

Anti-Diabetic Activity of Glycyrrhetinic Acid Derivatives FC-114 and FC-122: Scale-Up, In Silico, In Vitro, and In Vivo Studies

 ,
, 
 , , ,
, , ,  , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
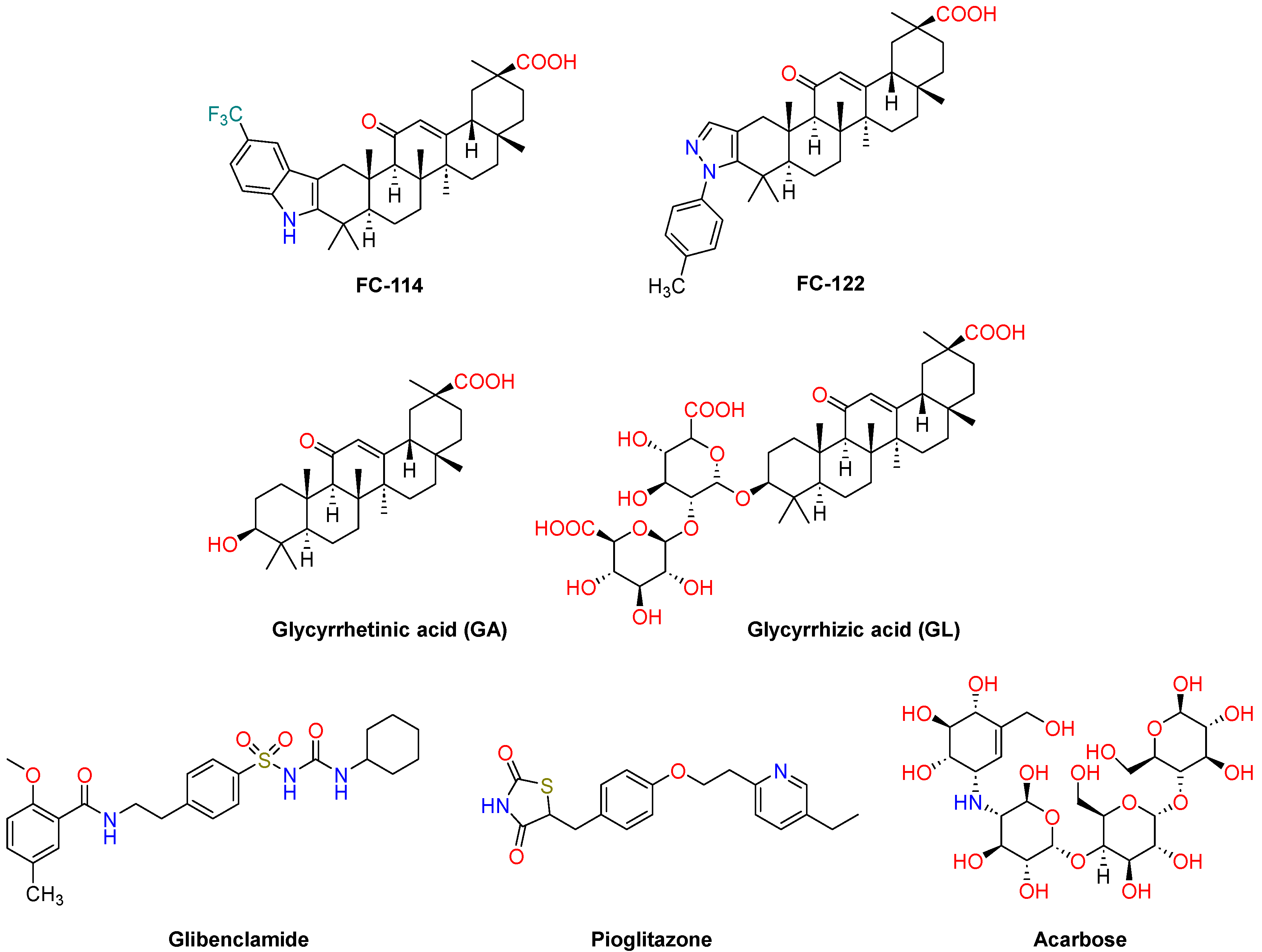
2.1. Chemistry
2.2. Enzymatic Kinetics of α-Glucosidase and PTP1B Inhibition
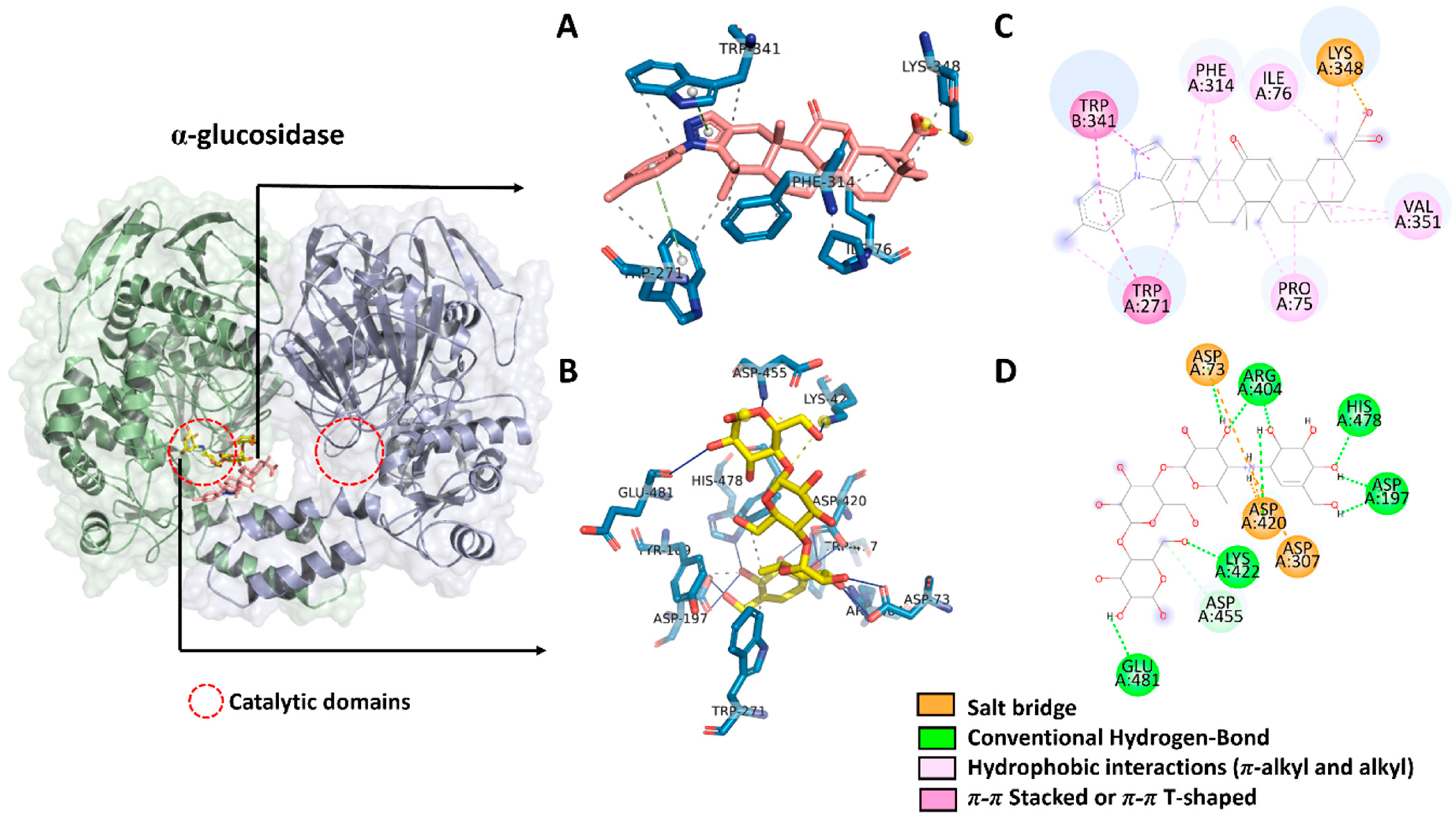
2.3. Molecular Docking
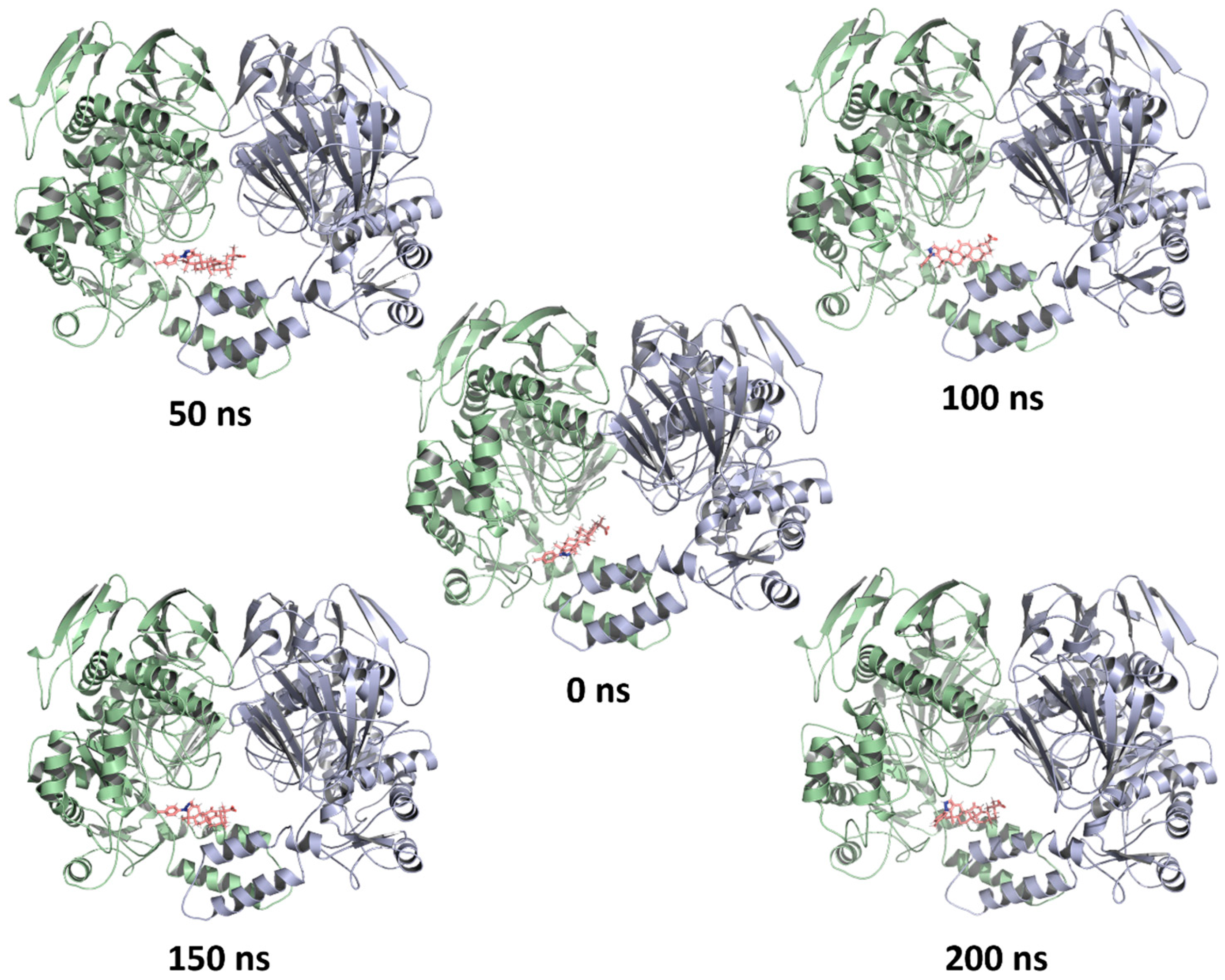
2.4. Molecular Dynamics Simulations
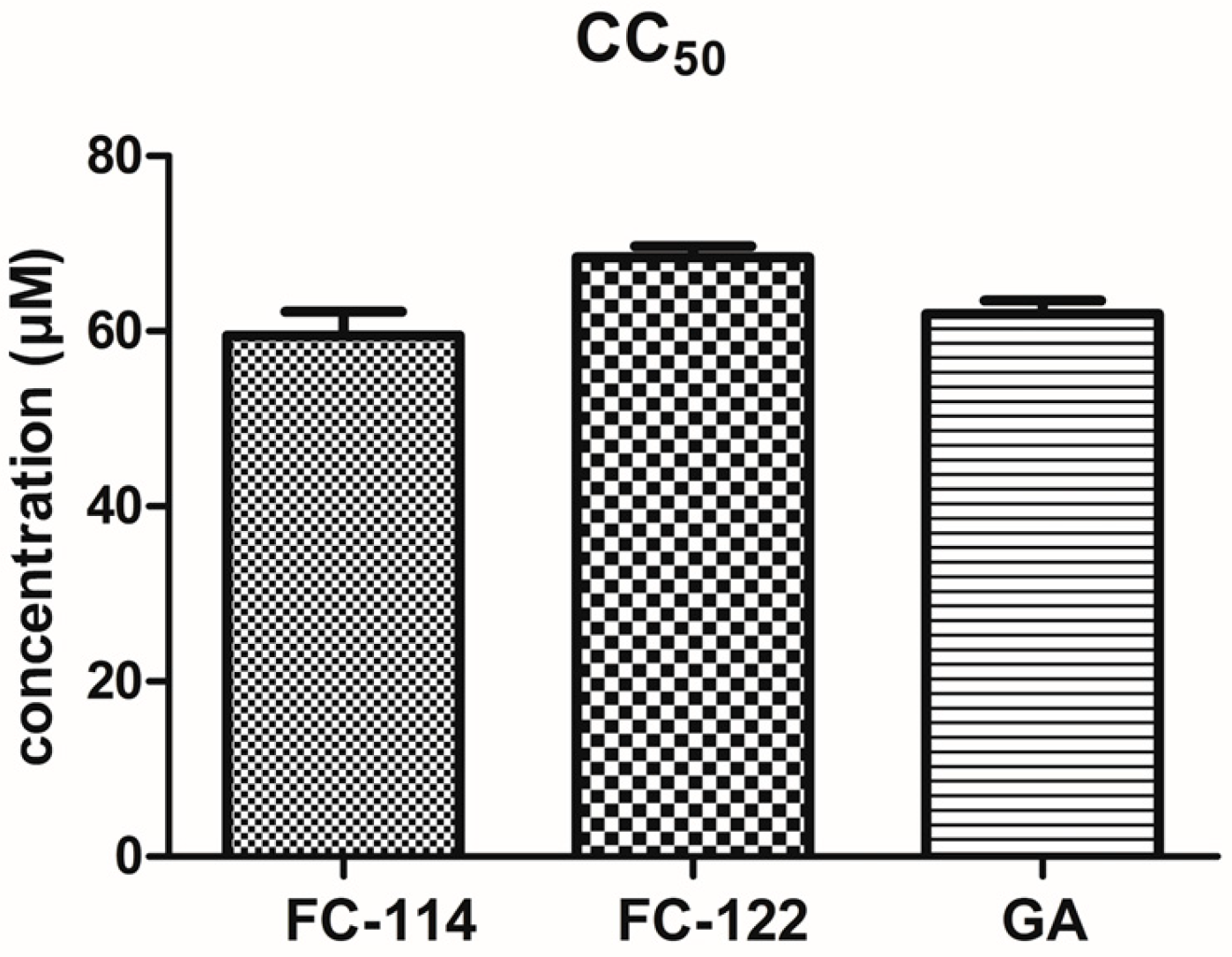
2.5. Cytotoxic Assay and Acute Oral Toxicity
2.6. In Vivo Administration of FC-114 and FC-122 in a Wistar Rat Diabetes Model
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. Synthesis of GA
3.2.2. Synthesis of 3-Oxo-Glycyrrhethinic Acid (2)
3.2.3. Synthesis of 3,11-Dioxo-Olean-12-En-30-Oic Acid (3)
3.2.4. Synthesis of FC-114
3.2.5. Synthesis of FC-122
3.3. In Vitro Assays
3.3.1. Enzyme Kinetics
3.3.2. Cytotoxic Effect on Human Foreskin Fibroblasts
3.4. In Silico Studies
3.4.1. Molecular Docking
3.4.2. MD Simulations
3.5. In Vivo Studies
3.5.1. Animals
3.5.2. Blood Sample Collection and Processing
3.5.3. Evaluation of Acute Oral Toxicity
3.5.4. Rat Model of Diabetes Induced by Streptozotocin
3.5.5. Administration of Treatments
3.5.6. The Oral Glucose Tolerance Test
3.5.7. Blood Determinations
3.5.8. Statistical Analysis
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magliano, D.J.; Boyko, E.J.; Balkau, B.; Barengo, N.; Barr, E.; Basit, A.; Bhata, D.; Bommer, C.; Booth, G.; Cariou, B.; et al. International Diabetes Federation IDF Diabetes Atlas, 10th ed.; Berkeley Communications: Reading, UK, 2021; Volume 102, ISBN 9782930229980. [Google Scholar]
- Cole, J.B.; Florez, J.C. Genetics of diabetes mellitus and diabetes complications. Nat. Rev. Nephrol. 2020, 16, 377–390. [Google Scholar] [CrossRef]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef]
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of Diabetes and Diabetes-Related Complications. Phys. Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Tomic, D.; Shaw, J.E.; Magliano, D.J. The burden and risks of emerging complications of diabetes mellitus. Nat. Rev. Endocrinol. 2022, 18, 525–539. [Google Scholar] [CrossRef]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- University of Oxford Our World in Data. Diabetes Prevalence. 2021. Available online: https://ourworldindata.org/search?q=diabetes (accessed on 5 March 2023).
- Bello-Chavolla, O.Y.; Antonio-Villa, N.E.; Fermín-Martínez, C.A.; Fernández-Chirino, L.; Vargas-Vázquez, A.; Ramírez-García, D.; Basile-Alvarez, M.R.; Hoyos-Lázaro, A.E.; Carrillo-Larco, R.M.; Wexler, D.J.; et al. Diabetes-Related Excess Mortality in Mexico: A Comparative Analysis of National Death Registries Between 2017–2019 and 2020. Diabetes Care 2022, 45, 2957–2966. [Google Scholar] [CrossRef]
- U.S. Department of Health & Human Services What is diabetes? Available online: https://www.cdc.gov/diabetes/basics/diabetes.html (accessed on 22 June 2023).
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Maria, Z.; Campolo, A.R.; Lacombe, V.A. Diabetes alters the expression and translocation of the insulin-sensitive glucose transporters 4 and 8 in the atria. PLoS ONE 2015, 10, e0146033. [Google Scholar] [CrossRef]
- Klip, A.; Marette, A.; Dlmitrakoudis, D.; Ramlal, T.; Giacca, A.; Shi, Z.Q.; Vranic, M. Effect of diabetes on glucoregulation. Diabetes Care 1992, 15, 1747–1766. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Almazán, S.; Navarrete-Vázquez, G.; Padilla-Martínez, I.I.; Correa-Basurto, J.; Alemán-González-duhart, D.; Tamay-Cach, F.; Mendieta-Wejebe, J.E. A new symmetrical thiazolidinedione derivative: In silico design, synthesis, and in vivo evaluation on a streptozotocin-induced rat model of diabetes. Processes 2021, 9, 1294. [Google Scholar] [CrossRef]
- Sandoval-Muñiz, R.d.J.; Vargas-Guerrero, B.; Flores-Alvarado, L.J.; Gurrola-Díaz, C.M. Glucotransportadores (GLUT): Aspectos clínicos, moleculares y genéticos. Gac. Med. Mex. 2016, 152, 547–557. [Google Scholar] [PubMed]
- Kouznetsova, V.L.; Hauptschein, M.; Tsigelny, I.F. Glucose and Lipid Transporters Roles in Type 2 Diabetes. Integr. Obes. Diabetes 2017, 3, 4. [Google Scholar] [CrossRef]
- Kanwal, A.; Kanwar, N.; Bharati, S.; Srivastava, P.; Singh, S.P.; Amar, S. Exploring New Drug Targets for Type 2 Diabetes: Success, Challenges and Opportunities. Biomedicines 2022, 10, 331. [Google Scholar] [CrossRef]
- Kerru, N.; Singh-Pillay, A.; Awolade, P.; Singh, P. Current anti-diabetic agents and their molecular targets: A review. Eur. J. Med. Chem. 2018, 152, 436–488. [Google Scholar] [CrossRef]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Anti-diabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Elangwe, A.; Katte, J.C.; Tchapmi, D.; Figueras, A.; Mbanya, J.C. Adverse drug reactions to anti-diabetic drugs are commonest in patients whose treatment do not adhere to diabetes management clinical guidelines: Cross-sectional study in a tertiary care service in sub-Saharan Africa. Eur. J. Clin. Pharmacol. 2020, 76, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Lingvay, I.; Legendre, J.L.; Kaloyanova, P.F.; Zhang, S.; Adams-Huet, B.; Raskin, P. Insulin-based versus triple oral therapy for newly diagnosed type 2 diabetes: Which is better? Diabetes Care 2009, 32, 1789–1796. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Desai, C.; Patel, P.; Shah, A.; Dikshit, R.K. An evaluation of the impact of anti-diabetic medication on treatment satisfaction and quality of life in patients of diabetes mellitus. Perspect. Clin. Res. 2018, 9, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, Y.; Dai, Y.; Peng, J. Natural products for the treatment of type 2 diabetes mellitus: Pharmacology and mechanisms. Pharmacol. Res. 2018, 130, 451–465. [Google Scholar] [CrossRef]
- Ríos, J.L.; Francini, F.; Schinella, G.R. Natural products for the treatment of type 2 diabetes mellitus. Planta Med. 2015, 81, 975–994. [Google Scholar] [CrossRef]
- Sun, L.; Warren, F.J.; Gidley, M.J. Natural products for glycaemic control: Polyphenols as inhibitors of alpha-amylase. Trends Food Sci. Technol. 2019, 91, 262–273. [Google Scholar] [CrossRef]
- He, J.H.; Chen, L.X.; Li, H. Progress in the discovery of naturally occurring anti-diabetic drugs and in the identification of their molecular targets. Fitoterapia 2019, 134, 270–289. [Google Scholar] [CrossRef] [PubMed]
- Lauren, D.R.; Jensen, D.J.; Douglas, J.A.; Follett, J.M. Efficient Method for Determining the Glycyrrhizin Content of Fresh and Dried Roots, and Root Extracts, of Glycyrrhiza Species. Phytochem. Anal. 2001, 12, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Baltina, L.A. Chemical Modification of Glycyrrhizic Acid as A Route to New Bioactive Compounds for Medicine. Curr. Med. Chem. 2003, 10, 155–171. [Google Scholar] [CrossRef]
- Karaogul, E.; Parlar, P.; Parlar, H.; Alma, M.H. Enrichment of the Glycyrrhizic Acid from Licorice Roots (Glycyrrhiza glabra L.) by Isoelectric Focused Adsorptive Bubble Chromatography. J. Anal. Methods Chem. 2016, 2016, 7201740. [Google Scholar] [CrossRef]
- Tan, D.; Hui, H.; Tseng, L.; Zhong, Z.; Wang, S.; Vong, C.T.; Wang, Y. Glycyrrhizic Acid and Its Derivatives: Promising Candidates for the Management of Type 2 Diabetes Mellitus and Its Complications. Int. J. Mol. Sci. 2022, 23, 10988. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Green, I.R.; Shamraiz, U.; Saleem, M.; Abbas, G.; Rehman, N.U.; Irshad, M. Therapeutic potential of glycyrrhetinic acids: A patent review (2010-2017). Expert Opin. Ther. Pat. 2018, 28, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Kalaiarasi, P.; Pugalendi, K.V. Antihyperglycemic effect of 18β-glycyrrhetinic acid, aglycone of glycyrrhizin, on streptozotocin-diabetic rats. Eur. J. Pharmacol. 2009, 606, 269–273. [Google Scholar] [CrossRef]
- Kalaiarasi, P.; Kaviarasan, K.; Pugalendi, K.V. Hypolipidemic activity of 18β-glycyrrhetinic acid on streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2009, 612, 93–97. [Google Scholar] [CrossRef]
- Na, M.; Cui, L.; Min, B.S.; Bae, K.; Yoo, J.K.; Kim, B.Y.; Oh, K.; Ahn, J.S. Protein tyrosine phosphatase 1B inhibitory activity of triterpenes isolated from Astilbe koreana. Bioorganic Med. Chem. 2006, 16, 3273–3276. [Google Scholar] [CrossRef]
- Seong, S.H.; Nguyen, D.H.; Wagle, A.; Woo, M.H.; Jung, H.A.; Choi, J.S. Experimental and Computational Study to Reveal the Potential of Non-Polar Constituents from Hizikia fusiformis as Dual Protein Tyrosine Phosphatase 1B and a-Glucosidase Inhivitors. Mar. Drugs 2019, 17, 302. [Google Scholar] [CrossRef]
- Tonkss, N.K.; Diltz, C.D.; Fischer, E.H. Purification of the Major Protein-tyrosine-phosphatases of Human Placenta. J. Biol. Chem. 1988, 263, 6722–6730. [Google Scholar] [CrossRef]
- Tonks, N.K. PTP1B: From the sidelines to the front lines! FEBS Lett. 2003, 546, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Picardi, P.K.; Calegari, V.C.; Prada, P.D.O.; Moraes, J.C.; Cristina, M.; Gomes, C.; Ueno, M.; Carvalheira, B.C.; Velloso, L.A.; Abdalla Saad, J.M. Reduction of Hypothalamic Protein Tyrosine Phosphatase Improves Insulin and Leptin Resistance in Diet-Induced Obese Rats. Endocrinology 2014, 149, 3870–3880. [Google Scholar] [CrossRef]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.; et al. Increased Insulin Sensitivity and Obesity Resistance in Mice Lacking the Protein Tyrosine Phosphatase-1B Gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2010, 12, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Lee, S.; Andersen, J.N.; Waters, S.; Shen, K.; Guo, X.; Moller, N.P.H.; Olefsky, J.M.; Lawrence, O.D.S.; Zhang, Z. Cellular Effects of Small Molecule PTP1B Inhibitors on Insulin Signaling. Biochemistry 2003, 42, 12792–12804. [Google Scholar] [CrossRef] [PubMed]
- Villamar-cruz, O.; Loza-mej, M.A.; Arias-romero, L.E.; Camacho-arroyo, I. Recent advances in PTP1B signaling in metabolism and cancer. Biosci. Rep. 2021, 41, BSR20211994. [Google Scholar] [CrossRef]
- Liu, R.; Mathieu, C.; Zhang, W.; Dupret, J.; Rodrigues Lima, F. Human Protein Tyrosine Phosphatase 1B (PTP1B): From Structure to Clinical Inhibitor Perspectives. Int. J. Mol. Sci. 2022, 23, 7027. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, H.; Zhao, Z.; Huang, M.; Wang, S.; Zhan, J. Status of research on natural protein tyrosine phosphatase 1B inhibitors as potential anti-diabetic agents: Update. Biomed. Pharmacother. 2023, 157, 113990. [Google Scholar] [CrossRef]
- Campos-Almazán, M.I.; Hernández-Campos, A.; Castillo, R.; Sierra-Campos, E.; Valdez-Solana, M.; Avitia-Domínguez, C.; Téllez-Valencia, A. Computational Methods in Cooperation with Experimental Approaches to Design Protein Tyrosine Phosphatase 1B Inhibitors in Type 2 Diabetes Drug Design: A Review of the Achievements of This Century. Pharmaceuticals 2022, 15, 866. [Google Scholar] [CrossRef] [PubMed]
- Tomasik, P.; Horton, D. Enzymatic Conversions of Starch. In Advances in Carbohydrate Chemistry and Biochemistry; Press, A., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 68, pp. 59–436. ISBN 9780123965233. [Google Scholar]
- Lebovitz, H.E. Alpha-glucosidase inhibitors. Endocrinol. Metab. Clin. N. Am. 1997, 26, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Hedrington, M.S.; Davis, S.N. Expert Opinion on Pharmacotherapy Considerations when using alpha-glucosidase inhibitors in the treatment of type 2 diabetes diabetes. Expert Opin. Pharmacother. 2019, 20, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Feng, R.; Yang, M.; Qian, C.; Wang, Z.; Liu, W.; Ma, J. Effects of metformin, acarbose, and sitagliptin monotherapy on gut microbiota in Zucker diabetic fatty rats. BMJ Open Diabetes Res. Care 2019, 7, e000717. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Yoon, J.-H.; Hahn, S.; Cho, Y.M. Efficacy and safety of combination therapy with an α-glucosidase inhibitor and a dipeptidyl peptidase-4 inhibitor in patients with type 2 diabetes mellitus: A systematic review with meta-analysis. J. Diabetes Investig. 2019, 9, 893–902. [Google Scholar] [CrossRef]
- Assefa, S.T.; Yang, E.; Chae, S.; Song, M.; Lee, J.; Cho, M.; Jang, S. Alpha Glucosidase Inhibitory Activities of Plants with Focus on Common Vegetables. Plants 2020, 9, 2. [Google Scholar] [CrossRef]
- Kumar, S.; Narwal, S.; Kumar, V.; Prakash, O. α-glucosidase inhibitors from plants: A natural approach to treat diabetes. Pharmacogn. Rev. 2011, 5, 19–30. [Google Scholar] [CrossRef]
- Joshi, S.R.; Standl, E.; Tong, N.; Shah, P.; Kalra, S.; Rathod, R. Therapeutic potential of a-glucosidase inhibitors in type 2 diabetes mellitus: An evidence-based review. Expert Opin. Pharmacother. 2015, 16, 1959–1981. [Google Scholar] [CrossRef]
- De-La-Cruz-Martínez, L.; Duran-Becerra, C.; González-Andrade, M.; Páez-Franco, J.C.; Germán-Acacio, J.M.; Espinosa-Chávez, J.; Torres-Valencia, J.M.; Pérez-Villanueva, J.; Palacios-Espinosa, J.F.; Soria-Arteche, O.; et al. Indole-and pyrazole-glycyrrhetinic acid derivatives as PTP1B inhibitors: Synthesis, in vitro and in silico studies. Molecules 2021, 26, 4375. [Google Scholar] [CrossRef]
- Flores-Bocanegra, L.; González-Andrade, M.; Bye, R.; Linares, E.; Mata, R. α-Glucosidase Inhibitors from Salvia circinata. J. Nat. Prod. 2017, 80, 1584–1593. [Google Scholar] [CrossRef]
- Son, H.-U.; Lee, S.-H. Comparison of α-glucosidase inhibition by Cudrania tricuspidata according to harvesting time. Biomed. Rep. 2013, 1, 624–628. [Google Scholar] [CrossRef]
- Calder, P.C.; Geddes, R. Acarbose is a competitive inhibitor of mammalian lysosomal acid alpha-D-glucosidases. Carbohydr. Res. 1989, 191, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Simamora, A.; Santoso, A.W.; Timotius, K.H. α-Glucosidase Inhibitory Effect of Fermented Fruit Juice of Morinda Citrifolia L and Combination Effect with Acarbose. Curr. Res. Nutr. Food Sci. 2019, 7, 218–226. [Google Scholar] [CrossRef]
- Peytam, F.; Adib, M.; Shourgeshty, R.; Mohammadi-Khanaposhtani, M.; Jahani, M.; Imanparast, S.; Faramarzi, M.A.; Mahdavi, M.; Moghadamnia, A.A.; Rastegar, H.; et al. Design and synthesis of new imidazo [1,2-b]pyrazole derivatives, in vitro α-glucosidase inhibition, kinetic and docking studies. Mol. Divers. 2019, 29, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Rojas, M.; Raja, H.; González-Andrade, M.; Rivera-Chávez, J.; Rangel-Grimaldo, M.; Rivero-Cruz, I.; Mata, R. Phytochemistry Protein tyrosine phosphatase 1B inhibitors from the fungus Malbranchea albolutea. Phytochemistry 2021, 184, 112664. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Grimaldo, M.; Macías-Rubalcava, M.L.; González-Andrade, M.; Raja, H.; Figueroa, M.; Mata, R. α-Glucosidase and Protein Tyrosine Phosphatase 1B Inhibitors from Malbranchea circinata. J. Nat. Prod. 2019, 83, 675–683. [Google Scholar] [CrossRef]
- Coronell-Tovar, A.; Cortés-Benítez, F.; González-Andrade, M. The importance of including the C-terminal domain of PTP1B1-400 to identify potential anti-diabetic inhibitors. J. Enzyme Inhib. Med. Chem. 2023, 38, 2170369. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Why is uncompetitive inhibition so rare? A possible explanation, with implications for the design of drugs and pesticides. FEBS Lett. 1986, 203, 3–6. [Google Scholar] [CrossRef]
- Whiteley, C.G. Enzyme kinetics: Partial and complete uncompetitive inhibition. Biochem. Educ. 2000, 28, 144–147. [Google Scholar] [CrossRef]
- Genovese, M.; Nesi, I.; Caselli, A.; Paoli, P. Natural α-glucosidase and protein tyrosine phosphatase 1B inhibitors: A source of scaffold molecules for synthesis of new multitarget anti-diabetic drugs. Molecules 2021, 26, 4818. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Y.; Gan, J.; Xiao, Z.-X.; Cao, Y. FitDock: Protein–ligand docking by template fitting. Brief. Bioinform. 2022, 23, bbac087. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software News and Updates Gabedit—A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human Intestinal Maltase-Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Cosmetic Ingredient Review Expert Panel. Final report on the safety assessment of glycyrrhetinic acid, potassium glycyrrhetinate, disodium succinoyl glycyrrhetinate, glyceryl glycyrrhetinate, glycyrrhetinyl stearate, stearyl glycyrrhetinate, glycyrrhizic acid, ammonium glycyrrhizate, dipotassium glycyrrhizate, disodium glycyrrhizate, trisodium glycyrrhizate, methyl glycyrrhizate, and potassium glycyrrhizinate. Int. J. Toxicol. 2007, 26, 79–112. [Google Scholar] [CrossRef]
- Szkudelski, T.; Zywert, A.; Szkudelska, K. Metabolic disturbances and defects in insulin secretion in rats with streptozotocin-nicotinamide-induced diabetes. Physiol. Res. 2013, 62, 663–670. [Google Scholar] [CrossRef]
- Diab, R.A.H.; Fares, M.; Abedi-Valugerdi, M.; Kumagai-Braesch, M.; Holgersson, J.; Hassan, M. Immunotoxicological effects of streptozotocin and alloxan: In vitro and in vivo studies. Immunol. Lett. 2015, 163, 193–198. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Ashour, M.B.; Abdel-Moneim, A.; Ahmed, O.M. Hesperidin and naringin attenuate hyperglycemia-mediated oxidative stress and proinflammatory cytokine production in high fat fed/streptozotocin-induced type 2 diabetic rats. J. Diabetes Complications 2012, 26, 483–490. [Google Scholar] [CrossRef]
- Goyal, S.N.; Reddy, N.M.; Patil, K.R.; Nakhate, K.T.; Ojha, S.; Patil, C.R.; Agrawal, Y.O. Challenges and issues with streptozotocin-induced diabetes—A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem. Biol. Interact. 2016, 244, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Al-Faris, N.A.; Al-sawadi, A.D.; Alokail, M.S. Effect of samh seeds supplementation (Mesembryanthemum forsskalei Hochst) on liver enzymes and lipid profiles of streptozotocin (STZ)-induced diabetic Wistar rats. Saudi J. Biol. Sci. 2010, 17, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Chatterjea, M.; Shinde, R. Textbook of Medical Biochemistry, 8th ed.; Jaypee Brothers Medical Publishers: New Delhi, India, 2012; ISBN 9789350254844. [Google Scholar]
- Nesti, L.; Tricò, D.; Mengozzi, A.; Natali, A. Rethinking pioglitazone as a cardioprotective agent: A new perspective on an overlooked drug. Cardiovasc. Diabetol. 2021, 20, 109. [Google Scholar] [CrossRef]
- Bellary, S.; Kyrou, I.; Brown, J.E.; Bailey, C.J. Type 2 diabetes mellitus in older adults: Clinical considerations and management. Nat. Rev. Endocrinol. 2021, 17, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.D.; Viscoli, C.; Kernan, W.N.; Young, L.H.; Furie, K.; DeFronzo, R.; Abdul-Ghani, M.; Dandona, P.; Inzucchi, S.E. Efficacy of lower doses of pioglitazone after stroke or transient ischaemic attack in patients with insulin resistance. Diabetes Obes. Metab. 2022, 24, 1150–1158. [Google Scholar] [CrossRef]
- González-Ortiz, M.; Hernández-Salazar, E.; Kam-Ramos, A.M.; Martínez-Abundis, E. Effect of pioglitazone on insulin secretion in patients with both impaired fasting glucose and impaired glucose tolerance. Diabetes Res. Clin. Pract. 2007, 75, 115–118. [Google Scholar] [CrossRef]
- Derosa, G.; Cicero, A.F.G.; Gaddi, A.; Ragonesi, P.D.; Fogari, E.; Bertone, G.; Ciccarelli, L.; Piccinni, M.N. Metabolic effects of pioglitazone and rosiglitazone in patients with diabetes and metabolic syndrome treated with glimepiride: A twelve-month, multicenter, double-blind, randomized, controlled, parallel-group trial. Clin. Ther. 2004, 26, 744–754. [Google Scholar] [CrossRef]
- Álvarez-Almazán, S.; Filisola-Villaseñor, J.G.; Alemán-González-Duhart, D.; Tamay-Cach, F.; Mendieta-Wejebe, J.E. Current molecular aspects in the development and treatment of diabetes. J. Physiol. Biochem. 2020, 76, 13–35. [Google Scholar] [CrossRef]
- Ko, B.-S.; Jang, J.S.; Hong, S.M.; Sung, S.R.; Lee, J.E.; Lee, M.Y.; Jeon, W.K.; Park, S. Changes in components, glycyrrhizin and glycyrrhetinic acid, in raw Glycyrrhiza uralensis fisch, modify insulin sensitizing and insulinotropic actions. Biosci. Biotechnol. Biochem. 2007, 71, 1452–1461. [Google Scholar] [CrossRef][Green Version]
- Gutierréz-Hernández, A.; Galván-Ciprés, Y.; Domínguez-Mendoza, E.A.; Aguirre-Vidal, Y.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Navarrete-Vázquez, G. Design, Synthesis, Antihyperglycemic Studies, and Docking Simulations of Benzimidazole-Thiazolidinedione Hybrids. J. Chem. 2019, 2019, 1650145. [Google Scholar] [CrossRef]
- Gao, C.; Dai, F.J.; Cui, H.W.; Peng, S.H.; He, Y.; Wang, X.; Yi, Z.F.; Qiu, W.W. Synthesis of novel heterocyclic ring-fused 18β-glycyrrhetinic acid derivatives with antitumor and antimetastatic activity. Chem. Biol. Drug Des. 2014, 84, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systemes. Discovery Studio Visualizer v21.1.0.20298; Dassault Systemes: San Diego, CA, USA, 2021. [Google Scholar]
- Velázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2005, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Stümpel, E.; Hartmann, H. Acute actions of insulin-like growth factor II on glucose metabolism in adult rats. Diabetology 1992, 35, 932–938. [Google Scholar] [CrossRef][Green Version]
- Elmazar, M.M.; El-abhar, H.S.; Schaalan, M.F.; Farag, N.A. Phytol/Phytanic Acid and Insulin Resistance: Potential Role of Phytanic Acid Proven by Docking Simulation and Modulation of Biochemical Alterations. PLoS ONE 2013, 8, e45638. [Google Scholar] [CrossRef] [PubMed]
- OECD Guidelines for the Testing of Chemicals Acute Oral Toxicity—Up-and-Down-Procedure (UDP). 2008, p. 27. Available online: https://www.oecd.org/env/test-no-425-acute-oral-toxicity-up-and-down-procedure-9789264071049-en.htm (accessed on 5 January 2023).
- Zafar, M.; Naeem-ul-Hassan Naqvi, S.; Ahmed, M.; Kaimkhani, Z.A. Altered Kidney Morphology and Enzymes in Streptozotocin Induced Diabetic Rats. Int. J. Morphol. 2009, 27, 719–725. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, S.; Ramírez-Espinosa, J.J.; Estrada-Soto, S.; Almanza-Pérez, J.C.; Román-Ramos, R.; Alarcón-Aguilar, F.J.; Hernández-Rosado, J.V.; Moreno-Díaz, H.; Díaz-Coutiño, D.; Navarrete-Vázquez, G. Discovery of Thiazolidine-2,4-Dione/Biphenylcarbonitrile Hybrid as Dual PPAR α/γ Modulator with Antidiabetic Effect: In vitro, In Silico and In Vivo Approaches. Chem. Biol. Drug Des. 2013, 81, 474–483. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Autodock | GOLD | |
|---|---|---|---|
| kcal/mol (RMSD) a | CS (RMSD) b | GS (RMSD) c | |
| VOG | −8.7 (0.85) | 63.3 (0.65) | 41.2 (0.65) |
| FC-122 | −8.7 | 54.2 | 34.8 |
| Acarbose | −8.9 | 79.3 | 4.9 |
| Dose (mg/kg) | Weight Gain for 2 Weeks (g) | Toxic Effects |
|---|---|---|
| Compound FC-114 | ||
| 2000 | 21.5 | None |
| 550 | 23.4 | |
| 175 | 22.8 | |
| Compound FC-122 | ||
| 2000 | 26.4 | None |
| 550 | 25.2 | |
| 175 | 23.5 |
| Vehicle | T2D | T2D + Glibenclamide | T2D + FC-114 | T2D + FC-122 | T2D + PIO | T2D + Acarbose |
|---|---|---|---|---|---|---|
| Week 5 (day 1 of treatment) | ||||||
| 21,681 | 75,891 | 62,022 | 53,895 | 58,941 | 49,677 | 81,327 |
| Week 6 (day 7 of treatment) | ||||||
| 21,228 | 72,447 | 67,311 | 62,944 | 57,003 | 43,584 | 81,507 |
| Week 7 (day 14 of treatment) | ||||||
| 21,831 | 66,504 | 50,874 | 57,131 | 53,076 | 40,044 | 71,240 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Almazán, S.; Solís-Domínguez, L.C.; Duperou-Luna, P.; Fuerte-Gómez, T.; González-Andrade, M.; Aranda-Barradas, M.E.; Palacios-Espinosa, J.F.; Pérez-Villanueva, J.; Matadamas-Martínez, F.; Miranda-Castro, S.P.; et al. Anti-Diabetic Activity of Glycyrrhetinic Acid Derivatives FC-114 and FC-122: Scale-Up, In Silico, In Vitro, and In Vivo Studies. Int. J. Mol. Sci. 2023, 24, 12812. https://doi.org/10.3390/ijms241612812
Álvarez-Almazán S, Solís-Domínguez LC, Duperou-Luna P, Fuerte-Gómez T, González-Andrade M, Aranda-Barradas ME, Palacios-Espinosa JF, Pérez-Villanueva J, Matadamas-Martínez F, Miranda-Castro SP, et al. Anti-Diabetic Activity of Glycyrrhetinic Acid Derivatives FC-114 and FC-122: Scale-Up, In Silico, In Vitro, and In Vivo Studies. International Journal of Molecular Sciences. 2023; 24(16):12812. https://doi.org/10.3390/ijms241612812
Chicago/Turabian StyleÁlvarez-Almazán, Samuel, Luz Cassandra Solís-Domínguez, Paulina Duperou-Luna, Teresa Fuerte-Gómez, Martin González-Andrade, María E. Aranda-Barradas, Juan Francisco Palacios-Espinosa, Jaime Pérez-Villanueva, Félix Matadamas-Martínez, Susana Patricia Miranda-Castro, and et al. 2023. "Anti-Diabetic Activity of Glycyrrhetinic Acid Derivatives FC-114 and FC-122: Scale-Up, In Silico, In Vitro, and In Vivo Studies" International Journal of Molecular Sciences 24, no. 16: 12812. https://doi.org/10.3390/ijms241612812
APA StyleÁlvarez-Almazán, S., Solís-Domínguez, L. C., Duperou-Luna, P., Fuerte-Gómez, T., González-Andrade, M., Aranda-Barradas, M. E., Palacios-Espinosa, J. F., Pérez-Villanueva, J., Matadamas-Martínez, F., Miranda-Castro, S. P., Mercado-Márquez, C., & Cortés-Benítez, F. (2023). Anti-Diabetic Activity of Glycyrrhetinic Acid Derivatives FC-114 and FC-122: Scale-Up, In Silico, In Vitro, and In Vivo Studies. International Journal of Molecular Sciences, 24(16), 12812. https://doi.org/10.3390/ijms241612812