

A Novel Lipid Metabolism and Endoplasmic Reticulum Stress-Related Risk Model for Predicting Immune Infiltration and Prognosis in Colorectal Cancer

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
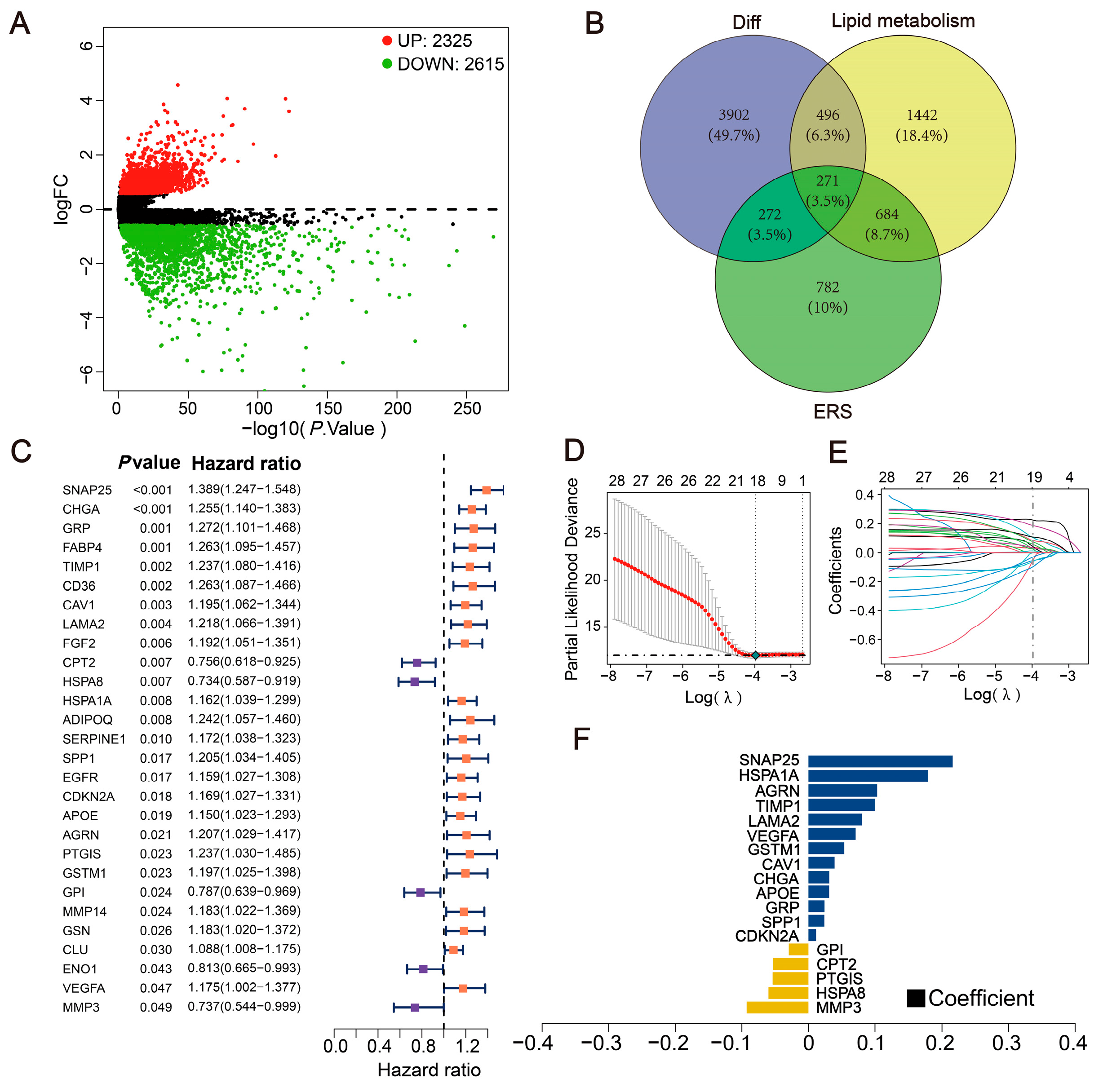
2.1. Exploration of Differentially Expressed LERGs in CRC
2.2. Construction of the Prognostic Model
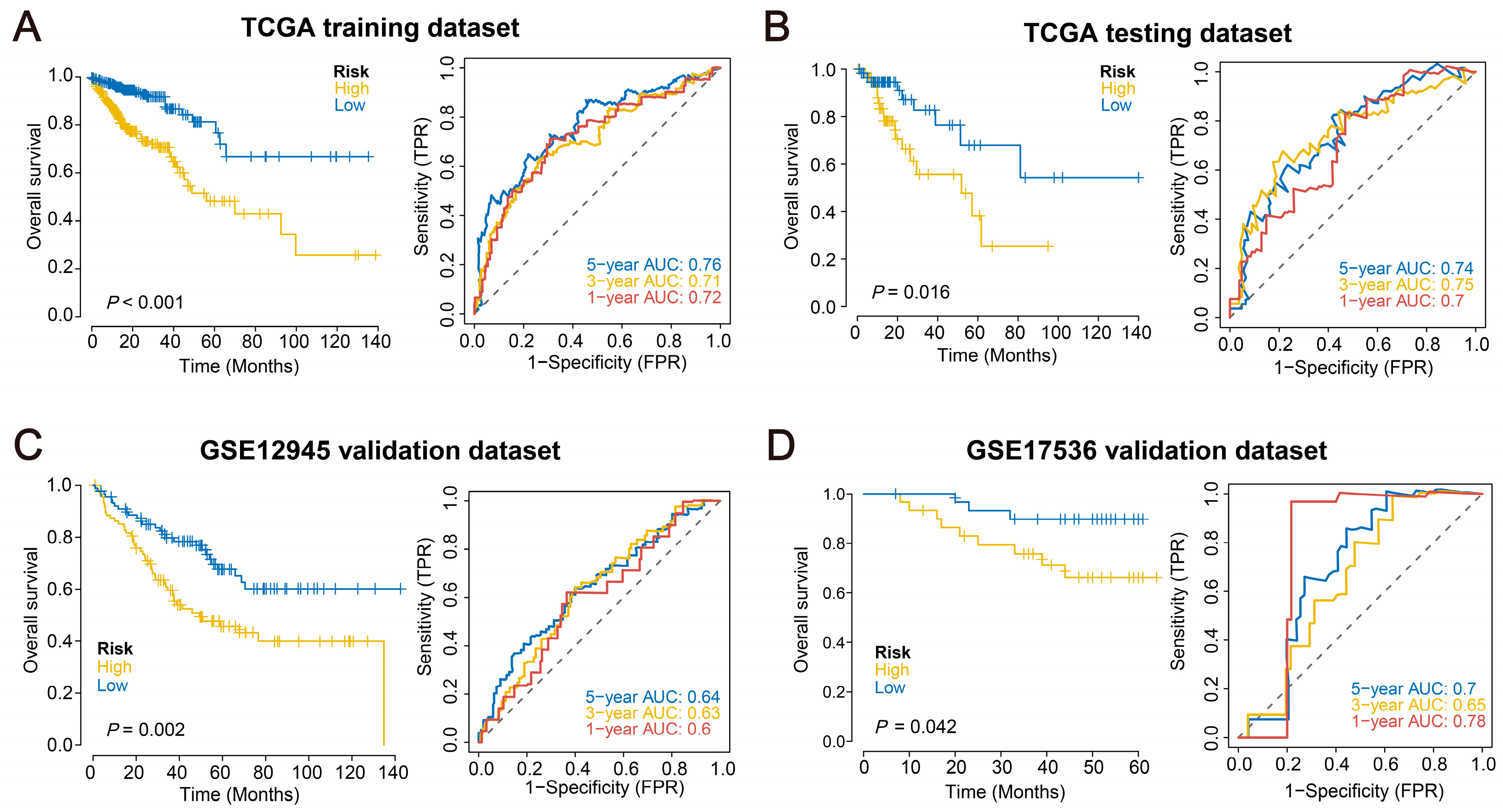
2.3. Validation of the Prognostic Model across Multiple External Datasets
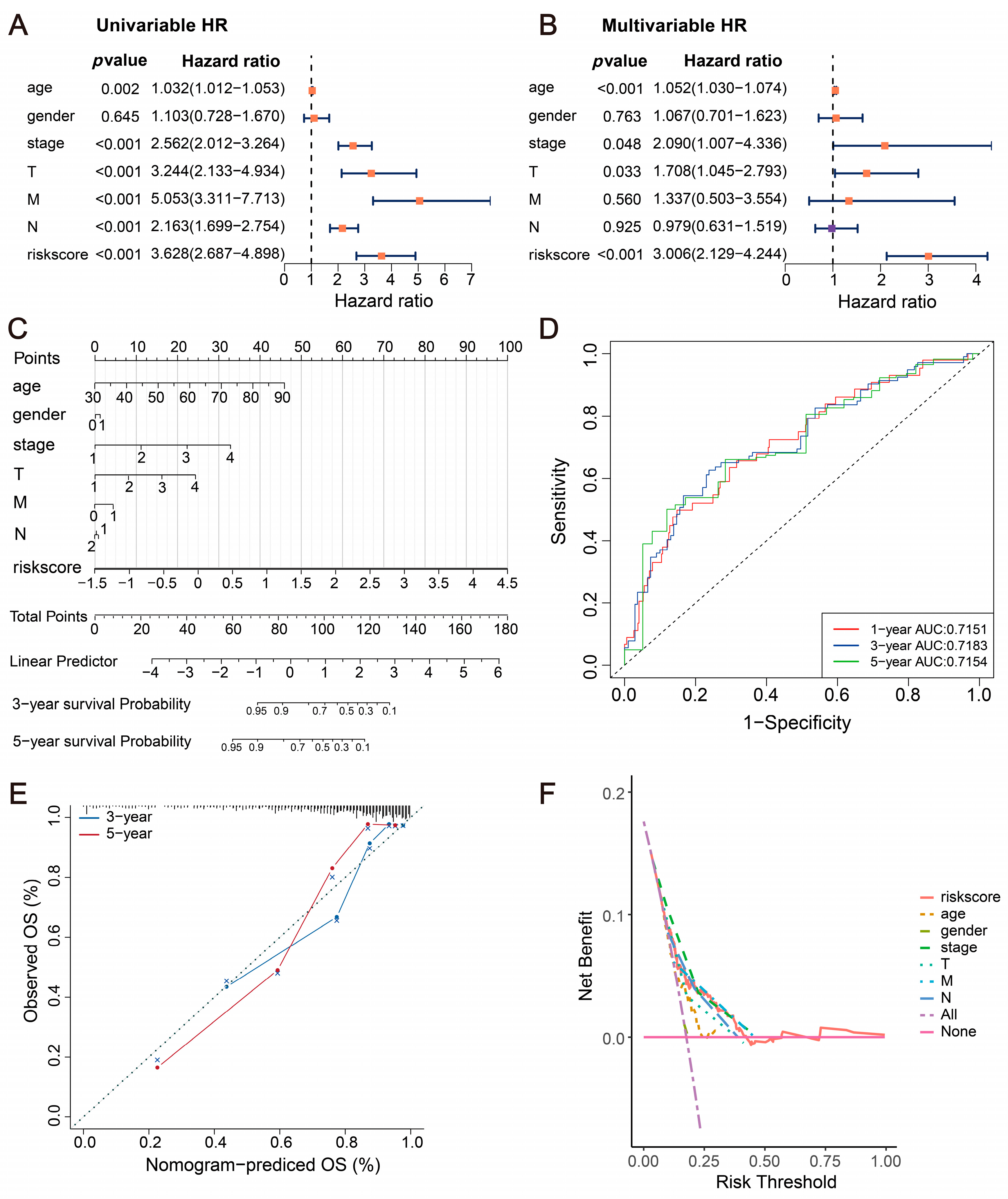
2.4. Incidence Risk and Independent Prognostic Analysis
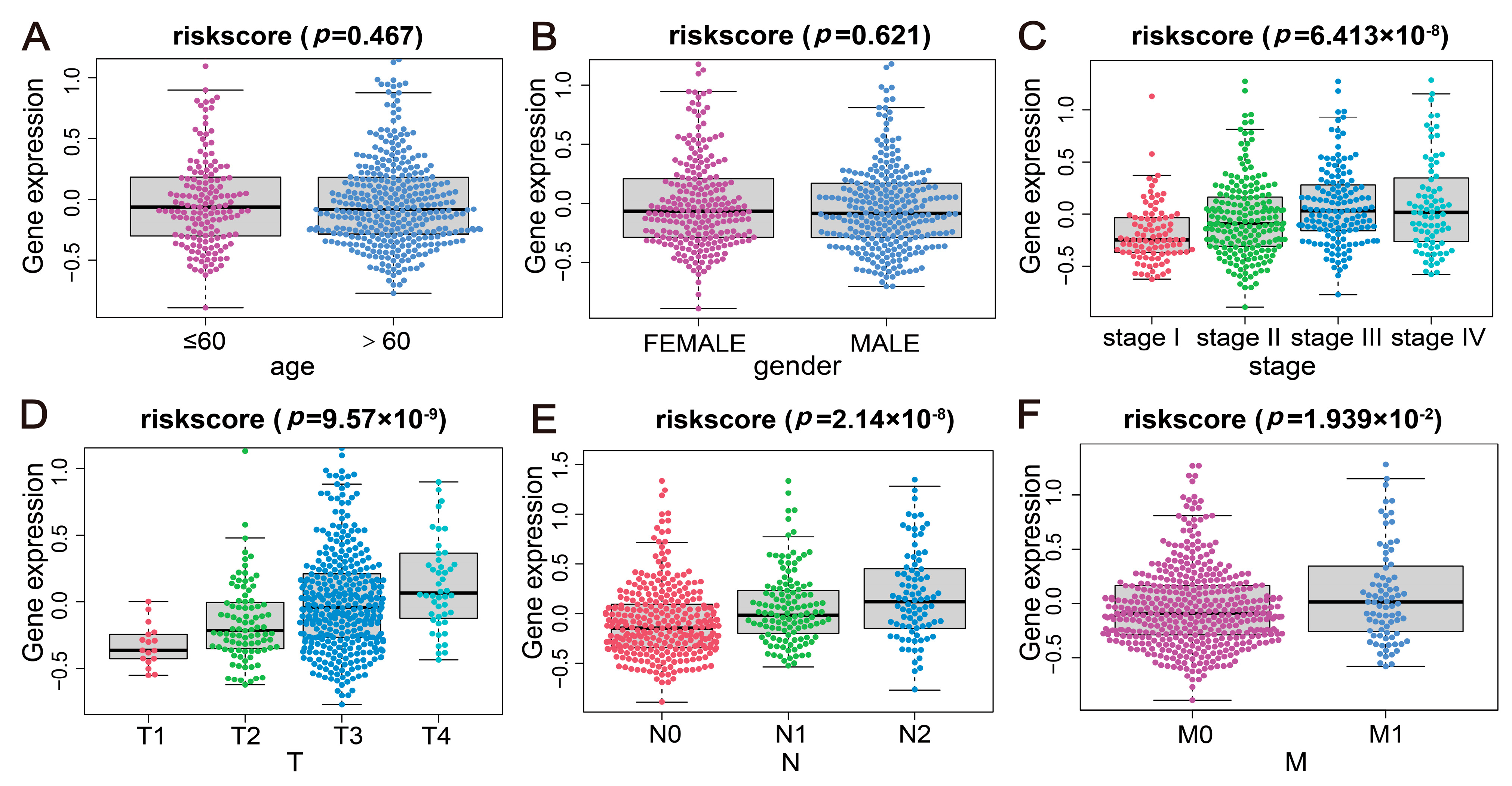
2.5. Correlation Analysis of the Disease Risk Score with Multiple Clinical Indicators
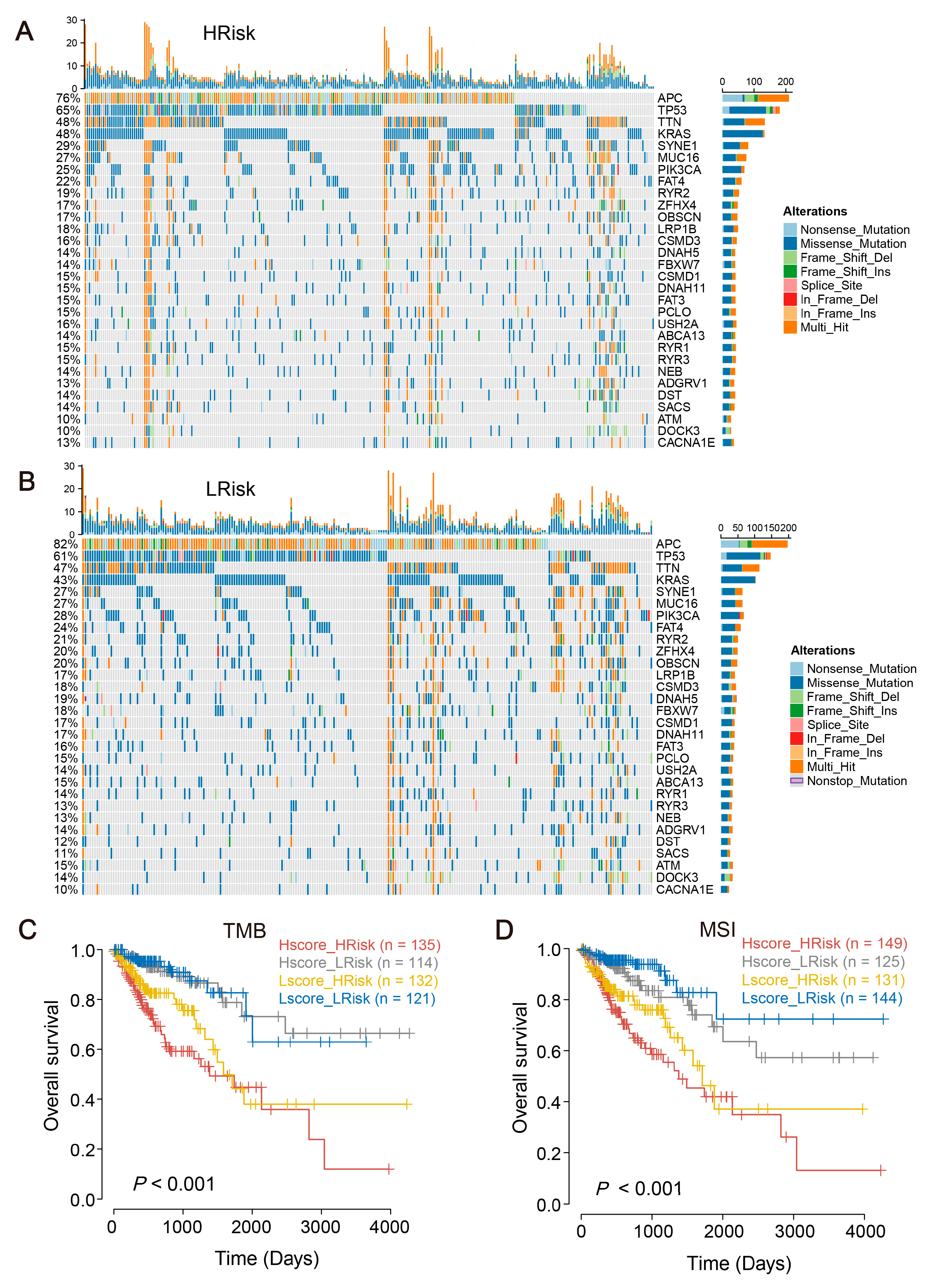
2.6. The Mutational Landscape for CRC and Tumor Mutational Burden (TMB)
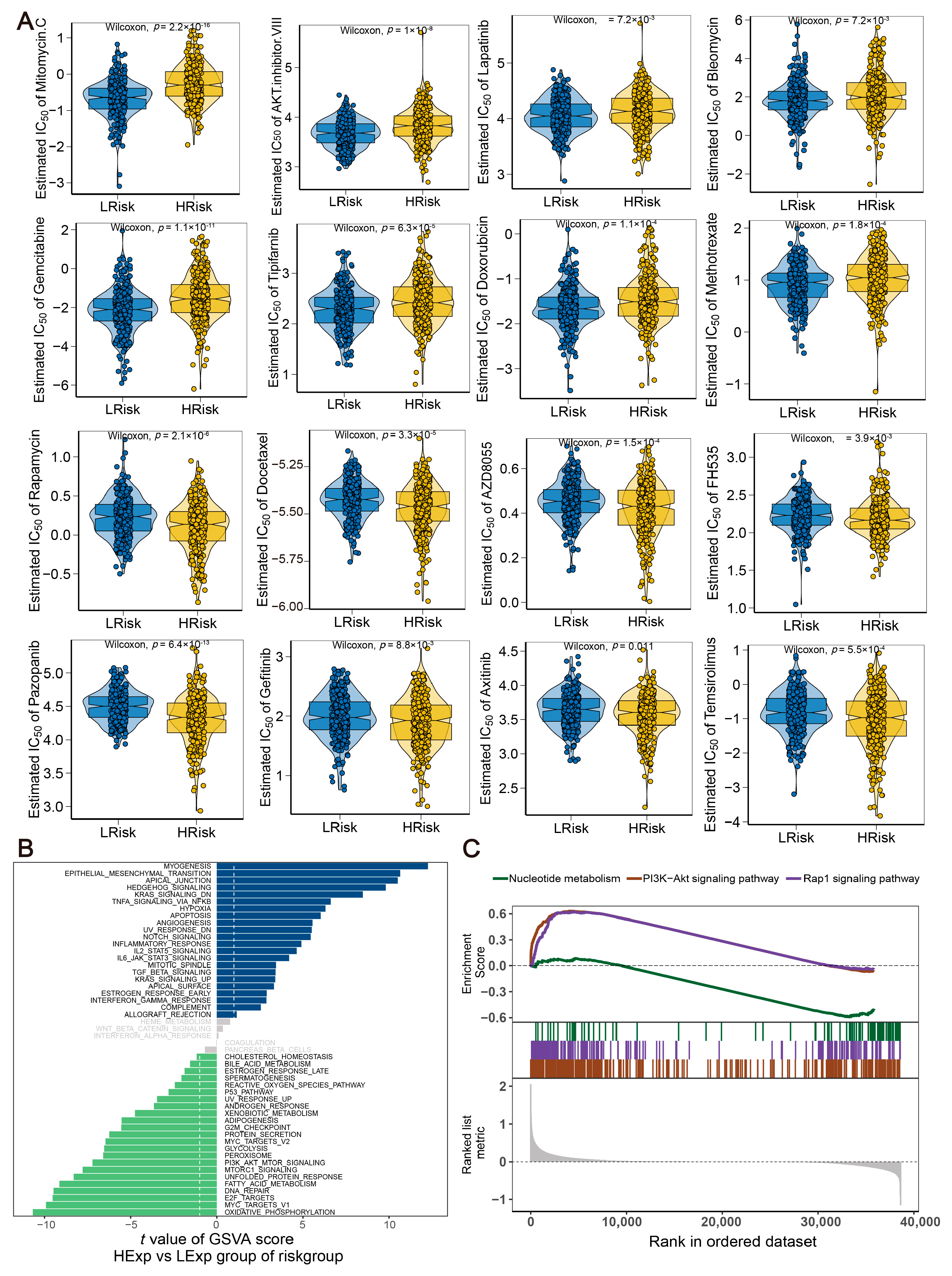
2.7. Drug Sensitivity and Molecular Pathway for the Risk Model
2.8. Relationship between the Risk Model and the Immune Microenvironment
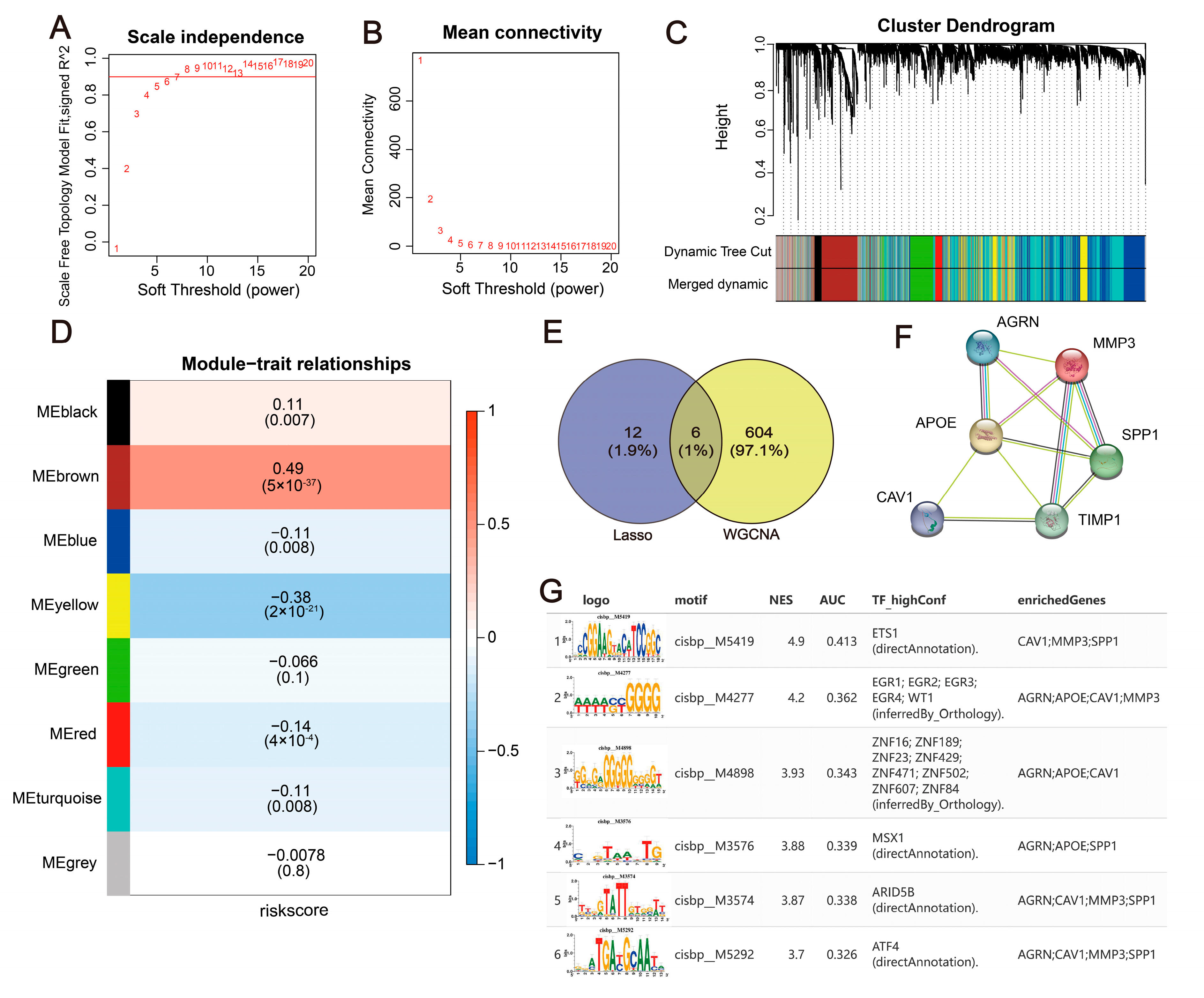
2.9. Construction of a WGCNA Co-Expression Network and Upstream Mechanism Analysis of Hub Genes
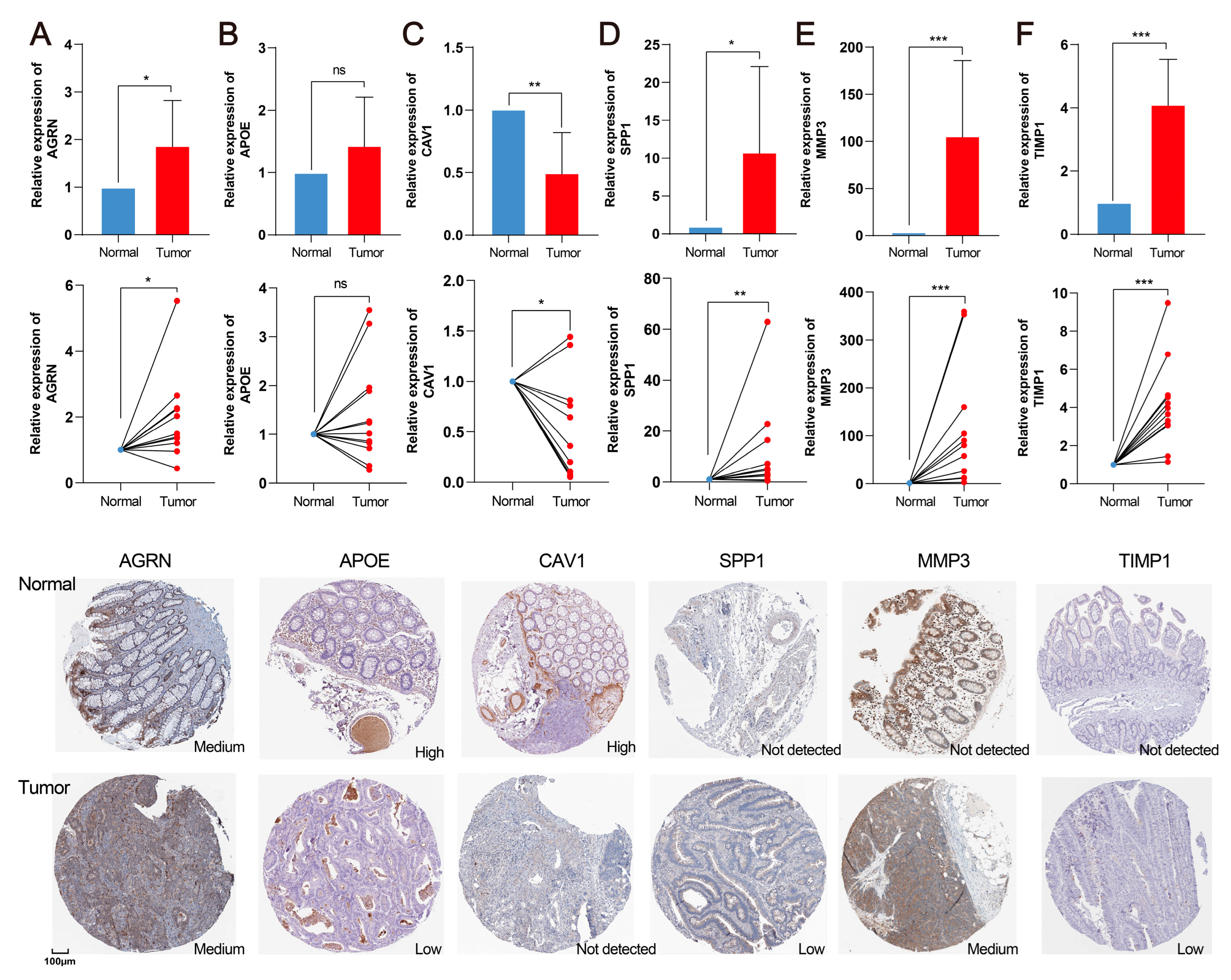
2.10. Validation of Hub Genes in the CRC Clinical Sample
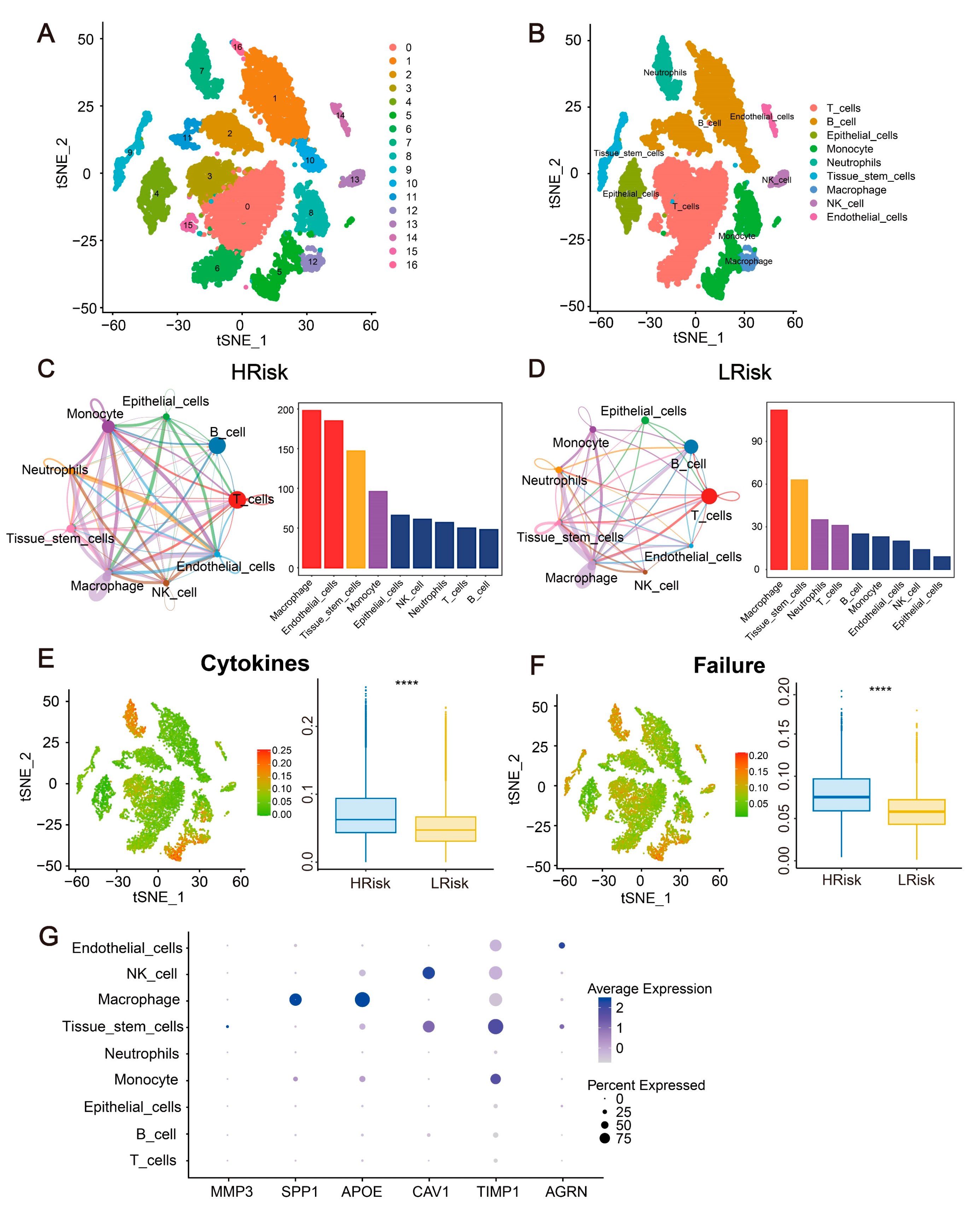
2.11. Single-Cell Analysis for the Immune Landscape and Gene Expression in CRC
3. Discussion
4. Materials and Methods
4.1. Data Sources and Collection
4.2. Construction and Validation of a Risk Model
4.3. Construction of a Predictive Nomogram
4.4. TMB Analysis
4.5. Drug Sensitivity Analysis
4.6. Gene Set Variation Analysis (GSVA) and Enrichment Analysis (GSEA)
4.7. Immune Infiltration Analysis
4.8. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.9. Construction of a PPI Network and Prediction of the Transcription Factor (TF)
4.10. Clinical Samples’ Verification of Hub Genes
4.11. Single-Cell RNA Sequencing Dataset Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Ciardiello, D.; Martini, G.; Napolitano, S.; Tabernero, J.; Cervantes, A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J. Clin. 2022, 72, 372–401. [Google Scholar] [CrossRef]
- Andrei, P.; Battuello, P.; Grasso, G.; Rovera, E.; Tesio, N.; Bardelli, A. Integrated approaches for precision oncology in colorectal cancer: The more you know, the better. Semin. Cancer Biol. 2022, 84, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.; Wang, B.; Wang, X.; Nie, Y.; Fan, D.; Zhao, X.; Lu, Y. Immunotherapy in colorectal cancer: Current achievements and future perspective. Int. J. Biol. Sci. 2021, 17, 3837–3849. [Google Scholar] [CrossRef]
- He, S.; Hu, D.; Feng, H.; Xue, Y.; Jin, J.; Wang, X. Efficacy of immunotherapy with PD-1 inhibitor in colorectal cancer: A meta-analysis. J. Comp. Eff. Res. 2020, 9, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Buikhuisen, J.Y.; Torang, A.; Medema, J.P. Exploring and modelling colon cancer inter-tumour heterogeneity: Opportunities and challenges. Oncogenesis 2020, 9, 66. [Google Scholar] [CrossRef]
- Becht, E.; de Reynies, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Liu, P.H.; Wu, K.; Ng, K.; Zauber, A.G.; Nguyen, L.H.; Song, M.; He, X.; Fuchs, C.S.; Ogino, S.; Willett, W.C.; et al. Association of Obesity with Risk of Early-Onset Colorectal Cancer among Women. JAMA Oncol. 2019, 5, 37–44. [Google Scholar] [CrossRef]
- Yang, M.H.; Rampal, S.; Sung, J.; Choi, Y.H.; Son, H.J.; Lee, J.H.; Kim, Y.H.; Chang, D.K.; Rhee, P.L.; Kim, J.J.; et al. The association of serum lipids with colorectal adenomas. Am. J. Gastroenterol. 2013, 108, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, J. The association of diet, gut microbiota and colorectal cancer: What we eat may imply what we get. Protein Cell 2018, 9, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Ecker, J.; Benedetti, E.; Kindt, A.S.D.; Horing, M.; Perl, M.; Machmuller, A.C.; Sichler, A.; Plagge, J.; Wang, Y.; Zeissig, S.; et al. The Colorectal Cancer Lipidome: Identification of a Robust Tumor-Specific Lipid Species Signature. Gastroenterology 2021, 161, 910–923.e19. [Google Scholar] [CrossRef] [PubMed]
- Ocvirk, S.; O’Keefe, S.J.D. Dietary fat, bile acid metabolism and colorectal cancer. Semin. Cancer Biol. 2021, 73, 347–355. [Google Scholar] [CrossRef]
- Jun, S.Y.; Brown, A.J.; Chua, N.K.; Yoon, J.Y.; Lee, J.J.; Yang, J.O.; Jang, I.; Jeon, S.J.; Choi, T.I.; Kim, C.H.; et al. Reduction of Squalene Epoxidase by Cholesterol Accumulation Accelerates Colorectal Cancer Progression and Metastasis. Gastroenterology 2021, 160, 1194–1207.e28. [Google Scholar] [CrossRef] [PubMed]
- Martin-Perez, M.; Urdiroz-Urricelqui, U.; Bigas, C.; Benitah, S.A. The role of lipids in cancer progression and metastasis. Cell Metab. 2022, 34, 1675–1699. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Liu, B.; Yin, X.; Jiang, G.; Li, Y.; Jiang, Z.; Qiang, L.; Chen, N.; Fan, Y.; Shen, C.; Dai, L.; et al. Identification of Endoplasmic Reticulum Stress-Related Subtypes, Infiltration Analysis of Tumor Microenvironment, and Construction of a Prognostic Model in Colorectal Cancer. Cancers 2022, 14, 3326. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Pillai, S.; Mahmud, I.; Mahar, R.; Griffith, C.; Langsen, M.; Nguyen, J.; Wojtkowiak, J.W.; Swietach, P.; Gatenby, R.A.; Bui, M.M.; et al. Lipogenesis mediated by OGR1 regulates metabolic adaptation to acid stress in cancer cells via autophagy. Cell Rep. 2022, 39, 110796. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fang, D. Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma. Int. J. Mol. Sci. 2021, 22, 1799. [Google Scholar] [CrossRef]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell. Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef]
- Todd, J.L.; Neely, M.L.; Overton, R.; Durham, K.; Gulati, M.; Huang, H.; Roman, J.; Newby, L.K.; Flaherty, K.R.; Vinisko, R.; et al. Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF-PRO Registry. Respir. Res. 2019, 20, 227. [Google Scholar] [CrossRef] [PubMed]
- Costas-Chavarri, A.; Nandakumar, G.; Temin, S.; Lopes, G.; Cervantes, A.; Cruz Correa, M.; Engineer, R.; Hamashima, C.; Ho, G.F.; Huitzil, F.D.; et al. Treatment of Patients With Early-Stage Colorectal Cancer: ASCO Resource-Stratified Guideline. J. Glob. Oncol. 2019, 5, 1–19. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- He, J.; Chen, Z.; Xue, Q.; Sun, P.; Wang, Y.; Zhu, C.; Shi, W. Identification of molecular subtypes and a novel prognostic model of diffuse large B-cell lymphoma based on a metabolism-associated gene signature. J. Transl. Med. 2022, 20, 186. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Li, Z.; Zhong, Y.; Wang, H.; Cheng, X.; Zhao, Y.; Mo, X.; Horbinski, C.; Duan, W.; et al. Ammonia stimulates SCAP/Insig dissociation and SREBP-1 activation to promote lipogenesis and tumour growth. Nat. Metab. 2022, 4, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Meyerhardt, J.A.; Kirkner, G.J.; Chan, A.T.; Kawasaki, T.; Giovannucci, E.L.; Loda, M.; Fuchs, C.S. Cohort study of fatty acid synthase expression and patient survival in colon cancer. J. Clin. Oncol. 2008, 26, 5713–5720. [Google Scholar] [CrossRef] [PubMed]
- Little, J.L.; Wheeler, F.B.; Fels, D.R.; Koumenis, C.; Kridel, S.J. Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Res. 2007, 67, 1262–1269. [Google Scholar] [CrossRef]
- Jin, H.R.; Du, C.H.; Wang, C.Z.; Yuan, C.S.; Du, W. Ginseng metabolite protopanaxadiol interferes with lipid metabolism and induces endoplasmic reticulum stress and p53 activation to promote cancer cell death. Phytother. Res. 2019, 33, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, W.; Ma, Y.; Zhao, X.; He, L.; Sun, P.; Wang, H. High-fat diet aggravates colitis-associated carcinogenesis by evading ferroptosis in the ER stress-mediated pathway. Free Radic. Biol. Med. 2021, 177, 156–166. [Google Scholar] [CrossRef]
- Patel, O.; Clyde, D.; Chang, M.; Nordlund, M.S.; Steel, R.; Kemp, B.E.; Pritchard, D.M.; Shulkes, A.; Baldwin, G.S. Pro-GRP-derived peptides are expressed in colorectal cancer cells and tumors and are biologically active in vivo. Endocrinology 2012, 153, 1082–1092. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, W.; Shang, W. Identification and validation of a prognostic risk-scoring model based on sphingolipid metabolism-associated cluster in colon adenocarcinoma. Front. Endocrinol. 2022, 13, 1045167. [Google Scholar] [CrossRef]
- Lichao, S.; Liang, P.; Chunguang, G.; Fang, L.; Zhihua, Y.; Yuliang, R. Overexpression of PTGIS could predict liver metastasis and is correlated with poor prognosis in colon cancer patients. Pathol. Oncol. Res. 2012, 18, 563–569. [Google Scholar] [CrossRef]
- Liu, F.; Li, X.; Yan, H.; Wu, J.; Yang, Y.; He, J.; Chen, J.; Jiang, Z.; Wu, F.; Jiang, Z. Downregulation of CPT2 promotes proliferation and inhibits apoptosis through p53 pathway in colorectal cancer. Cell. Signal. 2022, 92, 110267. [Google Scholar] [CrossRef]
- Song, L.; Yang, C.; He, X.F. Individual and combined effects of GSTM1 and GSTT1 polymorphisms on colorectal cancer risk: An updated meta-analysis. Biosci. Rep. 2020, 40, BSR20201927. [Google Scholar] [CrossRef]
- Lepine, S.; Allegood, J.C.; Park, M.; Dent, P.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2011, 18, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e18. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Yang, X.; Jia, X.; Rong, Y.; Chen, L.; Zeng, T.; Deng, X.; Li, W.; Wu, G.; Wang, L.; et al. CAV1-CAVIN1-LC3B-mediated autophagy regulates high glucose-stimulated LDL transcytosis. Autophagy 2020, 16, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; Ortiz-Sandoval, C.; Navarro-Marquez, M.; Rodriguez, A.E.; Diaz-Valdivia, N.; Sanhueza, C.; Lopez-Crisosto, C.; Tahbaz, N.; Rothermel, B.A.; et al. Caveolin-1 impairs PKA-DRP1-mediated remodelling of ER-mitochondria communication during the early phase of ER stress. Cell Death Differ. 2019, 26, 1195–1212. [Google Scholar] [CrossRef]
- Revel, M.; Sautes-Fridman, C.; Fridman, W.H.; Roumenina, L.T. C1q+ macrophages: Passengers or drivers of cancer progression. Trends Cancer 2022, 8, 517–526. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, Z.; Li, B.; Li, J.; Ou, Y.; Yu, X.; Zhang, Z.; Liu, S.; Fu, X.; Jin, H.; et al. Lipid-associated macrophages in the tumor-adipose microenvironment facilitate breast cancer progression. Oncoimmunology 2022, 11, 2085432. [Google Scholar] [CrossRef]
- Dai, X.Y.; Song, A.Y.; Mu, L.; Wang, L.J.; Huang, L.Q. Potential Function of MMP3Gene in Degradation of Extracellular Matrix Complex in Colorectal Carcinoma. Biomed. Environ. Sci. 2021, 34, 66–70. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Chen, L. TIMP1 represses sorafenib-triggered ferroptosis in colorectal cancer cells by activating the PI3K/Akt signaling pathway. Immunopharmacol. Immunotoxicol. 2023, 45, 419–425. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Sun, X.L.; Wang, Y.L.; Miao, Y.L. Agrin promotes the proliferation, invasion and migration of rectal cancer cells via the WNT signaling pathway to contribute to rectal cancer progression. J. Recept. Signal Transduct. Res. 2021, 41, 363–370. [Google Scholar] [CrossRef]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2022, 80, 1–17. [Google Scholar] [CrossRef]
- Shah, S.; Brock, E.J.; Ji, K.; Mattingly, R.R. Ras and Rap1: A tale of two GTPases. Semin. Cancer Biol. 2019, 54, 29–39. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Shin, S.J.; Kim, S.Y.; Lee, S.H.; Park, Y.S.; Park, S.H.; Lee, K.H.; Kim, T.W.; Hong, Y.S.; Ahn, J.B. Combining capecitabine, oxaliplatin, and gemcitabine (XELOXGEM) for colorectal carcinoma patients pretreated with irinotecan: A multicenter phase I/II trial. Cancer Chemother. Pharmacol. 2012, 69, 91–97. [Google Scholar] [CrossRef]
- Narayanankutty, A. PI3K/Akt/mTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Di Conza, G.; Tsai, C.H.; Gallart-Ayala, H.; Yu, Y.R.; Franco, F.; Zaffalon, L.; Xie, X.; Li, X.; Xiao, Z.; Raines, L.N.; et al. Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat. Immunol. 2021, 22, 1403–1415. [Google Scholar] [CrossRef]
- Tumor-Secreted Lipids Induce ER Stress Response to Polarize Macrophages. Cancer Discov. 2022, 12, OF5. [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Xia, B.; Wang, J.; Qi, S.; Jing, W.; Deng, K.; Yang, J. A Novel Lipid Metabolism and Endoplasmic Reticulum Stress-Related Risk Model for Predicting Immune Infiltration and Prognosis in Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 13854. https://doi.org/10.3390/ijms241813854
Jin H, Xia B, Wang J, Qi S, Jing W, Deng K, Yang J. A Novel Lipid Metabolism and Endoplasmic Reticulum Stress-Related Risk Model for Predicting Immune Infiltration and Prognosis in Colorectal Cancer. International Journal of Molecular Sciences. 2023; 24(18):13854. https://doi.org/10.3390/ijms241813854
Chicago/Turabian StyleJin, Haoran, Bihan Xia, Jin Wang, Shaochong Qi, Weina Jing, Kai Deng, and Jinlin Yang. 2023. "A Novel Lipid Metabolism and Endoplasmic Reticulum Stress-Related Risk Model for Predicting Immune Infiltration and Prognosis in Colorectal Cancer" International Journal of Molecular Sciences 24, no. 18: 13854. https://doi.org/10.3390/ijms241813854
APA StyleJin, H., Xia, B., Wang, J., Qi, S., Jing, W., Deng, K., & Yang, J. (2023). A Novel Lipid Metabolism and Endoplasmic Reticulum Stress-Related Risk Model for Predicting Immune Infiltration and Prognosis in Colorectal Cancer. International Journal of Molecular Sciences, 24(18), 13854. https://doi.org/10.3390/ijms241813854