15 VEC, rev | –0.03 | –0.12 | –0.08 | |
| 15 VEC to 16 VEC, irrev | – | –0.80 | – | |||
| [3]Br2 | DCM | 14 VEC to 15 VEC, rev | 0.38 | 0.29 | 0.33 | |
| 15 VEC to 16 VEC, qrev | –0.34 | –0.42 | –0.38 | |||
| [4]Br | DCM | 14 VEC to 15 VEC, rev | 0.17 | 0.09 | 0.13 | |
| 15 VEC to 16 VEC, qrev | –0.51 | –0.60 | –0.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savina, I.V.; Ivanov, A.A.; Eltsov, I.V.; Yanshole, V.V.; Kuratieva, N.V.; Komarovskikh, A.Y.; Syrokvashin, M.M.; Shestopalov, M.A. Chemical Diversity of Mo5S5 Clusters with Pyrazole: Synthesis, Redox and UV-vis-NIR Absorption Properties. Int. J. Mol. Sci. 2023, 24, 13879. https://doi.org/10.3390/ijms241813879
Savina IV, Ivanov AA, Eltsov IV, Yanshole VV, Kuratieva NV, Komarovskikh AY, Syrokvashin MM, Shestopalov MA. Chemical Diversity of Mo5S5 Clusters with Pyrazole: Synthesis, Redox and UV-vis-NIR Absorption Properties. International Journal of Molecular Sciences. 2023; 24(18):13879. https://doi.org/10.3390/ijms241813879
Chicago/Turabian StyleSavina, Iulia V., Anton A. Ivanov, Ilia V. Eltsov, Vadim V. Yanshole, Natalia V. Kuratieva, Andrey Y. Komarovskikh, Mikhail M. Syrokvashin, and Michael A. Shestopalov. 2023. "Chemical Diversity of Mo5S5 Clusters with Pyrazole: Synthesis, Redox and UV-vis-NIR Absorption Properties" International Journal of Molecular Sciences 24, no. 18: 13879. https://doi.org/10.3390/ijms241813879
APA StyleSavina, I. V., Ivanov, A. A., Eltsov, I. V., Yanshole, V. V., Kuratieva, N. V., Komarovskikh, A. Y., Syrokvashin, M. M., & Shestopalov, M. A. (2023). Chemical Diversity of Mo5S5 Clusters with Pyrazole: Synthesis, Redox and UV-vis-NIR Absorption Properties. International Journal of Molecular Sciences, 24(18), 13879. https://doi.org/10.3390/ijms241813879