
Amyloid Fibrils of Pisum sativum L. Vicilin Inhibit Pathological Aggregation of Mammalian Proteins

 ,
,  , , ,
, , ,  ,
,  , and
, and 
Abstract
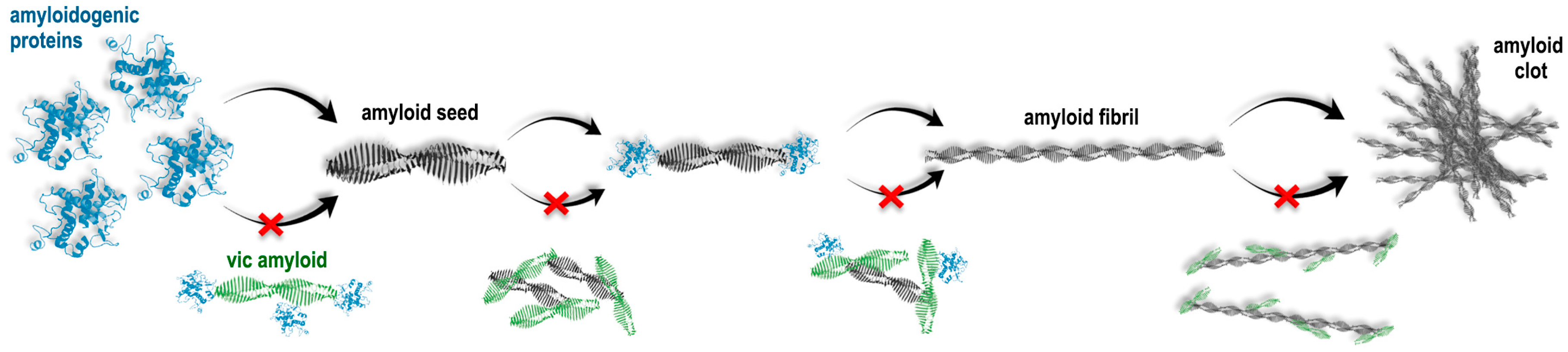
:1. Introduction
2. Results
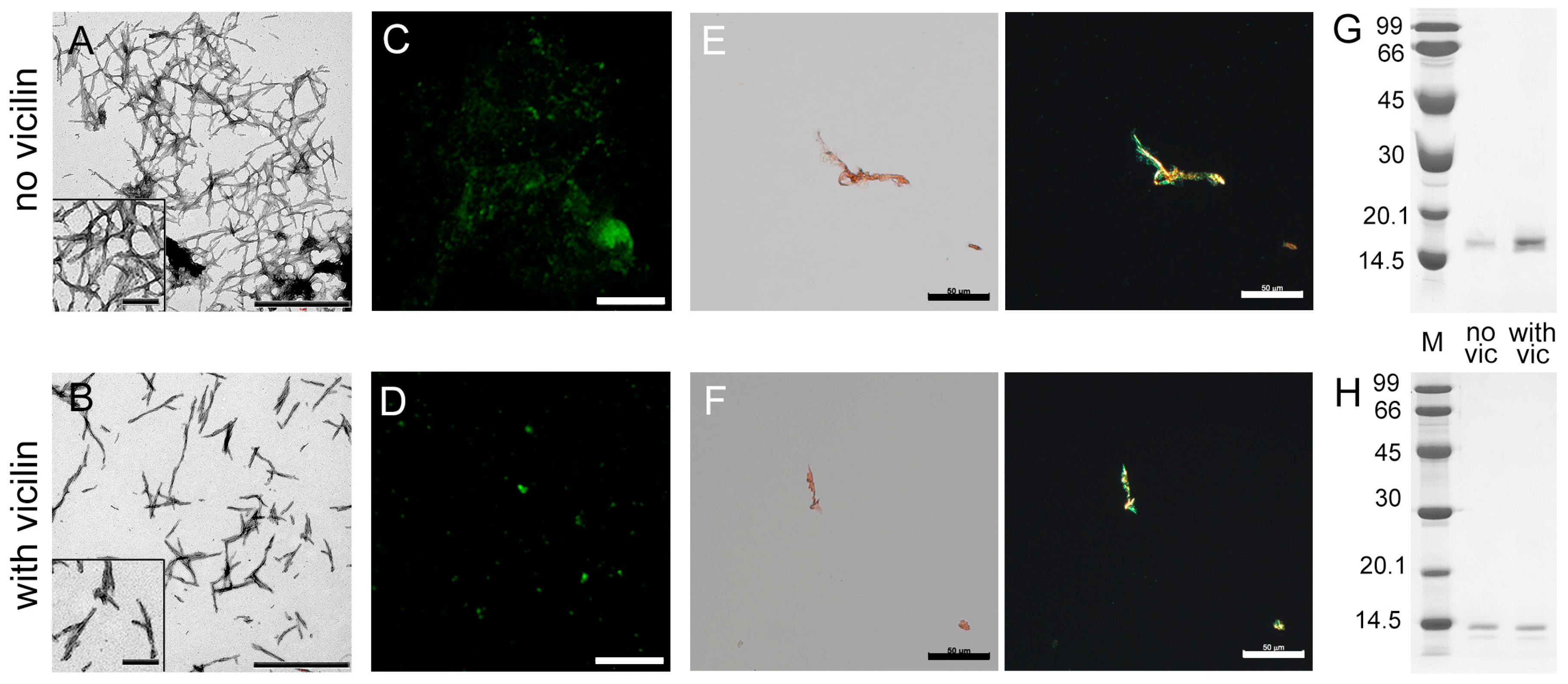
2.1. Amyloids Formed from Vicilin and Its Fragments Effectively Inhibit the Growth of Lysozyme Amyloid Fibrils
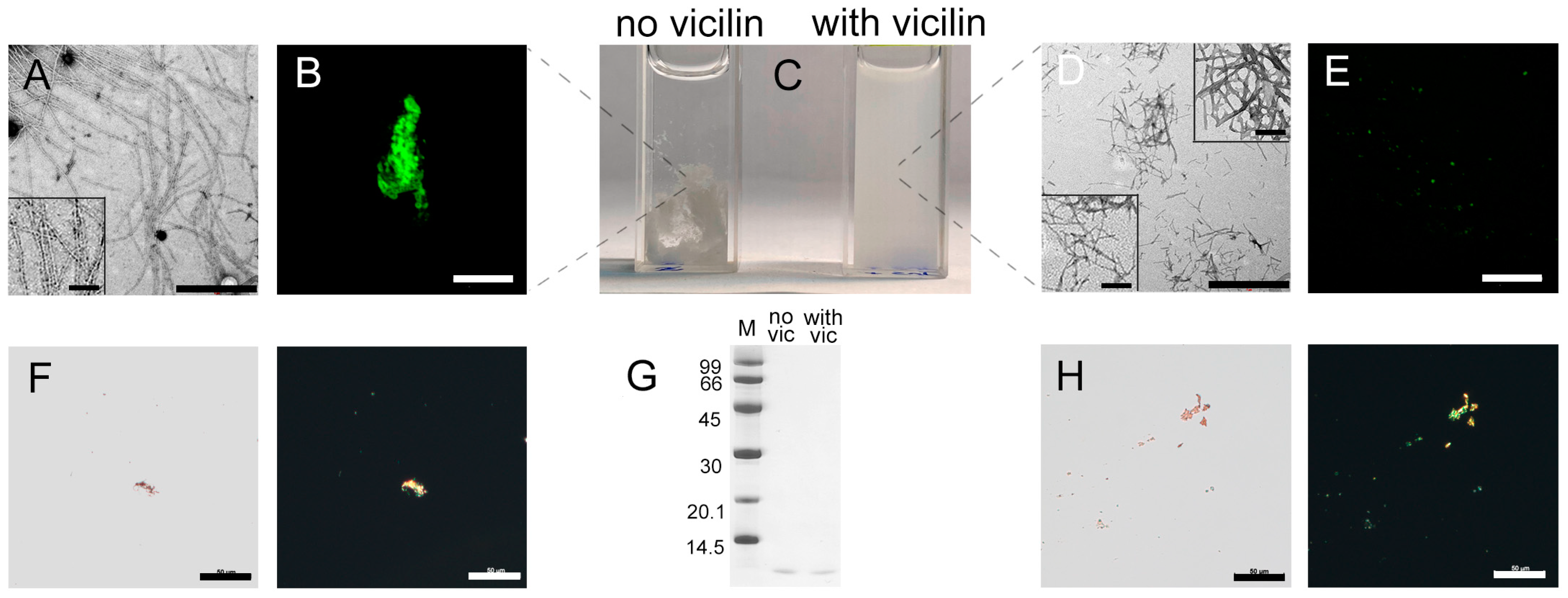
2.2. Vicilin Amyloids Cause a Decrease in the Length of β-2-Microglobulin Fibrils
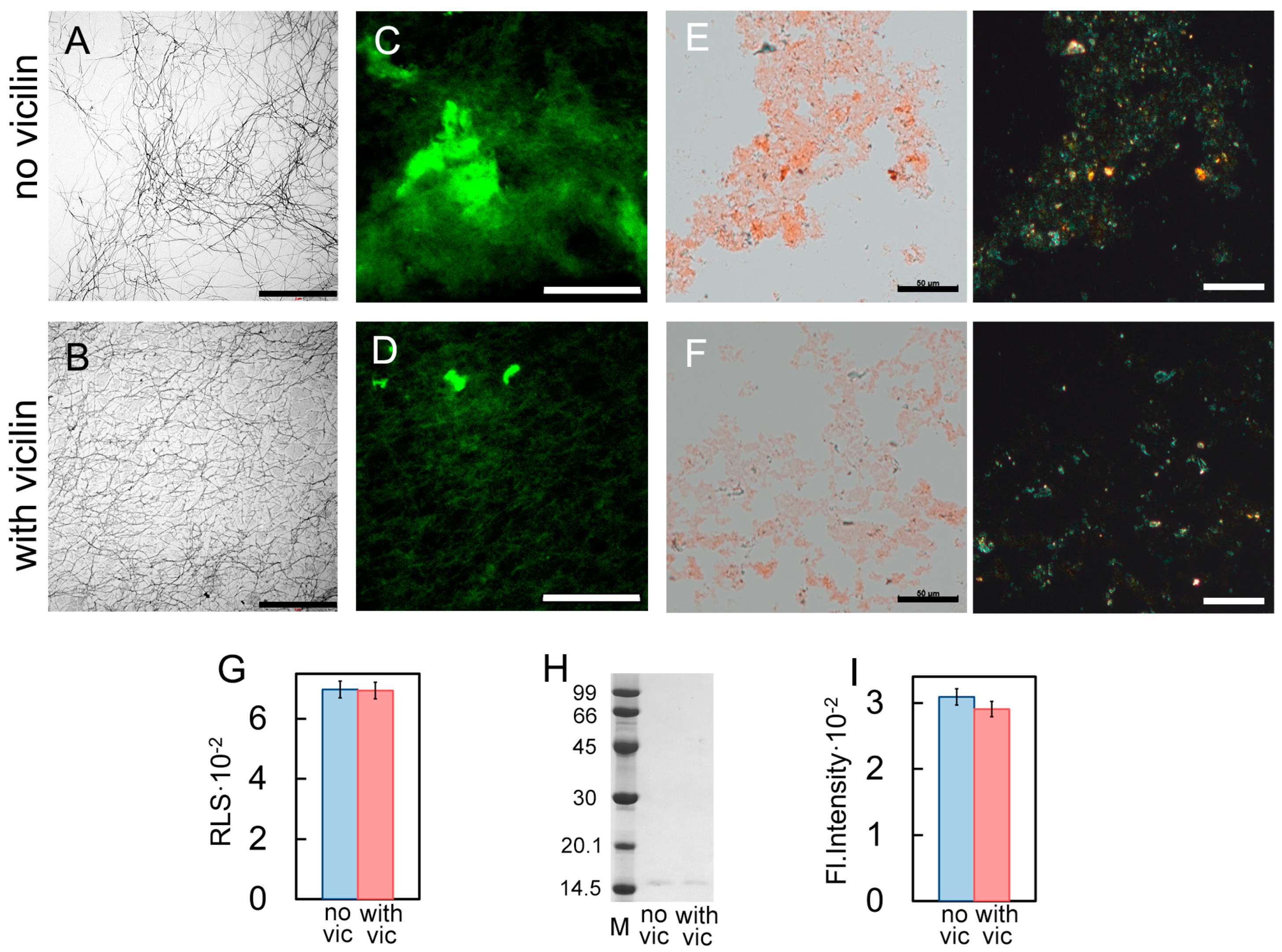
2.3. Vicilin Amyloids Inhibit the β-Amyloid Fibril Clustering
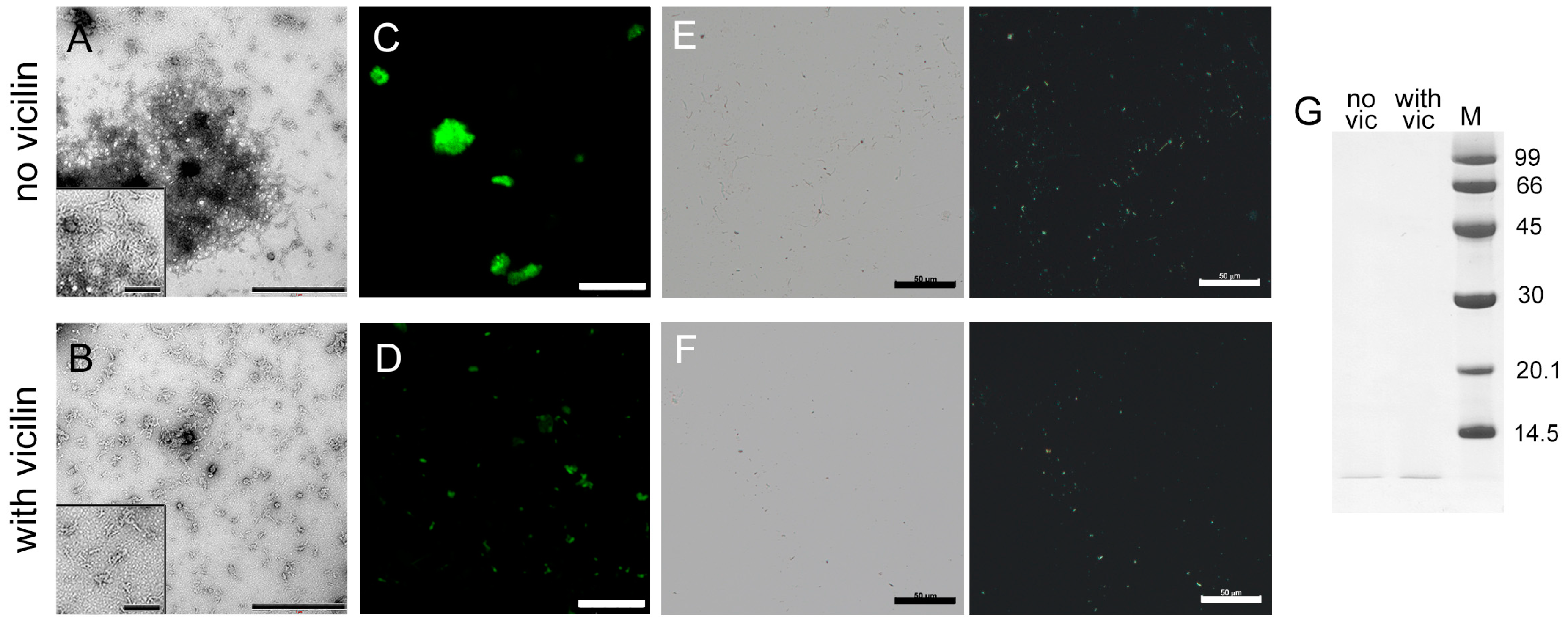
2.4. Vicilin Amyloids Alter the Structure of Insulin Fibrils
2.5. The Effect of Vicilin Amyloids on the Fibrillogenesis Depends on the Duration of the Lag Phase
2.6. Vicilin Amyloids Reduce the Cytotoxicity of Lysozyme, β2m and Aβ42 Fibrils and Increase the Cytotoxicity of Insulin Amyloids
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Recombinant Protein Production and Purification
4.3. Amyloid Fibrils’ Preparation
4.4. Transmission Electron Microscopy
4.5. ThT–Amyloid Fibril Sample Preparation
4.6. Spectral Measurements
4.7. Time-Resolved Fluorescence Measurements
4.8. Confocal Microscopy
4.9. SDS-PAGE
4.10. Study of Interaction between Vicilin Amyloids and Mammalian Proteins in Different Forms
4.11. Cell Viability Assessment
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Prusiner, S.B.; McKinley, M.P.; Bowman, K.A.; Bolton, D.C.; Bendheim, P.E.; Groth, D.F.; Glenner, G.G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 1983, 35 Pt 1, 349–358. [Google Scholar] [CrossRef]
- Warby, S.C.; Montpetit, A.; Hayden, A.R.; Carroll, J.B.; Butland, S.L.; Visscher, H.; Collins, J.A.; Semaka, A.; Hudson, T.J.; Hayden, M.R. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am. J. Hum. Genet. 2009, 84, 351–366. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Valentine, J.S.; Doucette, P.A.; Zittin Potter, S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 2005, 74, 563–593. [Google Scholar] [CrossRef]
- Pham, C.L.; Kwan, A.H.; Sunde, M. Functional amyloid: Widespread in Nature, diverse in purpose. Essays Biochem. 2014, 56, 207–219. [Google Scholar]
- Otzen, D.; Riek, R. Functional Amyloids. Cold Spring Harb. Perspect. Biol. 2019, 11, a033860. [Google Scholar] [CrossRef]
- Fowler, D.M.; Koulov, A.V.; Balch, W.E.; Kelly, J.W. Functional amyloid—From bacteria to humans. Trends Biochem. Sci. 2007, 32, 217–224. [Google Scholar] [CrossRef]
- Avni, A.; Swasthi, H.M.; Majumdar, A.; Mukhopadhyay, S. Intrinsically disordered proteins in the formation of functional amyloids from bacteria to humans. Prog. Mol. Biol. Transl. Sci. 2019, 166, 109–143. [Google Scholar]
- Antonets, K.S.; Belousov, M.V.; Sulatskaya, A.I.; Belousova, M.E.; Kosolapova, A.O.; Sulatsky, M.I.; Andreeva, E.A.; Zykin, P.A.; Malovichko, Y.V.; Shtark, O.Y.; et al. Accumulation of storage proteins in plant seeds is mediated by amyloid formation. PLoS Biol. 2020, 18, e3000564. [Google Scholar] [CrossRef]
- Buchanan, J.A.; Varghese, N.R.; Johnston, C.L.; Sunde, M. Functional amyloids: Where supramolecular amyloid assembly controls biological activity or generates new functionality. J. Mol. Biol. 2023, 435, 167919. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.M.; Chapman, M.R. Curli biogenesis and function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Collinson, S.K.; Emody, L.; Muller, K.H.; Trust, T.J.; Kay, W.W. Purification and characterization of thin, aggregative fimbriae from Salmonella Enteritidis. J. Bacteriol. 1991, 173, 4773–4781. [Google Scholar] [CrossRef]
- Levkovich, S.A.; Gazit, E.; Bar-Yosef, D.L. Two decades of studying functional amyloids in microorganisms. Trends Microbiol. 2021, 29, 251–265. [Google Scholar] [CrossRef]
- Kosolapova, A.O.; Antonets, K.S.; Belousov, M.V.; Nizhnikov, A.A. Biological Functions of Prokaryotic Amyloids in Interspecies Interactions: Facts and Assumptions. Int. J. Mol. Sci. 2020, 21, 7240. [Google Scholar] [CrossRef]
- Kosolapova, A.O.; Belousov, M.V.; Sulatskaya, A.I.; Belousova, M.E.; Sulatsky, M.I.; Antonets, K.S.; Volkov, K.V.; Lykholay, A.N.; Shtark, O.Y.; Vasileva, E.N.; et al. Two Novel Amyloid Proteins, RopA and RopB, from the Root Nodule Bacterium Rhizobium leguminosarum. Biomolecules 2019, 9, 694. [Google Scholar] [CrossRef]
- Kosolapova, A.O.; Belousov, M.V.; Sulatsky, M.I.; Tsyganova, A.V.; Sulatskaya, A.I.; Bobylev, A.G.; Shtark, O.Y.; Tsyganov, V.E.; Volkov, K.V.; Zhukov, V.A.; et al. RopB protein of Rhizobium leguminosarum bv. viciae adopts amyloid state during symbiotic interactions with pea (Pisum sativum L.). Front. Plant Sci. 2022, 13, 1014699. [Google Scholar] [CrossRef]
- Sengupta, U.; Kayed, R. Amyloid beta, Tau, and alpha-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef]
- Werner, T.; Horvath, I.; Wittung-Stafshede, P. Crosstalk between Alpha-Synuclein and Other Human and Non-Human Amyloidogenic Proteins: Consequences for Amyloid Formation in Parkinson’s Disease. J. Park. Dis. 2020, 10, 819–830. [Google Scholar] [CrossRef]
- Han, J.; Fan, Y.; Wu, P.; Huang, Z.; Li, X.; Zhao, L.; Ji, Y.; Zhu, M. Parkinson’s Disease Dementia: Synergistic Effects of Alpha-Synuclein, Tau, Beta-Amyloid, and Iron. Front. Aging Neurosci. 2021, 13, 743754. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.N.; Yang, Z.H.; Wang, X.K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Friesen, M.; Meyer-Luehmann, M. Abeta Seeding as a Tool to Study Cerebral Amyloidosis and Associated Pathology. Front. Mol. Neurosci. 2019, 12, 233. [Google Scholar] [CrossRef]
- Ren, B.; Zhang, Y.; Zhang, M.; Liu, Y.; Zhang, D.; Gong, X.; Feng, Z.; Tang, J.; Chang, Y.; Zheng, J. Fundamentals of cross-seeding of amyloid proteins: An introduction. J. Mater. Chem. B 2019, 7, 7267–7282. [Google Scholar] [CrossRef]
- Subedi, S.; Sasidharan, S.; Nag, N.; Saudagar, P.; Tripathi, T. Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition. Molecules 2022, 27, 1776. [Google Scholar] [CrossRef]
- Do, H.Q.; Hewetson, A.; Borcik, C.G.; Hastert, M.C.; Whelly, S.; Wylie, B.J.; Sutton, R.B.; Cornwall, G.A. Cross-seeding between the functional amyloidogenic CRES and CRES3 family members and their regulation of Abeta assembly. J. Biol. Chem. 2021, 296, 100250. [Google Scholar] [CrossRef]
- Zhou, Y.; Smith, D.; Leong, B.J.; Brannstrom, K.; Almqvist, F.; Chapman, M.R. Promiscuous cross-seeding between bacterial amyloids promotes interspecies biofilms. J. Biol. Chem. 2012, 287, 35092–35103. [Google Scholar] [CrossRef]
- Perera, S.; Uddin, M.; Hayes, J.A. Salivary lysozyme: A noninvasive marker for the study of the effects of stress of natural immunity. Int. J. Behav. Med. 1997, 4, 170–178. [Google Scholar] [CrossRef]
- Granel, B.; Serratrice, J.; Valleix, S.; Grateau, G.; Droz, D.; Lafon, J.; Sault, M.C.; Chaudier, B.; Disdier, P.; Laugier, R.; et al. A family with gastrointestinal amyloidosis associated with variant lysozyme. Gastroenterology 2002, 123, 1346–1349. [Google Scholar] [CrossRef]
- Pleyer, C.; Flesche, J.; Saeed, F. Lysozyme amyloidosis—A case report and review of the literature. Clin. Nephrol. Case Stud. 2015, 3, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Garza, A.S.; Khan, S.H.; Moure, C.M.; Edwards, D.P.; Kumar, R. Binding-folding induced regulation of AF1 transactivation domain of the glucocorticoid receptor by a cofactor that binds to its DNA binding domain. PLoS ONE 2011, 6, e25875. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Verkhusha, V.V.; Shavlovsky, M.M.; Biktashev, A.G.; Povarova, O.I.; Kuznetsova, I.M. Kinetics of actin unfolding induced by guanidine hydrochloride. Biochemistry 2002, 41, 1014–1019. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Q.; Hu, W.-J.; Mu, H.; Lin, Z.; Ma, L.; Park, Y.-D.; Zhou, H.-M. Effect of SNPs on creatine kinase structure and function: Identifying potential molecular mechanisms for possible creatine kinase deficiency diseases. PLoS ONE 2012, 7, e45949. [Google Scholar] [CrossRef]
- Krebs, M.R.; Bromley, E.H.; Donald, A.M. The binding of thioflavin-T to amyloid fibrils: Localisation and implications. J. Struct. Biol. 2005, 149, 30–37. [Google Scholar] [CrossRef]
- Sidhu, A.; Vaneyck, J.; Blum, C.; Segers-Nolten, I.; Subramaniam, V. Polymorph-specific distribution of binding sites determines thioflavin-T fluorescence intensity in α-synuclein fibrils. Amyloid 2018, 25, 189–196. [Google Scholar] [CrossRef]
- Peccati, F.; Pantaleone, S.; Riffet, V.; Solans-Monfort, X.; Contreras-García, J.; Guallar, V.; Sodupe, M. Binding of thioflavin T and related probes to polymorphic models of amyloid-β fibrils. J. Phys. Chem. 2017, 121, 8926–8934. [Google Scholar] [CrossRef] [PubMed]
- Sulatskaya, A.I.; Rodina, N.P.; Polyakov, D.S.; Sulatsky, M.I.; Artamonova, T.O.; Khodorkovskii, M.A.; Shavlovsky, M.M.; Kuznetsova, I.M.; Turoverov, K.K. Structural Features of Amyloid Fibrils Formed from the Full-Length and Truncated Forms of Beta-2-Microglobulin Probed by Fluorescent Dye Thioflavin T. Int. J. Mol. Sci. 2018, 19, 2762. [Google Scholar] [CrossRef]
- Sulatskaya, A.I.; Rychkov, G.N.; Sulatsky, M.I.; Mikhailova, E.V.; Melnikova, N.M.; Andozhskaya, V.S.; Kuznetsova, I.M.; Turoverov, K.K. New evidence on a distinction between Aβ40 and Aβ42 amyloids: Thioflavin T binding modes, clustering tendency, degradation resistance, and cross-seeding. Int. J. Mol. Sci. 2022, 23, 5513. [Google Scholar] [CrossRef]
- Kuznetsova, I.M.; Sulatskaya, A.I.; Uversky, V.N.; Turoverov, K.K. A new trend in the experimental methodology for the analysis of the thioflavin T binding to amyloid fibrils. Mol. Neurobiol. 2012, 45, 488–498. [Google Scholar] [CrossRef]
- Maruyama, H.; Gejyo, F.; Arakawa, M. Clinical studies of destructive spondyloarthropathy in long-term hemodialysis patients. Nephron 1992, 61, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Assenat, H.; Calemard, E.; Charra, B.; Laurent, G.; Terrat, J.C.; Vanel, T. Hemodialysis: Carpal tunnel syndrome and amyloid substance. Nouv. Presse Med. 1980, 9, 1715. [Google Scholar]
- Kuntz, D.; Naveau, B.; Bardin, T.; Drueke, T.; Treves, R.; Dryll, A. Destructive spondylarthropathy in hemodialyzed patients. A new syndrome. Arthritis Rheum. 1984, 27, 369–375. [Google Scholar] [CrossRef]
- Zingraff, J.J.; Noel, L.H.; Bardin, T.; Atienza, C.; Zins, B.; Drueke, T.B.; Kuntz, D. Beta 2-microglobulin amyloidosis in chronic renal failure. N. Engl. J. Med. 1990, 323, 1070–1071. [Google Scholar]
- Campistol, J.M.; Sole, M.; Munoz-Gomez, J.; Lopez-Pedret, J.; Revert, L. Systemic involvement of dialysis-amyloidosis. Am. J. Nephrol. 1990, 10, 389–396. [Google Scholar] [CrossRef]
- Gal, R.; Korzets, A.; Schwartz, A.; Rath-Wolfson, L.; Gafter, U. Systemic distribution of beta 2-microglobulin-derived amyloidosis in patients who undergo long-term hemodialysis. Report of seven cases and review of the literature. Arch. Pathol. Lab. Med. 1994, 118, 718–721. [Google Scholar]
- Charra, B.C.E.; Uzan, M.; Terrat, J.C.; Vanel, T.; Laurent, G. Carpal tunnel syndrome, shoulder pain and amyloid deposits in longterm hemodialysis patients. Proc. Eur. Dial. Transpl. Assoc. 1984, 21, 291–295. [Google Scholar]
- Sprague, S.M.M.S.M. Clinical manifestations and pathogenesis of dialysis-related amyloidosis. Semin. Dial. 1996, 9, 360–369. [Google Scholar] [CrossRef]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- Wang, S.; Mims, P.N.; Roman, R.J.; Fan, F. Is Beta-Amyloid Accumulation a Cause or Consequence of Alzheimer’s Disease? J. Alzheimer’s Park. Dement. 2016, 1, 007. [Google Scholar]
- Storkel, S.; Schneider, H.M.; Muntefering, H.; Kashiwagi, S. Iatrogenic, insulin-dependent, local amyloidosis. Lab. Investig. J. Tech. Methods Pathol. 1983, 48, 108–111. [Google Scholar]
- Dische, F.E.; Wernstedt, C.; Westermark, G.T.; Westermark, P.; Pepys, M.B.; Rennie, J.A.; Gilbey, S.G.; Watkins, P.J. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia 1988, 31, 158–161. [Google Scholar] [CrossRef]
- Swift, B. Examination of insulin injection sites: An unexpected finding of localized amyloidosis. Diabet. Med. J. Br. Diabet. Assoc. 2002, 19, 881–882. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Reeves, W.; DeMay, R.M. Amyloid tumor: A clinical and cytomorphologic study. Diagn. Cytopathol. 2003, 28, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.G.; Obadiah, J.; Parseghian, S.A.; Yadira Hurley, M.; Mooradian, A.D. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res. Clin. Pract. 2007, 75, 374–376. [Google Scholar] [CrossRef]
- Yumlu, S.; Barany, R.; Eriksson, M.; Rocken, C. Localized insulin-derived amyloidosis in patients with diabetes mellitus: A case report. Hum. Pathol. 2009, 40, 1655–1660. [Google Scholar] [CrossRef]
- Shikama, Y.; Kitazawa, J.; Yagihashi, N.; Uehara, O.; Murata, Y.; Yajima, N.; Wada, R.; Yagihashi, S. Localized amyloidosis at the site of repeated insulin injection in a diabetic patient. Intern. Med. 2010, 49, 397–401. [Google Scholar] [CrossRef]
- Gupta, Y.; Singla, G.; Singla, R. Insulin-derived amyloidosis. Indian J. Endocrinol. Metab. 2015, 19, 174–177. [Google Scholar] [CrossRef]
- Dahiya, D.S.; Kichloo, A.; Singh, J.; Albosta, M.; Wani, F. Gastrointestinal amyloidosis: A focused review. World J. Gastrointest. Endosc. 2021, 13, 1–12. [Google Scholar] [CrossRef]
- Carll, T.; Antic, T. An abdominal wall mass of exogenous insulin amyloidosis in setting of metastatic sarcoma. J. Cutan. Pathol. 2020, 47, 406–408. [Google Scholar] [CrossRef]
- Shiba, M.; Kitazawa, T. Progressive insulin-derived amyloidosis in a patient with type 2 diabetes. Case Rep. Plast. Surg. Hand Surg. 2016, 3, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sommerville, N.R.; Liu, J.Y.H.; Ngan, M.P.; Poon, D.; Ponomarev, E.D.; Lu, Z.; Kung, J.S.C.; Rudd, J.A. Intra-gastrointestinal amyloid-beta1-42 oligomers perturb enteric function and induce Alzheimer’s disease pathology. J. Physiol. 2020, 598, 4209–4223. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Lee, D.D. Atypical presentation of localized insulin-derived amyloidosis as protruding brownish skin tumours. Clin. Exp. Dermatol. 2020, 45, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, E.; Boland, B.; Walsh, D.M.; Linse, S. Amyloid beta-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 2010, 1, 13–18. [Google Scholar] [CrossRef]
- Adamcik, J.; Mezzenga, R. Amyloid Polymorphism in the Protein Folding and Aggregation Energy Landscape. Angew. Chem. 2018, 57, 8370–8382. [Google Scholar] [CrossRef]
- Aliyan, A.; Cook, N.P.; Marti, A.A. Interrogating Amyloid Aggregates using Fluorescent Probes. Chem. Rev. 2019, 119, 11819–11856. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Solomon, T.; Sahoo, B.R.; Ignasiak, K.; Gaskin, S.; Rowles, J.; Verdile, G.; Howard, M.J.; Bond, C.S.; Ramamoorthy, A.; et al. Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells. Sci. Rep. 2020, 10, 10356. [Google Scholar] [CrossRef]
- Stepanenko, O.V.; Sulatsky, M.I.; Mikhailova, E.V.; Stepanenko, O.V.; Povarova, O.I.; Kuznetsova, I.M.; Turoverov, K.K.; Sulatskaya, A.I. Alpha-B-Crystallin Effect on Mature Amyloid Fibrils: Different Degradation Mechanisms and Changes in Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 7659. [Google Scholar] [CrossRef]
- Stepanenko, O.V.; Sulatsky, M.I.; Mikhailova, E.V.; Stepanenko, O.V.; Kuznetsova, I.M.; Turoverov, K.K.; Sulatskaya, A.I. Trypsin Induced Degradation of Amyloid Fibrils. Int. J. Mol. Sci. 2021, 22, 4828. [Google Scholar] [CrossRef]
- Nilsson, L.; Pamren, A.; Islam, T.; Brannstrom, K.; Golchin, S.A.; Pettersson, N.; Iakovleva, I.; Sandblad, L.; Gharibyan, A.L.; Olofsson, A. Transthyretin Interferes with Abeta Amyloid Formation by Redirecting Oligomeric Nuclei into Non-Amyloid Aggregates. J. Mol. Biol. 2018, 430, 2722–2733. [Google Scholar] [CrossRef]
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Transthyretin Inhibits Primary and Secondary Nucleations of Amyloid-beta Peptide Aggregation and Reduces the Toxicity of Its Oligomers. Biomacromolecules 2020, 21, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Wasana Jayaweera, S.; Surano, S.; Pettersson, N.; Oskarsson, E.; Lettius, L.; Gharibyan, A.L.; Anan, I.; Olofsson, A. Mechanisms of Transthyretin Inhibition of IAPP Amyloid Formation. Biomolecules 2021, 11, 411. [Google Scholar] [CrossRef] [PubMed]
- Serio, T.R.; Cashikar, A.G.; Moslehi, J.J.; Kowal, A.S.; Lindquist, S.L. Yeast prion [psi +] and its determinant, Sup35p. Methods Enzymol. 1999, 309, 649–673. [Google Scholar] [PubMed]
- Broersen, K.; Jonckheere, W.; Rozenski, J.; Vandersteen, A.; Pauwels, K.; Pastore, A.; Rousseau, F.; Schymkowitz, J. A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer’s disease. Protein Eng. Des. Sel. PEDS 2011, 24, 743–750. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef]
- Vladimirov, Y.A.; Litvin, F.F. Photobiology and spectroscopic methods. In Handbook of General Biophisics; High School: Moscow, Russia, 1964; Volume 8, pp. 88–91. [Google Scholar]
- Micsonai, A.; Wien, F.; Bulyaki, E.; Kun, J.; Moussong, E.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Jiang, L.; Wang, W.; Sang, J.; Wang, X.; Lu, F.; Liu, F. The food additive fast green FCF inhibits alpha-synuclein aggregation, disassembles mature fibrils and protects against amyloid-induced neurotoxicity. Food Funct. 2021, 12, 5465–5477. [Google Scholar] [CrossRef]
- Fonin, A.V.; Sulatskaya, A.I.; Kuznetsova, I.M.; Turoverov, K.K. Fluorescence of dyes in solutions with high absorbance. Inner filter effect correction. PLoS ONE 2014, 9, e103878. [Google Scholar] [CrossRef]
- Ahmad, B.; Lapidus, L.J. Curcumin prevents aggregation in alpha-synuclein by increasing reconfiguration rate. J. Biol. Chem. 2012, 287, 9193–9199. [Google Scholar] [CrossRef]
- Chaudhary, A.P.; Vispute, N.H.; Shukla, V.K.; Ahmad, B. A comparative study of fibrillation kinetics of two homologous proteins under identical solution condition. Biochimie 2017, 132, 75–84. [Google Scholar] [CrossRef]
- Lee, C.C.; Nayak, A.; Sethuraman, A.; Belfort, G.; McRae, G.J. A three-stage kinetic model of amyloid fibrillation. Biophys. J. 2007, 92, 3448–3458. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D. Time-Correlated Single Photon Counting; Academic Press: New York, NY, USA, 1984; pp. 37–54. [Google Scholar]
- Marquardt, D.W. An algorithm for least-squares estimation of non linear parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Yanushevich, Y.G.; Staroverov, D.B.; Savitsky, A.P.; Fradkov, A.F.; Gurskaya, N.G.; Bulina, M.E.; Lukyanov, K.A.; Lukyanov, S.A. A strategy for the generation of non-aggregating mutants of Anthozoa fluorescent proteins. FEBS Lett. 2002, 511, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amyloidogenic Protein/Peptide | Vicilin Amyloids | RLS | Turbidity | β-Sheets, % | FThT·10−4 | qThT·102 | <τThT>, ns |
|---|---|---|---|---|---|---|---|
| Lysozyme | – | 423 ± 16 | 0.53 ± 0.02 | 45.6 ± 1.4 | 12.2 ± 0.3 | 2.2 ± 0.1 | 1.1 ± 0.1 |
| + | 81 ± 6 | 0.26 ± 0.01 | 37.1 ± 1.2 | 5.0 ± 0.4 | 2.2 ± 0.2 | 1.1 ± 0.1 | |
| β2m | – | 233 ± 9 | 0.24 ± 0.01 | 35.3 ± 1.2 | 4.3 ± 0.2 | 4.6 ± 0.2 | 1.6 ± 0.1 |
| + | 209 ± 7 | 0.21 ± 0.02 | 35.2 ± 1.3 | 3.9 ± 0.1 | 4.8 ± 0.1 | 1.6 ± 0.1 | |
| Aβ42 | – | 399 ± 12 | 0.78 ± 0.02 | 42.2 ± 1.5 | 8.1 ± 0.2 | 1.3 ± 0.2 | 0.8 ± 0.1 |
| + | 267 ± 12 | 0.61 ± 0.03 | 40.8 ± 1.0 | 7.1 ± 0.2 | 1.3 ± 0.2 | 0.8 ± 0.1 | |
| Insulin | – | 460 ± 9 | 3.13 ± 0.04 | 39.3 ± 1.2 | 6.2 ± 0.1 | 2.2 ± 0.1 | 0.8 ± 0.1 |
| + | 422 ± 8 | 2.52 ± 0.07 | 33.7 ± 1.2 | 15.0 ± 0.3 | 1.4 ± 0.1 | 1.0 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulatsky, M.I.; Belousov, M.V.; Kosolapova, A.O.; Mikhailova, E.V.; Romanenko, M.N.; Antonets, K.S.; Kuznetsova, I.M.; Turoverov, K.K.; Nizhnikov, A.A.; Sulatskaya, A.I. Amyloid Fibrils of Pisum sativum L. Vicilin Inhibit Pathological Aggregation of Mammalian Proteins. Int. J. Mol. Sci. 2023, 24, 12932. https://doi.org/10.3390/ijms241612932
Sulatsky MI, Belousov MV, Kosolapova AO, Mikhailova EV, Romanenko MN, Antonets KS, Kuznetsova IM, Turoverov KK, Nizhnikov AA, Sulatskaya AI. Amyloid Fibrils of Pisum sativum L. Vicilin Inhibit Pathological Aggregation of Mammalian Proteins. International Journal of Molecular Sciences. 2023; 24(16):12932. https://doi.org/10.3390/ijms241612932
Chicago/Turabian StyleSulatsky, Maksim I., Mikhail V. Belousov, Anastasiia O. Kosolapova, Ekaterina V. Mikhailova, Maria N. Romanenko, Kirill S. Antonets, Irina M. Kuznetsova, Konstantin K. Turoverov, Anton A. Nizhnikov, and Anna I. Sulatskaya. 2023. "Amyloid Fibrils of Pisum sativum L. Vicilin Inhibit Pathological Aggregation of Mammalian Proteins" International Journal of Molecular Sciences 24, no. 16: 12932. https://doi.org/10.3390/ijms241612932
APA StyleSulatsky, M. I., Belousov, M. V., Kosolapova, A. O., Mikhailova, E. V., Romanenko, M. N., Antonets, K. S., Kuznetsova, I. M., Turoverov, K. K., Nizhnikov, A. A., & Sulatskaya, A. I. (2023). Amyloid Fibrils of Pisum sativum L. Vicilin Inhibit Pathological Aggregation of Mammalian Proteins. International Journal of Molecular Sciences, 24(16), 12932. https://doi.org/10.3390/ijms241612932