1. Introduction
A growing body of research indicates a significant increase in the number of genes implicated in neurodevelopmental conditions altering several molecular pathways that involve various cellular processes and signaling networks that play critical roles in brain development and function. Although some serine/threonine protein phosphatases play a critical role in the control of cell function, they have barely been associated with developmental disorders [
1]. Within the cell, there are two major families of serine/threonine (Ser/Thr) protein phosphatases: protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A). PP2A is a heterotrimeric enzyme [
2,
3], composed of a catalytic subunit (C), a substrate-binding regulatory subunit (B), and a scaffolding subunit (A) that links the regulatory and catalytic subunits. The scaffolding A and the regulatory B subunits are encoded by the
PPP2R1A and
PPP2R5D genes, respectively [
2,
3].
Recently, associations between de novo missense pathogenic variants of PPP2R5D, PPP2R1A, and PPP2CA have been reported, leading to autosomal-dominant forms of intellectual disability (MRD35 (OMIM#616355), MRD36 (OMIM#616362), and NDLBA (OMIM#618354), respectively, also known as Houge-Janssen syndrome 1,2, and 3) [
1,
4]. In 2015, in the context of the Deciphering Developmental Disorders (DDD) study, five de novo
PPP2R1A mutations were identified among 1133 parent–child trios [
1]. The PPP2R1A protein is composed of 15 HEAT (huntingtin, elongation factor 3, protein phosphatase 2A, and yeast kinase TOR1) repeat motifs, of which HEATs 1–8 mediate interactions with a specific regulatory B subunit [
4]. Among the 46 patients reported to date, forty-five showed pathogenic variants that accumulate in HEATs 4–7 [
1,
5,
6,
7,
8,
9,
10], generating a dominant negative effect, and causing biochemical dysfunction in the majority of them [
1,
7]. The subjects reported with
PPP2R1A-related neurodevelopmental disorder (NDD) show a consistent neurological phenotype (corpus callosum hypoplasia, epilepsy, moderate-to-severe intellectual disability, and ventriculomegaly) but the clinical spectrum is expanding and extraneurological features, such as congenital heart disease, have recently been reported [
8]. Remarkably, in two patients bearing the same
PPP2R1A variants at HEAT5, in the presence of severe ventriculomegaly and a very extensively affected central nervous system, abnormal posterior fossa structures were observed, showing cerebellar and brainstem hypoplasia [
5,
8]. Both infants died prematurely and showed associated perinatal epilepsy. However, isolated pontocerebellar hypoplasia (PCH) as a cardinal sign has not been reported.
PCH comprises a group of clinically and genetically heterogeneous rare neurodegenerative disorders with a prenatal onset, characterized by abnormal growth and survival of neurons in the cerebellum, inferior olives, and ventral pons. Radiologically and pathologically, all PCH subtypes are characterized by hypoplasia and variable atrophy of the cerebellum and pons. Currently, pathogenic variants in at least 19 different genes explain different clinical subtypes of PCH based on distinct clinical, biochemical, and radiological features [
11,
12,
13,
14,
15]. Proteins encoded by PCH-associated genes are involved in various cellular functions, predominantly RNA metabolism and protein expression, but also nucleotide metabolism, mitochondrial function, and vesicular transport [
13,
14,
15]. Individuals affected by PCH share clinical features, such as prenatal or perinatal onset of disease, progressive microcephaly and global developmental delay with severe intellectual and motor function impairments, epilepsy, and, frequently, death during childhood. However, recent subtypes have been related to a mild, non-degenerative disease course [
16]. To date, no mutations in the protein phosphatase have been reported as causing PCH.
The present study aims to report a novel variant in a region of PPP2R1A where mutations have not been reported so far, leading to an undescribed phenotype, which shows PCH, congenital microcephaly, optic and peripheral nerve abnormalities, and an absence of some expected features such as epilepsy and an abnormal corpus callosum. To further establish the pathogenicity of the variant, comprehensive investigations were conducted, including in silico studies, confocal microscopy, and computational super-resolution microscopy functional studies. These involved a comparative analysis of the patient’s fibroblasts with both healthy control cells and cells from an individual with the previously described phenotype.
3. Discussion
The neurodevelopmental disorders related to PP2A dysfunction encompass a group of overlapping syndromes characterized by severe PP2A dysfunction. The phenotypic features related to the described neurodevelopmental syndrome due to variants in
PPP2R1A are neurodevelopmental and language delay, hypotonia, behavior problems, frontal bossing, a long face, joint hypermobility, and hypoplasia or agenesis of the corpus callosum, which are present in more than 50% of patients [
7].
To date, two reports describe subjects with variants in
PPP2R1A and associated PCH. The first one was reported by Wallace et al. [
5] describing an infant bearing a previously reported
PPP2R1A variant (c.548G > A; p.Arg183Gln) with severe ventriculomegaly and severe neuroanatomical distortion of both supratentorial and infratentorial structures and presenting epilepsy during the first days of life. Baker et al. reported the second one [
8], also presenting with severe ventriculomegaly, corpus callosum agenesis, and hypoplastic brainstem with vermian hypoplasia. This second baby showed the same variant and presented with epilepsy during the first days of life, too. In fact, p.Arg183Gln has been associated with the most severe phenotype. Additionally, in a murine model, it has been associated with the most profound binding deficiency [
20], as also found in the biochemical characterization reported by Lenaerts et al. [
7]. Both subjects presented very premature death, and posterior fossa structure abnormalities at least partly affected by the severe ventriculomegaly. Unfortunately, a third patient with ventriculomegaly and posterior fossa abnormalities was incompletely described, since the pathogenic variant in
PPP2R1A is not detailed at all [
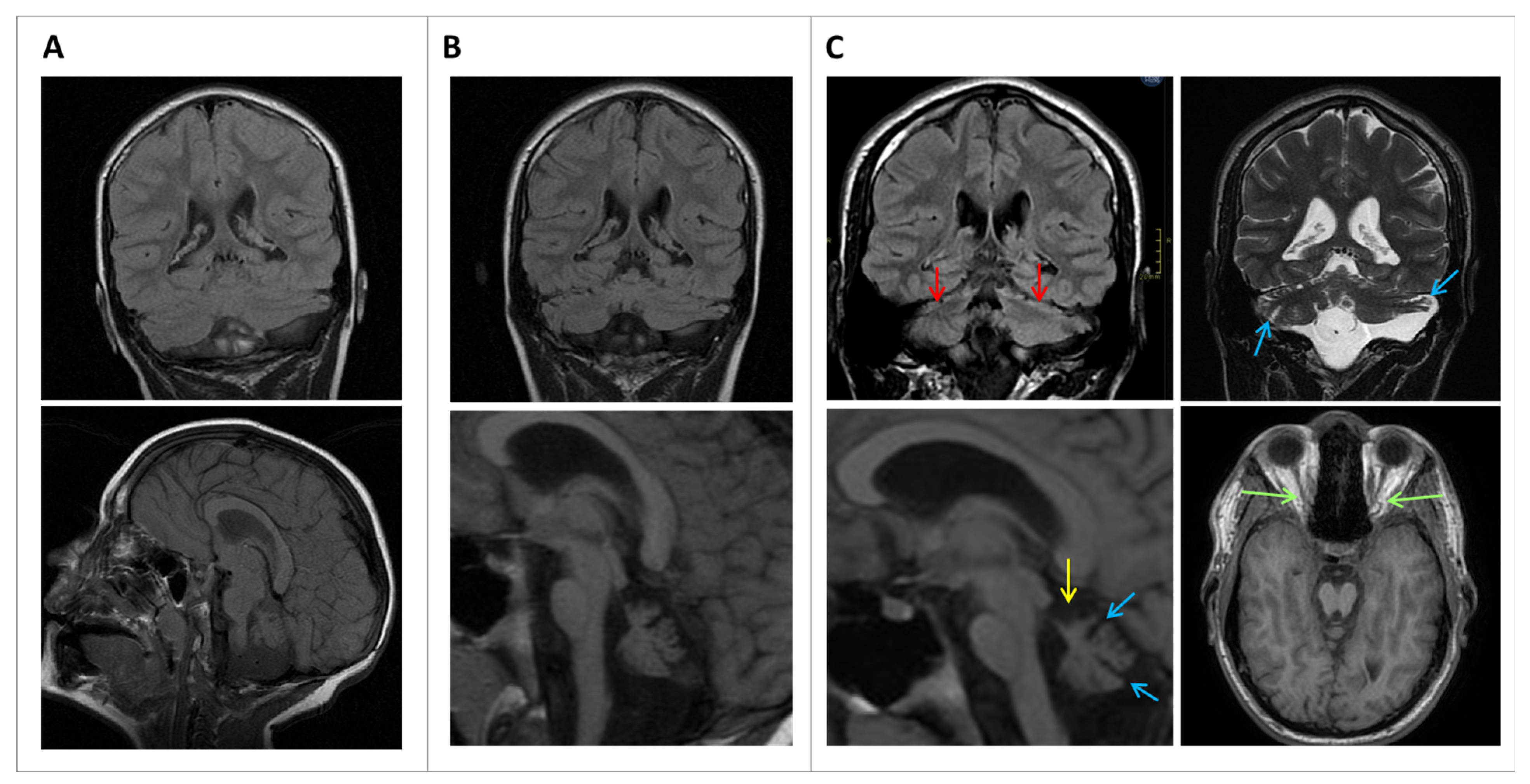
10]. In the present study, the S1 proband presented with isolated PCH on neuroimaging, pointing towards an initial malformative rhombencephalic basis and not a secondary disruptive abnormality, in the context of a generalized central nervous system alteration. Moreover, optic nerve atrophy and peripheral nerve implications have never been reported in relation to
PPP2R1A.
Cerebellar hypoplasia and atrophy are common in neuropediatrics, occurring in a very heterogeneous group of disorders and frequently as a nonspecific finding. However, the coexistence of midbrain hypoplasia narrows down the diagnostic options. Initially, since S1 presented with microcephaly and PCH, and in the absence of a sentinel event in their personal history explaining a possible non-genetic but disruptive cause, the diagnostic search was focused on classical PCH-related genes, without success. From a clinical perspective, the most similar PCH type is PCH3, which has been particularly associated with optic nerve atrophy and an increase in DTR. However, the molecular basis of PCH3 is biallelic variants in the
PCLO gene, encoding a protein related to the regulation of vesicle formation and synaptic vesicle trafficking, but the sequencing of
PCLO was normal in S1. PCH or neurogenetic disorders causing pontocerebellar hypoplasia are a growing group of genetic conditions [
11], and new genes or new phenotypes related to already known genes will probably be discovered in the near future. Since the finding of a
PPP2R1A variant in S1 was unexpected, a more detailed revision of the sequences of all those genes related to PCH and diseases associating cerebellar and brainstem hypoplasia was performed. Moreover, studies of repeat expansion mutations were also performed, including early-onset SCAs. All other filtered variants found in the WES were carefully reviewed, in an attempt to rule out any contribution explaining or distorting the phenotype.
All previously reported pathogenic variants in
PPP2R1A (seventeen to date) are clustered close to the regulatory subunit-binding region of the protein, accumulating in HEATs 4–7 [
1,
4,
5,
6,
7,
8,
9] (
Figure 2,
Supplementary Table S1) and causing single-amino-acid substitution. These variants generate a dominant negative effect and there is compelling evidence that the severity of the phenotypic spectrum correlates with biochemical dysfunctions [
7]. S1 presented a missense mutation outside of the reported regions, leading to a replacement of the amino acid arginine (basic side chain, positively charged) with the amino acid cysteine (polar, uncharged side chain) at residue 21. The high evolutionary conservation of this altered residue supports its pathogenicity. Notably, the whole
PPP2R1A protein sequence shows high conservation, especially among vertebrata and, for this particular residue, in other organisms, lysine (K), also basic and positively charged, is found. Moreover, in silico analysis of previous variants and the Arg21Cys mutation showed that it is located closer to the catalytic subunit, which is certainly outside of the described cluster of mutations, which are closer to the regulatory subunit. These in silico studies also show that Arg21 seems to form hydrogen bonds with the HEAT2 alpha helix, and could play a role in 3D structure maintenance. These bonds are lost in the presence of cysteine, suggesting that this change can affect protein folding, which could be in agreement with the increase in protein aggregates observed in the S1 patient’s fibroblasts. Moreover, in silico analysis of previous variants and the S1 mutation showed that it is located close to the catalytic subunit, which is certainly outside of the described cluster of mutations, which are closer to the regulatory subunit. This atypical localization may explain the unexpected clinical phenotype, but in silico studies are not enough to support pathogenicity. In this context, functional studies currently play a crucial role in establishing the connection between new variants and emerging phenotypes. Immunofluorescent labeling denoting the expression of
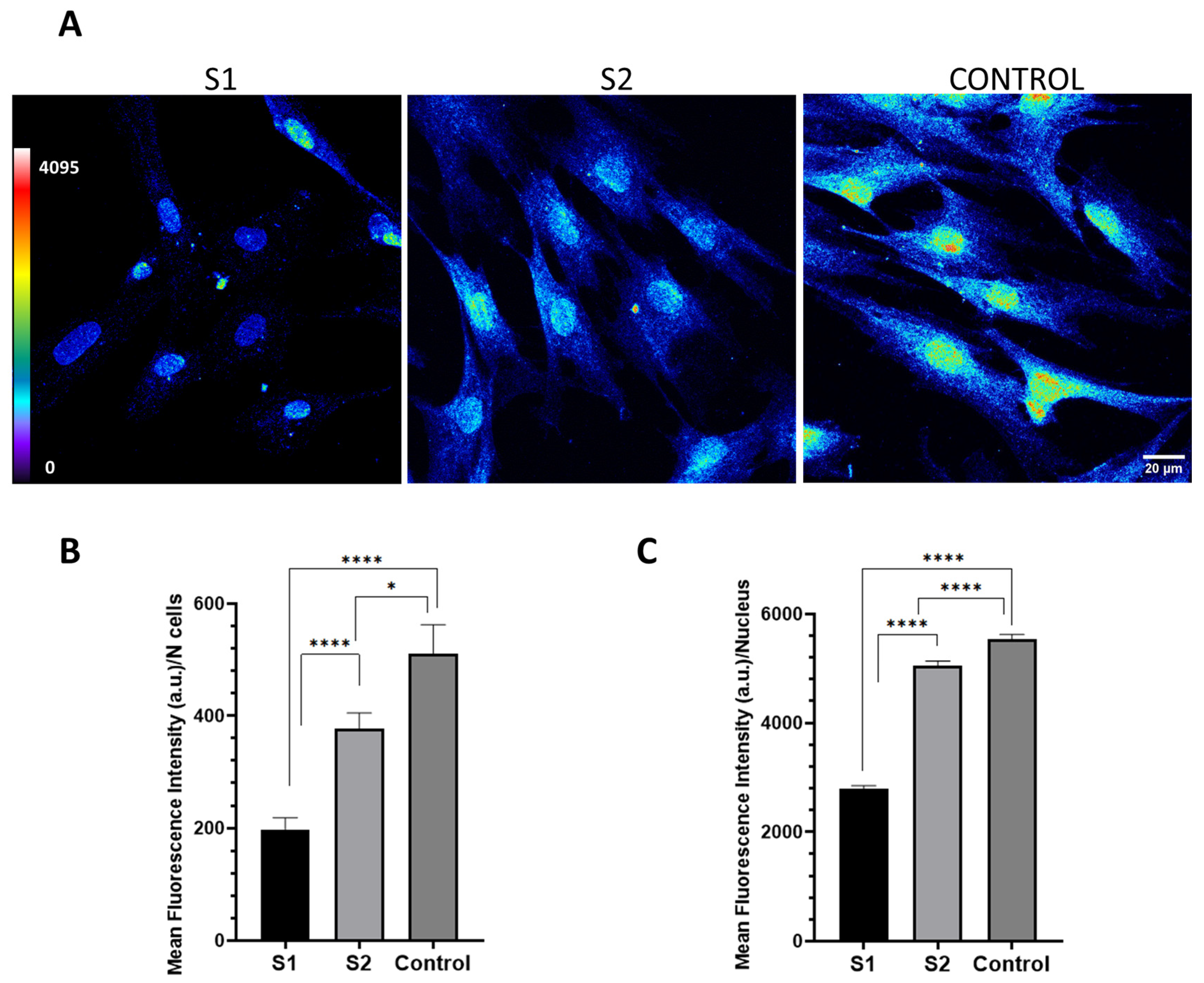
PPP2R1A in the cytoplasm depicts the abundance of PPP2R1A, which was clearly greater in the control cells than in S2, and in turn, greater than S1, bearing the unreported variant.
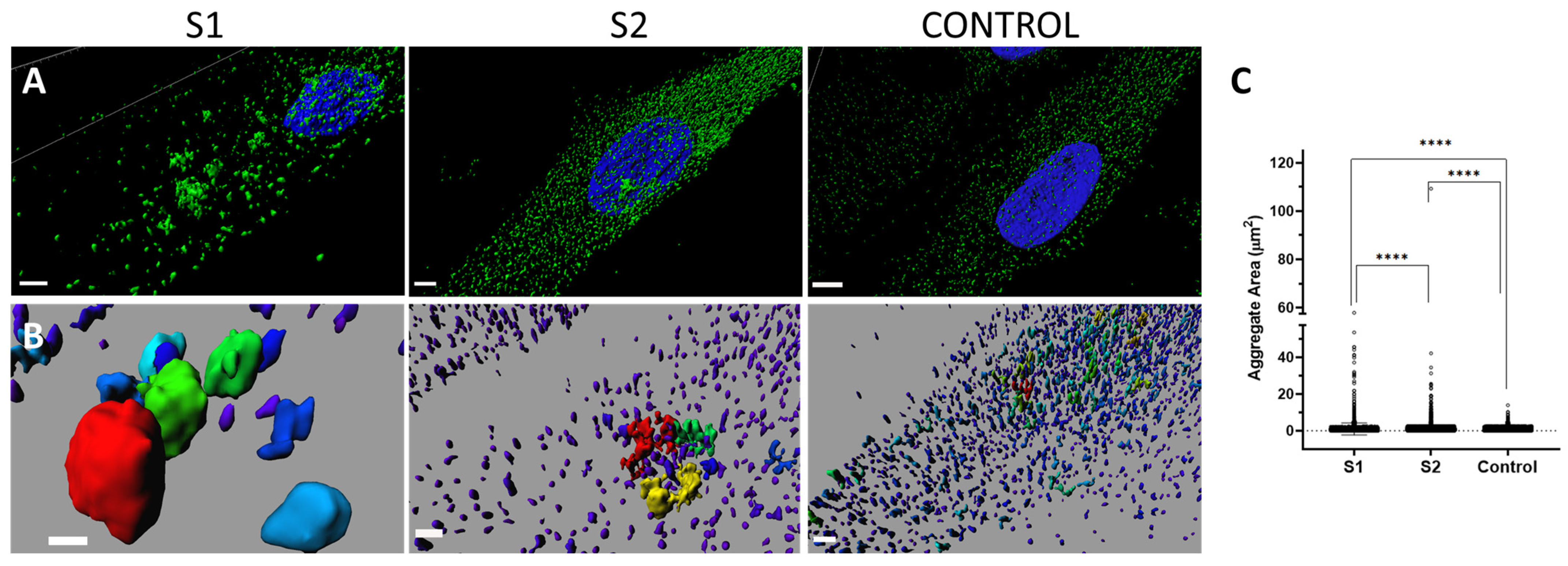
Computational super-resolution microscopy revealed the presence of aberrant protein aggregates in the cytoplasm of S1′s fibroblasts, abnormal due to their size (moderate and large aggregates) but also due to the huge variability in size. When they were quantified, progression among the cells of S1, S2, and the control was identified, showing that S1′s cells present greater abnormalities than S2′s cells, which showed, in turn, greater abnormalities than the control cells. Taken together, these findings point to a greater distortion or cellular effect related to the new variant. In summary, from a cellular point of view, greater abnormalities have been demonstrated in S1 than in S2, who presents the already known phenotype. Although global severity measured using Lanaerts’ score is similar for both [
7], S1 also has peripheral nerve implications and optic nerve atrophy, which were not included in the proposed score design.
Regarding the biological significance of protein aggregates, they are subcellular perinuclear structures in which misfolded proteins accumulate through retrograde transport on microtubules [
21], and highly dynamic processes are involved in their formation [
22]. The presence of relatively large protein aggregates in any cell type indicates an impairment of the protein elimination machinery, compromising proteostatic homeostasis and leading to cellular stress and dysfunction. Additionally, the fact that the aggregates are relatively large affects the localization and movement/dynamics of other proteins and cellular organelles (differences compared to the control might be found if we were to study different cellular organelles). This is likely to result in the disruption of cellular processes, particularly intracellular transport (which can have significant implications at the neurological level). Furthermore, the presence of aggregates that can affect cellular organelles will also induce oxidative stress on proteins, lipids, and nucleic acids, triggering specific cellular responses that may ultimately lead to inflammatory reactions during the course of the disease. In general, the presence of large aggresomes in cells can trigger a series of cellular responses and compensatory mechanisms to counteract their detrimental effects. However, if these responses are insufficient, or if aggresomes continue to accumulate, they can contribute to the deterioration of cellular function and may be associated with neurodegenerative diseases [
23] such as PCH, originally reported as a disorder of prenatal onset neurodegeneration, and as observed, from a neuroradiological point of view, in S1, showing greater cerebellar atrophy with age. Interestingly, S1 has not presented any regression or worsening from a clinical point of view. This is like some newly reported PCH types that behave in a stable manner, despite the common rule that the degenerative nature of PHC frequently leads to premature death [
11,
12,
13,
14,
15].
It is important to consider that increased cellular resistance to proteotoxicity in patients may not be due solely to a lower level of protein aggregates, but rather, to the more efficient management of protein aggregates by the cell through the formation of inclusion bodies resembling aggresomes [
23], as may be the case with our patient S1.
Although the functioning of PP2A is not well understood, it is well established as a regulator of cell division, growth, and differentiation, and the functions of PP2A in cancer and various neurodegenerative disorders, such as Alzheimer’s disease, have been studied in detail; for PPP2R1A, the crucial mechanism is determined by an alteration in the dependent dephosphorylation dynamics [
24].
Neurological disease diagnosis has progressively evolved towards a molecular definition, but gene characterization still presents a challenge, which complicates the diagnostic process. Advances in Next-Generation Sequencing (NGS) techniques have greatly facilitated the identification of novel phenotypes associated with previously known genes and new variants in genes, shedding light on previously unreported phenotypes. Currently, functional studies play a crucial role in establishing the connection between new variants and emerging phenotypes. Although advanced optical microscopy techniques have been used extensively in research, their potential in diagnostic applications is yet to be fully explored. Their speed, ease of use, simple sample preparation, and relatively affordable instrumentation compared to electron microscopy make them potentially more accessible to clinicians and suitable for routine diagnosis. Collaboration between clinicians and researchers performing exhaustive phenotyping, NGS, and functional validations may prove essential to uncovering new molecular causes of PCH, as well as new phenotypes related to recently reported pathogenic genes, such as PPP2R1A.
To summarize, hypoplasia of the cerebellum and the brainstem has been only rarely associated with PPP2R1A-associated NDD. When present, it has been linked to the most severe phenotype, presenting a recurrent variant. We report an individual with a novel PPP2R1A variant and PCH in the context of a stable clinical condition, and not showing other typical features of PPP2R1A-associated NDD, while presenting optic atrophy and peripheral neuropathy. These findings suggest a potential association between PPP2R1A variants and PCH, expanding the clinical spectrum of neurodevelopmental disorders associated with PPP2R1A. Further studies and descriptions of additional patients are needed to improve the understanding of the genotype–phenotype correlation and the underlying mechanisms of this novel phenotype.
4. Materials and Methods
4.1. Phenotype and Molecular Studies
Two individuals bearing variants in
PPP2R1A were studied. The first subject (S1) presented a variant in a not-typical region of
PPP2R1A, while the second subject (S2) had a variant in the region where previous variants have been reported and her clinical features were recently published [
7]. Exhaustive phenotyping was performed, including studies of dysmorphology and general and neurological exams. For Subject 1 (S1), conventional MRI (1.5 T scanner, GE Healthcare, Milwaukee, WI, USA) was performed at the ages of 6 years, 10 years, and 16 years. A nerve conduction velocity (NCV) study was performed at the age of 10 years via standard techniques using a Nantus Key Point Focus electromyograph with surface electrode recordings.
Blood samples were collected from both individuals and their parents, after obtaining written informed consent. Both individuals underwent chromosomal microarray analysis. Additionally, for S1, PCH panel gene, trio-based WES and testing for nucleotide repeat expansions (to rule out other potential variants in early-presenting spinocerebellar ataxias genes (SCA)) were performed. For S2, after normal chromosomal microarray analysis, a WES trio was performed.
4.2. In Silico Studies
In silico modeling of the multimeric protein was performed using the PyMol Molecular Graphics System (v.2.4.1, Schrödinger, LLC, 1540 Broadway, NY, USA) with the Protein Phosphatase 2A Holoenzyme model (2NPP) [
18]. For conservation analysis, human
PPP2R1A was used as a BLAST query and model organisms were selected. Multiple protein alignment was performed using Clustal Omega, EMBL-EBI ver. 1.2.4
https://www.ebi.ac.uk/Tools/msa/clustalo/ accessed on 9 April 2023.
4.3. Functional Studies
Immunofluorescent labeling of PPP2R1A in the patients’ fibroblasts to locate and quantify this protein was performed. Fibroblasts from a healthy control (12-year-old boy) were obtained from the Sant Joan de Déu Barcelona Children’s Hospital Biobank. These cells were cultured in Dulbecco’s modified Eagle medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% v/v fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, USA), 2 mmol/L L-glutamine (Sigma-Aldrich, St. Louis, MO, USA), and 100 mg/mL penicillin–streptomycin (Sigma-Aldrich, St. Louis, MO, USA). Cell cultures were maintained at 37 °C in a 5% CO2 humidified atmosphere. For immunofluorescence studies, fibroblasts were seeded onto glass coverslips and fixed in 4% formaldehyde for 20 min. Permeabilization was performed using 0.5% TritonX-100 diluted in PBS for 30 min at RT. Cells were blocked in PBS and 6% BSA for 45 min at RT. The anti-PPP2R1A primary antibody (1:100) was used and incubated for 1 h at 37 °C (ab154551, Abcam, Waltham, MA, USA).
After washing in PBS, cells were incubated at 37 °C for 1 h in the dark using the secondary antibody α-rabbit Alexa Fluor 594 (A21207, Thermo Fisher Scientific, Inc., Waltham, MA, USA). Nuclei were stained with Hoechst 33342 trihydrochloride trihydrate (Life Technologies, Carlsbad, CA, USA), and then, mounted with Prolong Diamond antifade (Life Technologies). Confocal and super-resolution microscopy analysis was performed using a Leica TCS SP8 equipped with a white light laser, a HyVolution super-resolution module, and hybrid spectral detectors (Leica Microsystems GmbH, Mannheim, Germany). For the quantification, confocal images of the fibroblast cultures were acquired using an HC × PL APO 63×/1.4 oil immersion objective. Hoechst 33342 was excited using a blue diode laser (405 nm) and detected in the 420–475 nm range. PPP2R1A was excited using a white light laser (594 nm) and detected in the 635–795 nm range. An image sequence (XYZ) comprising 10 sections with a step size of 0.7 μm was captured to visualize the fluorescence distribution of the PPP2R1A protein throughout the cell’s thickness. A total of 122 fields were acquired, each with a field of view (FOV) measuring 184.52 × 184.52 μm. To represent the variation in intensity for each spectral component, a 12-bit encoding scheme was employed.
For precise data collection, we implemented appropriate negative controls to calibrate the confocal settings and eliminate non-specific fluorescence artifacts. To ensure comparability across different samples, identical confocal settings were maintained during image acquisition. Subsequent to image capture, we carried out sum intensity projections and fluorescence quantification using the ImageJ Fiji software (Image J 1.52n) (National Institutes of Health in Bethesda, MD, USA). The mean fluorescence intensity was then normalized by dividing it by the total number of cells. In the case of nuclei, a mask was generated using the nucleus staining channel, and the mean fluorescence intensity for each nucleus was quantified.
Super-resolution images were acquired using an HC × PL APO 100x/1.4 oil immersion objective, an HyD detector, and mode HyVolution, with the pinhole set to 0.6 Airy units. To investigate the distribution of PPP2R1A in three dimensions, Z stacks were acquired at 0.2 μm intervals throughout the cell thickness, resulting in a total of 28 sections. These images had a resolution of 0.071 × 0.071 µm. Image deconvolution was performed using Huygens Professional software v17.10.0p7 64b (SVI, Leiden, The Netherlands) and stacks were reconstructed and visualized as three-dimensional (3D) volumes using Imaris software ver. 7.2.1 (Bitplane AG, Zürich, Switzerland) (Bitplane, Zürich, Switzerland). PPP2R1A aggregate area analysis was conducted using Imaris software (Surface module), and the aggregate areas were measured using the Surface Statistics function.
4.4. Statistical Analysis
The data are presented as means ± SEM and are displayed either as column bars or scatter plots, with error bars. p-values are indicated by asterisks as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001. Graphs were created and statistical analysis was performed using GraphPad Prism version 8.0.1 (GraphPad Software, Inc., La Jolla, CA, USA).
The data’s normality were assessed using a Kolmogorov–Smirnov test, which indicated that the data did not conform to a normal distribution. Consequently, a Kruskal–Wallis test was utilized to compare all samples, while a Mann–Whitney test was performed to compare the control data with individual patient data, both in pairwise comparisons and across patients. The significance levels are indicated as * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
4.5. Ethical Issues
Ethical permission for the study was obtained from the Research and Ethics Committee of the SJD Research Foundation. Parents gave their written informed consent and children/adolescents and adults gave their assent. Samples were obtained in accordance with the Helsinki Declaration of 1964, as revised in October 2013 in Fortaleza, Brazil.





 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}