

Genetic Mechanisms of Migraine: Insights from Monogenic Migraine Mutations



Abstract
1. Introduction
2. CSD, Genetics, and Animal Models
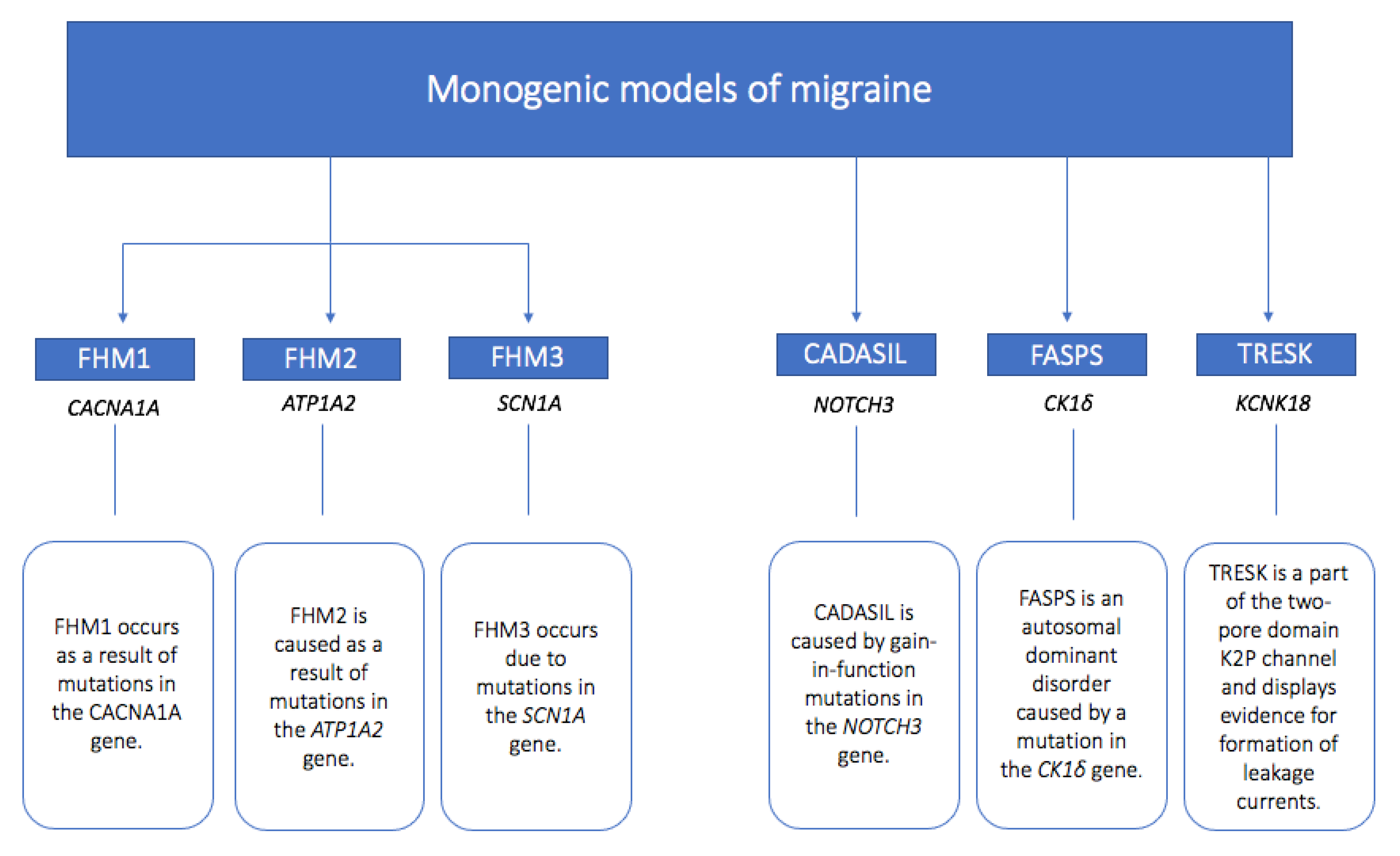
3. Monogenic Models of Migraine
3.1. Familial Hemiplegic Migraine Type 1 (FHM1)
3.2. Familial Hemiplegic Migraine Type 2 (FHM2)
3.3. Familial Hemiplegic Migraine Type 3 (FHM3)
3.4. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL)
3.5. Familial Advanced Sleep-Phase Syndrome (FASPS)
3.6. TWIK-Related Spinal Cord Potassium Channel (TRESK)
4. Insights and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef] [PubMed]
- Karsan, N.; Goadsby, P.J. Biological insights from the premonitory symptoms of migraine. Nat. Rev. Neurol. 2018, 14, 699–710. [Google Scholar] [CrossRef]
- Schwedt, T.J.; Dodick, D.W. Advanced neuroimaging of migraine. Lancet Neurol. 2009, 8, 560–568. [Google Scholar] [CrossRef]
- Petrusic, I.; Viana, M.; Dakovic, M.; Zidverc-Trajkovic, J. Application of the Migraine Aura Complexity Score (MACS): Clinical and Neuroimaging Study. Front. Neurol. 2019, 10, 1112. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M.; Hansen, J.M.; BO, A.D.; Olesen, J. Human models of migraine—Short-term pain for long-term gain. Nat. Rev. Neurol. 2017, 13, 713–724. [Google Scholar] [CrossRef]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the target of new migraine therapies—Successful translation from bench to clinic. Nat. Rev. Neurol. 2018, 14, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.F. Calcitonin gene-related peptide (CGRP): A new target for migraine. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 533–552. [Google Scholar] [CrossRef]
- Rasmussen, B.K.; Olesen, J. Migraine with aura and migraine without aura: An epidemiological study. Cephalalgia 1992, 12, 221–228, discussion 186. [Google Scholar] [CrossRef]
- Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia 2018, 38, 1–211. [Google Scholar] [CrossRef]
- Honkasalo, M.L.; Kaprio, J.; Winter, T.; Heikkila, K.; Sillanpaa, M.; Koskenvuo, M. Migraine and concomitant symptoms among 8167 adult twin pairs. Headache 1995, 35, 70–78. [Google Scholar] [CrossRef]
- Larsson, B.; Bille, B.; Pedersen, N.L. Genetic influence in headaches: A Swedish twin study. Headache 1995, 35, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Mulder, E.J.; Van Baal, C.; Gaist, D.; Kallela, M.; Kaprio, J.; Svensson, D.A.; Nyholt, D.R.; Martin, N.G.; MacGregor, A.J.; Cherkas, L.F.; et al. Genetic and environmental influences on migraine: A twin study across six countries. Twin Res. 2003, 6, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, V.; Gervil, M.; Kyvik, K.O.; Olesen, J.; Russell, M.B. Evidence of a genetic factor in migraine with aura: A population-based Danish twin study. Ann. Neurol. 1999, 45, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Polderman, T.J.; Benyamin, B.; de Leeuw, C.A.; Sullivan, P.F.; van Bochoven, A.; Visscher, P.M.; Posthuma, D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 2015, 47, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.B.; Ulrich, V.; Gervil, M.; Olesen, J. Migraine without aura and migraine with aura are distinct disorders. A population-based twin survey. Headache 2002, 42, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, M.; Hougaard, A.; Amin, F.M.; Ashina, M. Can migraine aura be provoked experimentally? A systematic review of potential methods for the provocation of migraine aura. Cephalalgia 2017, 37, 74–88. [Google Scholar] [CrossRef]
- Ashina, H.; Christensen, R.H.; Ashina, M. Provoked versus spontaneous migraine attacks: Pathophysiological similarities and differences. J. Headache Pain 2022, 23, 87. [Google Scholar] [CrossRef]
- Hansen, J.M.; Thomsen, L.L.; Olesen, J.; Ashina, M. Familial hemiplegic migraine type 1 shows no hypersensitivity to nitric oxide. Cephalalgia 2008, 28, 496–505. [Google Scholar] [CrossRef]
- Hansen, J.M.; Thomsen, L.L.; Olesen, J.; Ashina, M. Calcitonin gene-related peptide does not cause migraine attacks in patients with familial hemiplegic migraine. Headache 2011, 51, 544–553. [Google Scholar] [CrossRef]
- Hansen, J.M.; Thomsen, L.L.; Marconi, R.; Casari, G.; Olesen, J.; Ashina, M. Familial hemiplegic migraine type 2 does not share hypersensitivity to nitric oxide with common types of migraine. Cephalalgia 2008, 28, 367–375. [Google Scholar] [CrossRef]
- Anttila, V.; Winsvold, B.S.; Gormley, P.; Kurth, T.; Bettella, F.; McMahon, G.; Kallela, M.; Malik, R.; de Vries, B.; Terwindt, G.; et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet. 2013, 45, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Hautakangas, H.; Winsvold, B.S.; Ruotsalainen, S.E.; Bjornsdottir, G.; Harder, A.V.E.; Kogelman, L.J.A.; Thomas, L.F.; Noordam, R.; Benner, C.; Gormley, P.; et al. Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat. Genet. 2022, 54, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Gormley, P.; Anttila, V.; Winsvold, B.S.; Palta, P.; Esko, T.; Pers, T.H.; Farh, K.H.; Cuenca-Leon, E.; Muona, M.; Furlotte, N.A.; et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 2016, 48, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Schurks, M. Genetics of migraine in the age of genome-wide association studies. J. Headache Pain 2012, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.G.; Albury, C.L.; Griffiths, L.R. Advances in genetics of migraine. J. Headache Pain 2019, 20, 72. [Google Scholar] [CrossRef]
- Hiekkala, M.E.; Vuola, P.; Artto, V.; Happola, P.; Happola, E.; Vepsalainen, S.; Cuenca-Leon, E.; Lal, D.; Gormley, P.; Hamalainen, E.; et al. The contribution of CACNA1A, ATP1A2 and SCN1A mutations in hemiplegic migraine: A clinical and genetic study in Finnish migraine families. Cephalalgia 2018, 38, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, A.R.; Bhatia, K.P.; Stamelou, M.; Dale, R.C.; Kurian, M.A.; Schneider, S.A.; Wali, G.M.; Counihan, T.; Schapira, A.H.; Spacey, S.D.; et al. PRRT2 gene mutations: From paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology 2012, 79, 2115–2121. [Google Scholar] [CrossRef]
- Brennan, K.C.; Bates, E.A.; Shapiro, R.E.; Zyuzin, J.; Hallows, W.C.; Huang, Y.; Lee, H.Y.; Jones, C.R.; Fu, Y.H.; Charles, A.C.; et al. Casein kinase idelta mutations in familial migraine and advanced sleep phase. Sci. Transl. Med. 2013, 5, 183ra156. [Google Scholar] [CrossRef]
- Xu, Y.; Padiath, Q.S.; Shapiro, R.E.; Jones, C.R.; Wu, S.C.; Saigoh, N.; Saigoh, K.; Ptacek, L.J.; Fu, Y.H. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature 2005, 434, 640–644. [Google Scholar] [CrossRef]
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.G. Cadasil. Lancet Neurol. 2009, 8, 643–653. [Google Scholar] [CrossRef]
- Leao, A.A. Spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1944, 7, 359–390. [Google Scholar] [CrossRef]
- Leao, A.A. Further observations on the spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1947, 10, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J. Migraine, aura, and cortical spreading depression: Why are we still talking about it? Ann. Neurol. 2001, 49, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Akerman, S.; Goadsby, P.J. Topiramate inhibits cortical spreading depression in rat and cat: Impact in migraine aura. Neuroreport 2005, 16, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M. Pathophysiology of the migraine aura. The spreading depression theory. Brain 1994, 117 Pt 1, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Eikermann-Haerter, K.; Ayata, C. Cortical spreading depression and migraine. Curr. Neurol. Neurosci. Rep. 2010, 10, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Aurora, S.K.; Nagesh, V.; Patel, S.C.; Welch, K.M. Functional MRI-BOLD of brainstem structures during visually triggered migraine. Neurology 2002, 59, 72–78. [Google Scholar] [CrossRef]
- Hadjikhani, N.; Sanchez Del Rio, M.; Wu, O.; Schwartz, D.; Bakker, D.; Fischl, B.; Kwong, K.K.; Cutrer, F.M.; Rosen, B.R.; Tootell, R.B.; et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. USA 2001, 98, 4687–4692. [Google Scholar] [CrossRef]
- Olesen, J.; Larsen, B.; Lauritzen, M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann. Neurol. 1981, 9, 344–352. [Google Scholar] [CrossRef]
- Kraig, R.P.; Nicholson, C. Extracellular ionic variations during spreading depression. Neuroscience 1978, 3, 1045–1059. [Google Scholar] [CrossRef]
- Fabricius, M.; Jensen, L.H.; Lauritzen, M. Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res. 1993, 612, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Zeevalk, G.D.; Nicklas, W.J. Evidence that the loss of the voltage-dependent Mg2+ block at the N-methyl-D-aspartate receptor underlies receptor activation during inhibition of neuronal metabolism. J. Neurochem. 1992, 59, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Grafstein, B. Mechanism of spreading cortical depression. J. Neurophysiol. 1956, 19, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Piilgaard, H.; Lauritzen, M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J. Cereb. Blood Flow Metab. 2009, 29, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M. Long-lasting reduction of cortical blood flow of the brain after spreading depression with preserved autoregulation and impaired CO2 response. J. Cereb. Blood Flow Metab. 1984, 4, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, R.; Cui, L.; Yong, C.; Bowyer, S.; Klein, R.M.; Welch, K.M.; Berman, N.E. Cortical spreading depression and gene regulation: Relevance to migraine. Ann. Neurol. 2002, 51, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, R.C.; Kaja, S.; Plomp, J.J.; Frants, R.R.; van den Maagdenberg, A.M.; Ferrari, M.D. Genetic models of migraine. Arch. Neurol. 2007, 64, 643–646. [Google Scholar] [CrossRef]
- Tottene, A.; Pivotto, F.; Fellin, T.; Cesetti, T.; van den Maagdenberg, A.M.; Pietrobon, D. Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J. Biol. Chem. 2005, 280, 17678–17686. [Google Scholar] [CrossRef]
- Van den Maagdenberg, A.M.; Pizzorusso, T.; Kaja, S.; Terpolilli, N.; Shapovalova, M.; Hoebeek, F.E.; Barrett, C.F.; Gherardini, L.; van de Ven, R.C.; Todorov, B.; et al. High cortical spreading depression susceptibility and migraine-associated symptoms in Cav2.1 S218L mice. Ann. Neurol. 2010, 67, 85–98. [Google Scholar] [CrossRef]
- Pietrobon, D. Migraine: New molecular mechanisms. Neuroscientist 2005, 11, 373–386. [Google Scholar] [CrossRef]
- Tolner, E.A.; Houben, T.; Terwindt, G.M.; de Vries, B.; Ferrari, M.D.; van den Maagdenberg, A. From migraine genes to mechanisms. Pain 2015, 156 (Suppl. 1), S64–S74. [Google Scholar] [CrossRef] [PubMed]
- Vuralli, D.; Karatas, H.; Yemisci, M.; Bolay, H. Updated review on the link between cortical spreading depression and headache disorders. Expert Rev. Neurother. 2021, 21, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Karsan, N.; Palethorpe, D.; Rattanawong, W.; Marin, J.C.; Bhola, R.; Goadsby, P.J. Flunarizine in migraine-related headache prevention: Results from 200 patients treated in the UK. Eur. J. Neurol. 2018, 25, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef]
- De Fusco, M.; Marconi, R.; Silvestri, L.; Atorino, L.; Rampoldi, L.; Morgante, L.; Ballabio, A.; Aridon, P.; Casari, G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 2003, 33, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Kors, E.E.; Terwindt, G.M.; Vermeulen, F.L.; Fitzsimons, R.B.; Jardine, P.E.; Heywood, P.; Love, S.; van den Maagdenberg, A.M.; Haan, J.; Frants, R.R.; et al. Delayed cerebral edema and fatal coma after minor head trauma: Role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 2001, 49, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, M.; Ursu, S.; Lehmann-Horn, F.; Veneziano, L.; Antonini, G.; Giunti, P.; Frontali, M.; Jurkat-Rott, K. A G301R Na+/K+ -ATPase mutation causes familial hemiplegic migraine type 2 with cerebellar signs. Neurogenetics 2004, 5, 177–185. [Google Scholar] [CrossRef]
- Pietrobon, D. Familial hemiplegic migraine. Neurotherapeutics 2007, 4, 274–284. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell Calcium 1998, 24, 307–323. [Google Scholar] [CrossRef]
- Hans, M.; Luvisetto, S.; Williams, M.E.; Spagnolo, M.; Urrutia, A.; Tottene, A.; Brust, P.F.; Johnson, E.C.; Harpold, M.M.; Stauderman, K.A.; et al. Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. J. Neurosci. 1999, 19, 1610–1619. [Google Scholar] [CrossRef]
- Labrum, R.W.; Rajakulendran, S.; Graves, T.D.; Eunson, L.H.; Bevan, R.; Sweeney, M.G.; Hammans, S.R.; Tubridy, N.; Britton, T.; Carr, L.J.; et al. Large scale calcium channel gene rearrangements in episodic ataxia and hemiplegic migraine: Implications for diagnostic testing. J. Med. Genet. 2009, 46, 786–791. [Google Scholar] [CrossRef]
- Tottene, A.; Conti, R.; Fabbro, A.; Vecchia, D.; Shapovalova, M.; Santello, M.; van den Maagdenberg, A.M.; Ferrari, M.D.; Pietrobon, D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Cav2.1 knockin migraine mice. Neuron 2009, 61, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Tottene, A.; Fellin, T.; Pagnutti, S.; Luvisetto, S.; Striessnig, J.; Fletcher, C.; Pietrobon, D. Familial hemiplegic migraine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 13284–13289. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, N.; Haan, J.; Stam, A.H.; Vijfhuizen, L.S.; Koelewijn, S.C.; Smagge, A.; de Vries, B.; Ferrari, M.D.; van den Maagdenberg, A.; Terwindt, G.M. Clinical spectrum of hemiplegic migraine and chances of finding a pathogenic mutation. Neurology 2018, 90, e575–e582. [Google Scholar] [CrossRef] [PubMed]
- Van den Maagdenberg, A.M.; Pietrobon, D.; Pizzorusso, T.; Kaja, S.; Broos, L.A.; Cesetti, T.; van de Ven, R.C.; Tottene, A.; van der Kaa, J.; Plomp, J.J.; et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 2004, 41, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Marchenkova, A.; van den Maagdenberg, A.M.; Nistri, A. Loss of inhibition by brain natriuretic peptide over P2X3 receptors contributes to enhanced spike firing of trigeminal ganglion neurons in a mouse model of familial hemiplegic migraine type-1. Neuroscience 2016, 331, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Hullugundi, S.K.; Ansuini, A.; Ferrari, M.D.; van den Maagdenberg, A.M.; Nistri, A. A hyperexcitability phenotype in mouse trigeminal sensory neurons expressing the R192Q Cacna1a missense mutation of familial hemiplegic migraine type-1. Neuroscience 2014, 266, 244–254. [Google Scholar] [CrossRef]
- Chanda, M.L.; Tuttle, A.H.; Baran, I.; Atlin, C.; Guindi, D.; Hathaway, G.; Israelian, N.; Levenstadt, J.; Low, D.; Macrae, L.; et al. Behavioral evidence for photophobia and stress-related ipsilateral head pain in transgenic Cacna1a mutant mice. Pain 2013, 154, 1254–1262. [Google Scholar] [CrossRef]
- Khennouf, L.; Gesslein, B.; Lind, B.L.; van den Maagdenberg, A.M.; Lauritzen, M. Activity-dependent calcium, oxygen, and vascular responses in a mouse model of familial hemiplegic migraine type 1. Ann. Neurol. 2016, 80, 219–232. [Google Scholar] [CrossRef]
- Eikermann-Haerter, K.; Dilekoz, E.; Kudo, C.; Savitz, S.I.; Waeber, C.; Baum, M.J.; Ferrari, M.D.; van den Maagdenberg, A.M.; Moskowitz, M.A.; Ayata, C. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J. Clin. Investig. 2009, 119, 99–109. [Google Scholar] [CrossRef]
- Eikermann-Haerter, K.; Baum, M.J.; Ferrari, M.D.; van den Maagdenberg, A.M.; Moskowitz, M.A.; Ayata, C. Androgenic suppression of spreading depression in familial hemiplegic migraine type 1 mutant mice. Ann. Neurol. 2009, 66, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, R.; Zucca, C.; Tonelli, A.; Bonato, S.; Baschirotto, C.; Zanotta, N.; Epifanio, R.; Righini, A.; Bresolin, N.; Bassi, M.T.; et al. A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J. Neurol. Neurosurg. Psychiatry 2010, 81, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Eikermann-Haerter, K.; Yuzawa, I.; Qin, T.; Wang, Y.; Baek, K.; Kim, Y.R.; Hoffmann, U.; Dilekoz, E.; Waeber, C.; Ferrari, M.D.; et al. Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J. Neurosci. 2011, 31, 5755–5763. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.M.; Bohnet, B.; LeDue, J.; Yung, A.C.; Garcia, E.; Tyson, J.R.; Alles, S.R.; Han, H.; van den Maagdenberg, A.M.; Kozlowski, P.; et al. In vivo imaging reveals that pregabalin inhibits cortical spreading depression and propagation to subcortical brain structures. Proc. Natl. Acad. Sci. USA 2017, 114, 2401–2406. [Google Scholar] [CrossRef]
- Park, J.; Moon, H.; Akerman, S.; Holland, P.R.; Lasalandra, M.P.; Andreou, A.P.; Ferrari, M.D.; van den Maagdenberg, A.M.; Goadsby, P.J. Differential trigeminovascular nociceptive responses in the thalamus in the familial hemiplegic migraine 1 knock-in mouse: A Fos protein study. Neurobiol. Dis. 2014, 64, 1–7. [Google Scholar] [CrossRef]
- Fioretti, B.; Catacuzzeno, L.; Sforna, L.; Gerke-Duncan, M.B.; van den Maagdenberg, A.M.; Franciolini, F.; Connor, M.; Pietrobon, D. Trigeminal ganglion neuron subtype-specific alterations of CaV2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J. Physiol. 2011, 589, 5879–5895. [Google Scholar] [CrossRef] [PubMed]
- Verriello, L.; Pauletto, G.; Nilo, A.; Lonigro, I.; Betto, E.; Valente, M.; Curcio, F.; Gigli, G.L. Epilepsy and episodic ataxia type 2: Family study and review of the literature. J. Neurol. 2021, 268, 4296–4302. [Google Scholar] [CrossRef] [PubMed]
- Watase, K.; Barrett, C.F.; Miyazaki, T.; Ishiguro, T.; Ishikawa, K.; Hu, Y.; Unno, T.; Sun, Y.; Kasai, S.; Watanabe, M.; et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc. Natl. Acad. Sci. USA 2008, 105, 11987–11992. [Google Scholar] [CrossRef]
- Lipman, A.R.; Fan, X.; Shen, Y.; Chung, W.K. Clinical and genetic characterization of CACNA1A-related disease. Clin. Genet. 2022, 102, 288–295. [Google Scholar] [CrossRef]
- Manickam, A.H.; Ramasamy, S. Mutations in the Voltage Dependent Calcium Channel CACNA1A (P/Q type alpha 1A subunit) Causing Neurological Disorders—An Overview. Neurol. India 2021, 69, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; Tavraz, N.N.; Junghans, C. ATP1A2 Mutations in Migraine: Seeing through the Facets of an Ion Pump onto the Neurobiology of Disease. Front. Physiol. 2016, 7, 239. [Google Scholar] [CrossRef] [PubMed]
- Gritz, S.M.; Radcliffe, R.A. Genetic effects of ATP1A2 in familial hemiplegic migraine type II and animal models. Hum. Genom. 2013, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Tavraz, N.N.; Friedrich, T.; Durr, K.L.; Koenderink, J.B.; Bamberg, E.; Freilinger, T.; Dichgans, M. Diverse functional consequences of mutations in the Na+/K+-ATPase alpha2-subunit causing familial hemiplegic migraine type 2. J. Biol. Chem. 2008, 283, 31097–31106. [Google Scholar] [CrossRef] [PubMed]
- Tavraz, N.N.; Durr, K.L.; Koenderink, J.B.; Freilinger, T.; Bamberg, E.; Dichgans, M.; Friedrich, T. Impaired plasma membrane targeting or protein stability by certain ATP1A2 mutations identified in sporadic or familial hemiplegic migraine. Channels 2009, 3, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, N.; Blom, D.E.; Stam, A.H.; Vijfhuizen, L.S.; Hageman, A.; van Vliet, J.A.; Ferrari, M.D.; van den Maagdenberg, A.; Haan, J.; Terwindt, G.M. Recurrent coma and fever in familial hemiplegic migraine type 2. A prospective 15-year follow-up of a large family with a novel ATP1A2 mutation. Cephalalgia 2017, 37, 737–755. [Google Scholar] [CrossRef] [PubMed]
- Sampedro Castaneda, M.; Zanoteli, E.; Scalco, R.S.; Scaramuzzi, V.; Marques Caldas, V.; Conti Reed, U.; da Silva, A.M.S.; O’Callaghan, B.; Phadke, R.; Bugiardini, E.; et al. A novel ATP1A2 mutation in a patient with hypokalaemic periodic paralysis and CNS symptoms. Brain 2018, 141, 3308–3318. [Google Scholar] [CrossRef]
- Riant, F.; Ducros, A.; Ploton, C.; Barbance, C.; Depienne, C.; Tournier-Lasserve, E. De novo mutations in ATP1A2 and CACNA1A are frequent in early-onset sporadic hemiplegic migraine. Neurology 2010, 75, 967–972. [Google Scholar] [CrossRef]
- Imbrici, P.; Jaffe, S.L.; Eunson, L.H.; Davies, N.P.; Herd, C.; Robertson, R.; Kullmann, D.M.; Hanna, M.G. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain 2004, 127, 2682–2692. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xiao, H.; Qin, X.; Nong, Y.; Zou, D.; Wu, Y. The genetic relationship between epilepsy and hemiplegic migraine. Neuropsychiatr. Dis. Treat. 2017, 13, 1175–1179. [Google Scholar] [CrossRef]
- Haut, S.R.; Bigal, M.E.; Lipton, R.B. Chronic disorders with episodic manifestations: Focus on epilepsy and migraine. Lancet Neurol. 2006, 5, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.R.; Tolner, E.A.; Keezer, M.R.; Ferrari, M.D.; Sander, J.W. Headache in people with epilepsy. Nat. Rev. Neurol. 2021, 17, 529–544. [Google Scholar] [CrossRef]
- Deprez, L.; Weckhuysen, S.; Peeters, K.; Deconinck, T.; Claeys, K.G.; Claes, L.R.; Suls, A.; Van Dyck, T.; Palmini, A.; Matthijs, G.; et al. Epilepsy as part of the phenotype associated with ATP1A2 mutations. Epilepsia 2008, 49, 500–508. [Google Scholar] [CrossRef]
- Ikeda, K.; Onaka, T.; Yamakado, M.; Nakai, J.; Ishikawa, T.O.; Taketo, M.M.; Kawakami, K. Degeneration of the amygdala/piriform cortex and enhanced fear/anxiety behaviors in sodium pump alpha2 subunit (Atp1a2)-deficient mice. J. Neurosci. 2003, 23, 4667–4676. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, F.P.; Curry, C.J.; Hevner, R.; Elliott, S.; Fisher, J.H.; Turocy, J.; Dobyns, W.B.; Costa, L.A.; Freitas, E.; Kitajima, J.P.; et al. Biallelic loss of function variants in ATP1A2 cause hydrops fetalis, microcephaly, arthrogryposis and extensive cortical malformations. Eur. J. Med. Genet. 2020, 63, 103624. [Google Scholar] [CrossRef] [PubMed]
- Unekawa, M.; Ikeda, K.; Tomita, Y.; Kawakami, K.; Suzuki, N. Enhanced susceptibility to cortical spreading depression in two types of Na+,K+-ATPase alpha2 subunit-deficient mice as a model of familial hemiplegic migraine 2. Cephalalgia 2018, 38, 1515–1524. [Google Scholar] [CrossRef]
- Leo, L.; Gherardini, L.; Barone, V.; De Fusco, M.; Pietrobon, D.; Pizzorusso, T.; Casari, G. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLoS Genet. 2011, 7, e1002129. [Google Scholar] [CrossRef]
- Capuani, C.; Melone, M.; Tottene, A.; Bragina, L.; Crivellaro, G.; Santello, M.; Casari, G.; Conti, F.; Pietrobon, D. Defective glutamate and K+ clearance by cortical astrocytes in familial hemiplegic migraine type 2. EMBO Mol. Med. 2016, 8, 967–986. [Google Scholar] [CrossRef]
- Bottger, P.; Glerup, S.; Gesslein, B.; Illarionova, N.B.; Isaksen, T.J.; Heuck, A.; Clausen, B.H.; Fuchtbauer, E.M.; Gramsbergen, J.B.; Gunnarson, E.; et al. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci. Rep. 2016, 6, 22047. [Google Scholar] [CrossRef]
- Smith, S.E.; Chen, X.; Brier, L.M.; Bumstead, J.R.; Rensing, N.R.; Ringel, A.E.; Shin, H.; Oldenborg, A.; Crowley, J.R.; Bice, A.R.; et al. Astrocyte deletion of alpha2-Na/K ATPase triggers episodic motor paralysis in mice via a metabolic pathway. Nat. Commun. 2020, 11, 6164. [Google Scholar] [CrossRef]
- Rosenberg, L.; Butler, N.; Seng, E.K. Health Behaviors in Episodic Migraine: Why Behavior Change Matters. Curr. Pain Headache Rep. 2018, 22, 65. [Google Scholar] [CrossRef]
- Noruzzadeh, R.; Modabbernia, A.; Aghamollaii, V.; Ghaffarpour, M.; Harirchian, M.H.; Salahi, S.; Nikbakht, N.; Noruzi, N.; Tafakhori, A. Memantine for Prophylactic Treatment of Migraine Without Aura: A Randomized Double-Blind Placebo-Controlled Study. Headache 2016, 56, 95–103. [Google Scholar] [CrossRef]
- Bigal, M.; Rapoport, A.; Sheftell, F.; Tepper, D.; Tepper, S. Memantine in the preventive treatment of refractory migraine. Headache 2008, 48, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Jin, S.; Chen, X.; Yang, B. The Efficacy of Memantine for the Treatment of Migraine: A Meta-Analysis of Randomized Controlled Studies. Clin. Neuropharmacol. 2021, 44, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Freilinger, T.; Eckstein, G.; Babini, E.; Lorenz-Depiereux, B.; Biskup, S.; Ferrari, M.D.; Herzog, J.; van den Maagdenberg, A.M.; Pusch, M.; et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005, 366, 371–377. [Google Scholar] [CrossRef]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Meng, H.; Xu, H.Q.; Yu, L.; Lin, G.W.; He, N.; Su, T.; Shi, Y.W.; Li, B.; Wang, J.; Liu, X.R.; et al. The SCN1A mutation database: Updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum. Mutat. 2015, 36, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Scheffer, I.E.; Nabbout, R.; Suls, A.; De Jonghe, P.; Zara, F.; Guerrini, R. The genetics of Dravet syndrome. Epilepsia 2011, 52 (Suppl. 2), 24–29. [Google Scholar] [CrossRef]
- Castro, M.J.; Stam, A.H.; Lemos, C.; de Vries, B.; Vanmolkot, K.R.; Barros, J.; Terwindt, G.M.; Frants, R.R.; Sequeiros, J.; Ferrari, M.D.; et al. First mutation in the voltage-gated Nav1.1 subunit gene SCN1A with co-occurring familial hemiplegic migraine and epilepsy. Cephalalgia 2009, 29, 308–313. [Google Scholar] [CrossRef]
- Escayg, A.; Goldin, A.L. Sodium channel SCN1A and epilepsy: Mutations and mechanisms. Epilepsia 2010, 51, 1650–1658. [Google Scholar] [CrossRef]
- Bertelli, S.; Barbieri, R.; Pusch, M.; Gavazzo, P. Gain of function of sporadic/familial hemiplegic migraine-causing SCN1A mutations: Use of an optimized cDNA. Cephalalgia 2019, 39, 477–488. [Google Scholar] [CrossRef]
- Cestele, S.; Labate, A.; Rusconi, R.; Tarantino, P.; Mumoli, L.; Franceschetti, S.; Annesi, G.; Mantegazza, M.; Gambardella, A. Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia 2013, 54, 927–935. [Google Scholar] [CrossRef]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Jansen, N.A.; Dehghani, A.; Linssen, M.M.L.; Breukel, C.; Tolner, E.A.; van den Maagdenberg, A. First FHM3 mouse model shows spontaneous cortical spreading depolarizations. Ann. Clin. Transl. Neurol. 2020, 7, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Hadjikhani, N.; Vincent, M. Neuroimaging clues of migraine aura. J. Headache Pain 2019, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Desroches, M.; Faugeras, O.; Krupa, M.; Mantegazza, M. Modeling cortical spreading depression induced by the hyperactivity of interneurons. J. Comput. Neurosci. 2019, 47, 125–140. [Google Scholar] [CrossRef]
- Auffenberg, E.; Hedrich, U.B.; Barbieri, R.; Miely, D.; Groschup, B.; Wuttke, T.V.; Vogel, N.; Luhrs, P.; Zanardi, I.; Bertelli, S.; et al. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine model. J. Clin. Investig. 2021, 131, e142202. [Google Scholar] [CrossRef]
- Chever, O.; Zerimech, S.; Scalmani, P.; Lemaire, L.; Pizzamiglio, L.; Loucif, A.; Ayrault, M.; Krupa, M.; Desroches, M.; Duprat, F.; et al. Initiation of migraine-related cortical spreading depolarization by hyperactivity of GABAergic neurons and NaV1.1 channels. J. Clin. Investig. 2021, 131, e142203. [Google Scholar] [CrossRef]
- Joutel, A.; Vahedi, K.; Corpechot, C.; Troesch, A.; Chabriat, H.; Vayssiere, C.; Cruaud, C.; Maciazek, J.; Weissenbach, J.; Bousser, M.G.; et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 1997, 350, 1511–1515. [Google Scholar] [CrossRef]
- Chabriat, H.; Joutel, A.; Tournier-Lasserve, E.; Bousser, M.G. CADASIL: Yesterday, today, tomorrow. Eur. J. Neurol. 2020, 27, 1588–1595. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Q.; Wang, Q.; Luan, S.; Dong, X.; Cao, H.; Tao, D.; Dong, H.; Ji, X. A case of CADASIL caused by NOTCH3 c.512_605delinsA heterozygous mutation. J. Clin. Lab. Anal. 2021, 35, e24027. [Google Scholar] [CrossRef] [PubMed]
- Rutten, J.W.; Van Eijsden, B.J.; Duering, M.; Jouvent, E.; Opherk, C.; Pantoni, L.; Federico, A.; Dichgans, M.; Markus, H.S.; Chabriat, H.; et al. The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1–6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7–34 pathogenic variant. Genet. Med. 2019, 21, 676–682. [Google Scholar] [CrossRef]
- Papakonstantinou, E.; Bacopoulou, F.; Brouzas, D.; Megalooikonomou, V.; D’Elia, D.; Bongcam-Rudloff, E.; Vlachakis, D. NOTCH3 and CADASIL syndrome: A genetic and structural overview. EMBnet J. 2019, 24, e921. [Google Scholar] [CrossRef]
- Tan, R.Y.; Markus, H.S. CADASIL: Migraine, Encephalopathy, Stroke and Their Inter-Relationships. PLoS ONE 2016, 11, e0157613. [Google Scholar] [CrossRef]
- Dichgans, M.; Mayer, M.; Uttner, I.; Bruning, R.; Muller-Hocker, J.; Rungger, G.; Ebke, M.; Klockgether, T.; Gasser, T. The phenotypic spectrum of CADASIL: Clinical findings in 102 cases. Ann. Neurol. 1998, 44, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Liem, M.K.; Oberstein, S.A.; van der Grond, J.; Ferrari, M.D.; Haan, J. CADASIL and migraine: A narrative review. Cephalalgia 2010, 30, 1284–1289. [Google Scholar] [CrossRef]
- Oka, F.; Lee, J.H.; Yuzawa, I.; Li, M.; von Bornstaedt, D.; Eikermann-Haerter, K.; Qin, T.; Chung, D.Y.; Sadeghian, H.; Seidel, J.L.; et al. CADASIL mutations sensitize the brain to ischemia via spreading depolarizations and abnormal extracellular potassium homeostasis. J. Clin. Investig. 2022, 132, e149759. [Google Scholar] [CrossRef]
- Kruit, M.C.; van Buchem, M.A.; Hofman, P.A.; Bakkers, J.T.; Terwindt, G.M.; Ferrari, M.D.; Launer, L.J. Migraine as a risk factor for subclinical brain lesions. JAMA 2004, 291, 427–434. [Google Scholar] [CrossRef]
- Kruit, M.C.; van Buchem, M.A.; Launer, L.J.; Terwindt, G.M.; Ferrari, M.D. Migraine is associated with an increased risk of deep white matter lesions, subclinical posterior circulation infarcts and brain iron accumulation: The population-based MRI CAMERA study. Cephalalgia 2010, 30, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Toh, K.L.; Jones, C.R.; He, Y.; Eide, E.J.; Hinz, W.A.; Virshup, D.M.; Ptacek, L.J.; Fu, Y.H. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 2001, 291, 1040–1043. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Akashi, M.; Matsuda, M.; Goto, K.; Miyata, Y.; Node, K.; Nishida, E. Involvement of the protein kinase CK2 in the regulation of mammalian circadian rhythms. Sci. Signal 2009, 2, ra26. [Google Scholar] [CrossRef]
- Lee, H.; Chen, R.; Lee, Y.; Yoo, S.; Lee, C. Essential roles of CKIdelta and CKIepsilon in the mammalian circadian clock. Proc. Natl. Acad. Sci. USA 2009, 106, 21359–21364. [Google Scholar] [CrossRef] [PubMed]
- Stanyer, E.C.; Creeney, H.; Nesbitt, A.D.; Holland, P.R.; Hoffmann, J. Subjective Sleep Quality and Sleep Architecture in Patients with Migraine: A Meta-analysis. Neurology 2021, 97, e1620–e1631. [Google Scholar] [CrossRef]
- Pettingill, P.; Weir, G.A.; Wei, T.; Wu, Y.; Flower, G.; Lalic, T.; Handel, A.; Duggal, G.; Chintawar, S.; Cheung, J.; et al. A causal role for TRESK loss of function in migraine mechanisms. Brain 2019, 142, 3852–3867. [Google Scholar] [CrossRef] [PubMed]
- Grangeon, L.; Lange, K.S.; Waliszewska-Prosol, M.; Onan, D.; Marschollek, K.; Wiels, W.; Mikulenka, P.; Farham, F.; Gollion, C.; Ducros, A.; et al. Genetics of migraine: Where are we now? J. Headache Pain 2023, 24, 12. [Google Scholar] [CrossRef]
- Weir, G.A.; Pettingill, P.; Wu, Y.; Duggal, G.; Ilie, A.S.; Akerman, C.J.; Cader, M.Z. The Role of TRESK in Discrete Sensory Neuron Populations and Somatosensory Processing. Front. Mol. Neurosci. 2019, 12, 170. [Google Scholar] [CrossRef]
- Liu, P.; Xiao, Z.; Ren, F.; Guo, Z.; Chen, Z.; Zhao, H.; Cao, Y.Q. Functional analysis of a migraine-associated TRESK K+ channel mutation. J. Neurosci. 2013, 33, 12810–12824. [Google Scholar] [CrossRef]
- Dobler, T.; Springauf, A.; Tovornik, S.; Weber, M.; Schmitt, A.; Sedlmeier, R.; Wischmeyer, E.; Doring, F. TRESK two-pore-domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J. Physiol. 2007, 585, 867–879. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, P.; Ren, F.; Cao, Y.Q. Nonmigraine-associated TRESK K+ channel variant C110R does not increase the excitability of trigeminal ganglion neurons. J. Neurophysiol. 2014, 112, 568–579. [Google Scholar] [CrossRef]
- Royal, P.; Andres-Bilbe, A.; Avalos Prado, P.; Verkest, C.; Wdziekonski, B.; Schaub, S.; Baron, A.; Lesage, F.; Gasull, X.; Levitz, J.; et al. Migraine-Associated TRESK Mutations Increase Neuronal Excitability through Alternative Translation Initiation and Inhibition of TREK. Neuron 2019, 101, 232–245.e6. [Google Scholar] [CrossRef] [PubMed]
- Lafreniere, R.G.; Cader, M.Z.; Poulin, J.F.; Andres-Enguix, I.; Simoneau, M.; Gupta, N.; Boisvert, K.; Lafreniere, F.; McLaughlan, S.; Dube, M.P.; et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 2010, 16, 1157–1160. [Google Scholar] [CrossRef]
- Kullmann, D.M. The neuronal channelopathies. Brain 2002, 125, 1177–1195. [Google Scholar] [CrossRef]
- Afridi, S.K.; Giffin, N.J.; Kaube, H.; Goadsby, P.J. A randomized controlled trial of intranasal ketamine in migraine with prolonged aura. Neurology 2013, 80, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Tang, Y.; Zhu, H. Effectiveness and Safety of Memantine for Headache: A Meta-analysis of Randomized Controlled Studies. Clin. Neuropharmacol. 2022, 45, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Waung, M.W.; Akerman, S.; Wakefield, M.; Keywood, C.; Goadsby, P.J. Metabotropic glutamate receptor 5: A target for migraine therapy. Ann. Clin. Transl. Neurol. 2016, 3, 560–571. [Google Scholar] [CrossRef]
- Chabi, A.; Zhang, Y.; Jackson, S.; Cady, R.; Lines, C.; Herring, W.J.; Connor, K.M.; Michelson, D. Randomized controlled trial of the orexin receptor antagonist filorexant for migraine prophylaxis. Cephalalgia 2015, 35, 379–388. [Google Scholar] [CrossRef]
- Oliveira, M.M.; Akerman, S.; Tavares, I.; Goadsby, P.J. Neuropeptide Y inhibits the trigeminovascular pathway through NPY Y1 receptor: Implications for migraine. Pain 2016, 157, 1666–1673. [Google Scholar] [CrossRef]
- Yang, C.; Gong, Z.; Zhang, X.; Miao, S.; Li, B.; Xie, W.; Wang, T.; Han, X.; Wang, L.; Dong, Z.; et al. Neuropeptide Y in the medial habenula alleviates migraine-like behaviors through the Y1 receptor. J. Headache Pain 2023, 24, 61. [Google Scholar] [CrossRef]
- Riant, F.; Roos, C.; Roubertie, A.; Barbance, C.; Hadjadj, J.; Auvin, S.; Baille, G.; Beltramone, M.; Boulanger, C.; Cahn, A.; et al. Hemiplegic Migraine Associated With PRRT2 Variations: A Clinical and Genetic Study. Neurology 2022, 98, e51–e61. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| FHM1 | FHM2 | FHM3 | |
|---|---|---|---|
| Genes | CACNA1A | ATP1A2 | SCN1A |
| Chromosome location | 19p13 | 1q23 | 2q24 |
| Year of identification | 1996 | 2003 | 2005 |
| Mutation summary | Encodes the α-1A subunit of the P/Q type calcium channel | Encodes the α-2 subunit of the Na+,K+-ATPase | Encodes the α-1 subunit of the voltage-gated Na+ channel NaV1.1 |
| Typical functional modulation | Gain-of-function | Loss-of-function | Gain-of-function |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gosalia, H.; Karsan, N.; Goadsby, P.J. Genetic Mechanisms of Migraine: Insights from Monogenic Migraine Mutations. Int. J. Mol. Sci. 2023, 24, 12697. https://doi.org/10.3390/ijms241612697
Gosalia H, Karsan N, Goadsby PJ. Genetic Mechanisms of Migraine: Insights from Monogenic Migraine Mutations. International Journal of Molecular Sciences. 2023; 24(16):12697. https://doi.org/10.3390/ijms241612697
Chicago/Turabian StyleGosalia, Helin, Nazia Karsan, and Peter J. Goadsby. 2023. "Genetic Mechanisms of Migraine: Insights from Monogenic Migraine Mutations" International Journal of Molecular Sciences 24, no. 16: 12697. https://doi.org/10.3390/ijms241612697
APA StyleGosalia, H., Karsan, N., & Goadsby, P. J. (2023). Genetic Mechanisms of Migraine: Insights from Monogenic Migraine Mutations. International Journal of Molecular Sciences, 24(16), 12697. https://doi.org/10.3390/ijms241612697