
NMR-Chemical-Shift-Driven Protocol Reveals the Cofactor-Bound, Complete Structure of Dynamic Intermediates of the Catalytic Cycle of Oncogenic KRAS G12C Protein and the Significance of the Mg2+ Ion

 , , , and
, , , and 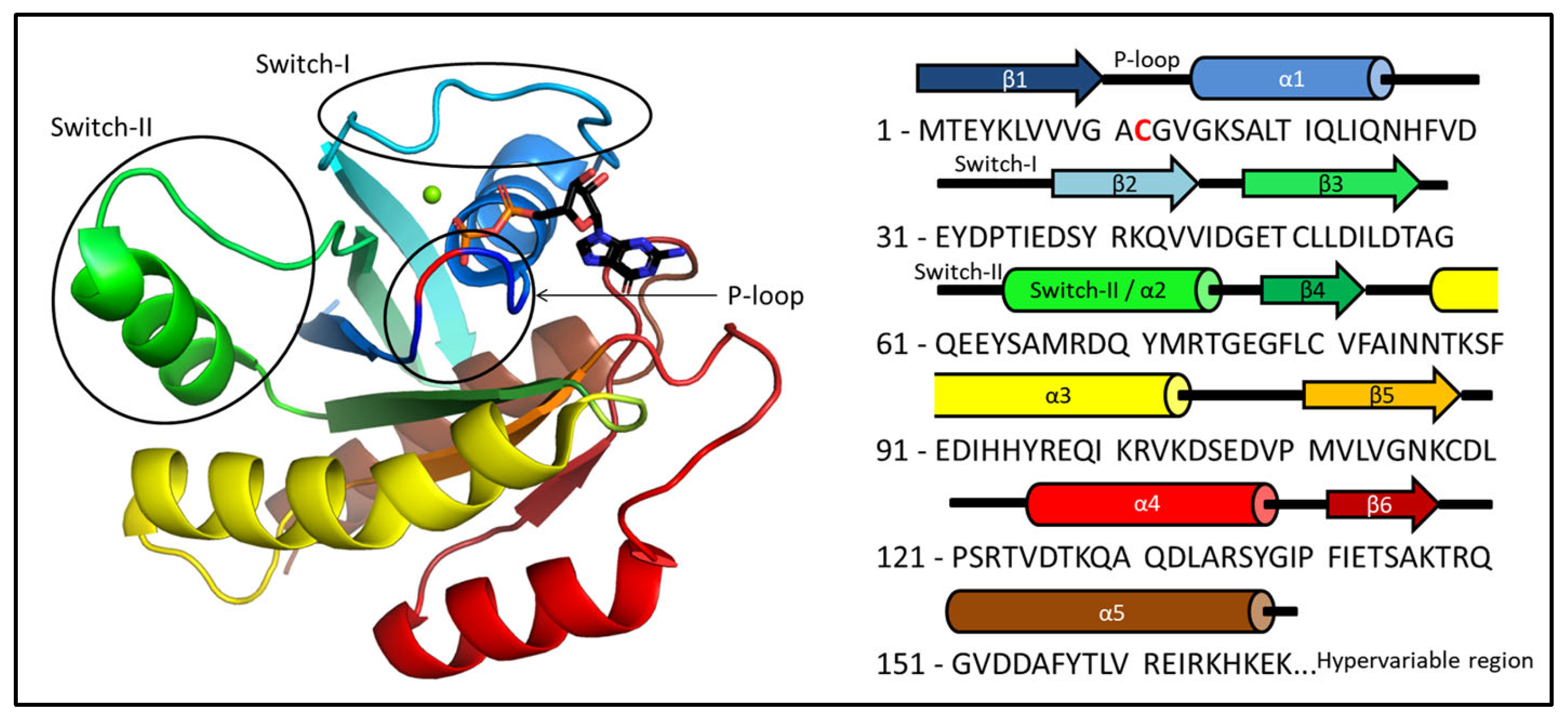{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
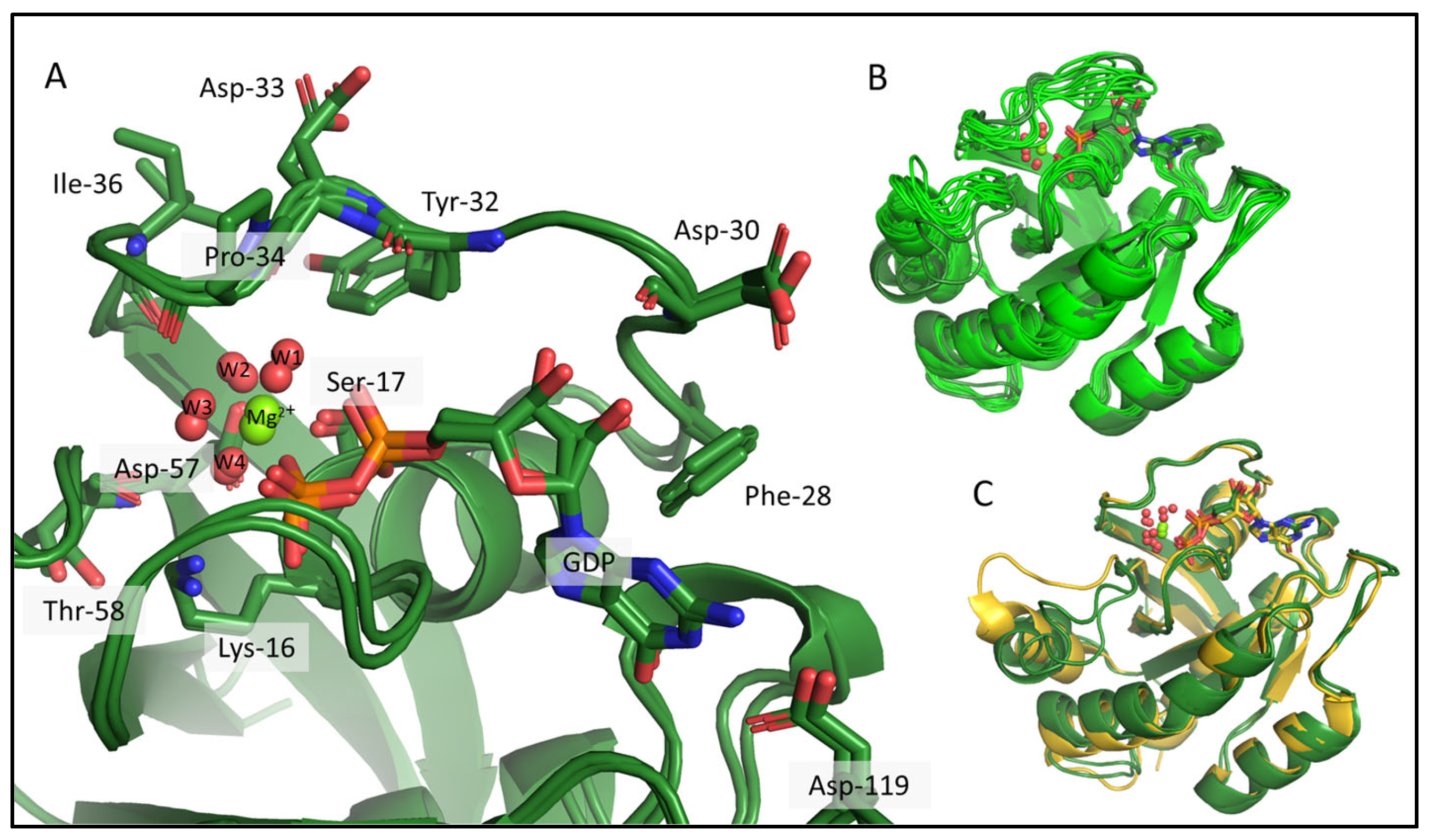
2.1. Ab Initio Structure Models of the Mg2+-Bound KRAS-G12C/GDP by Chemical-Shift-Rosetta
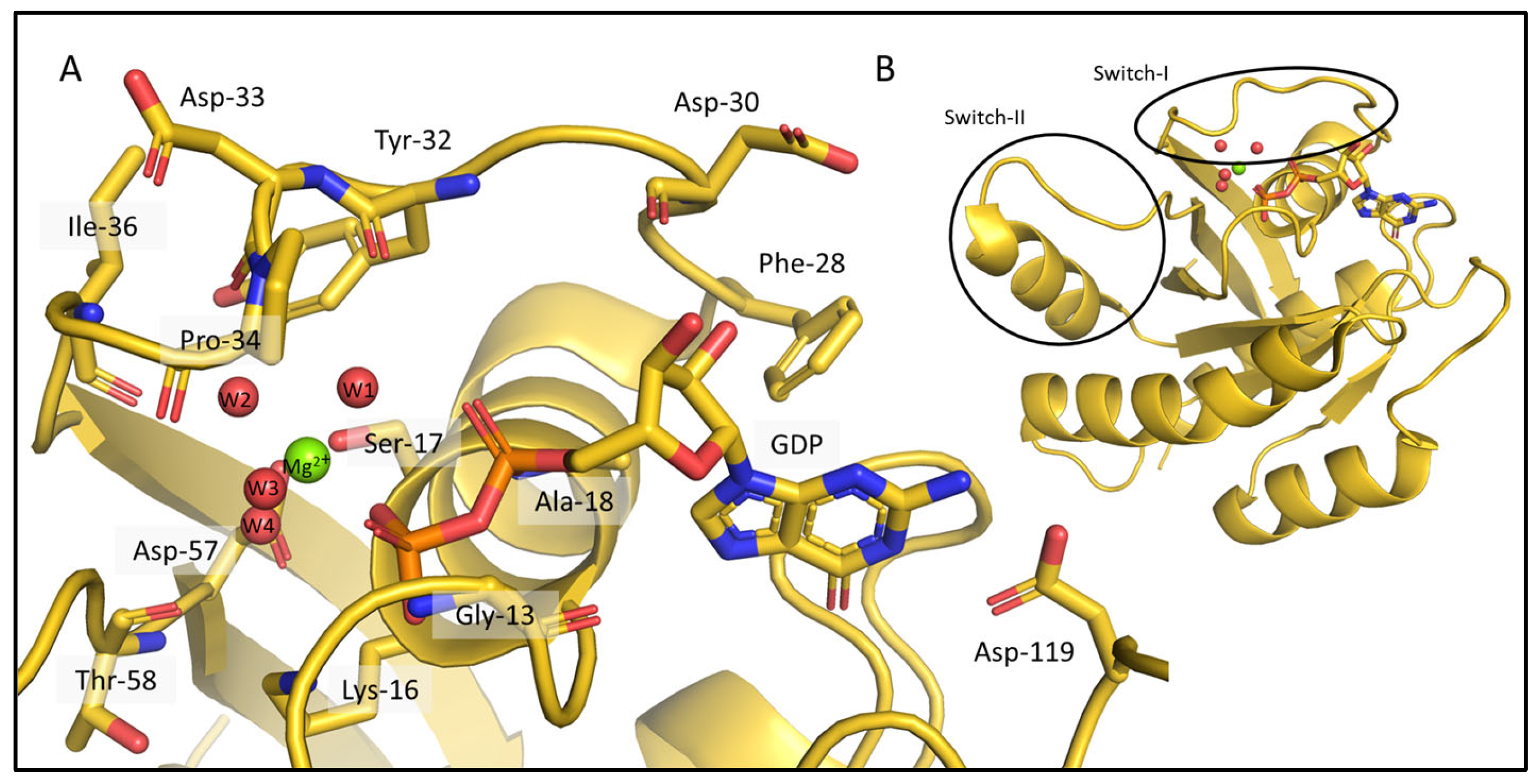
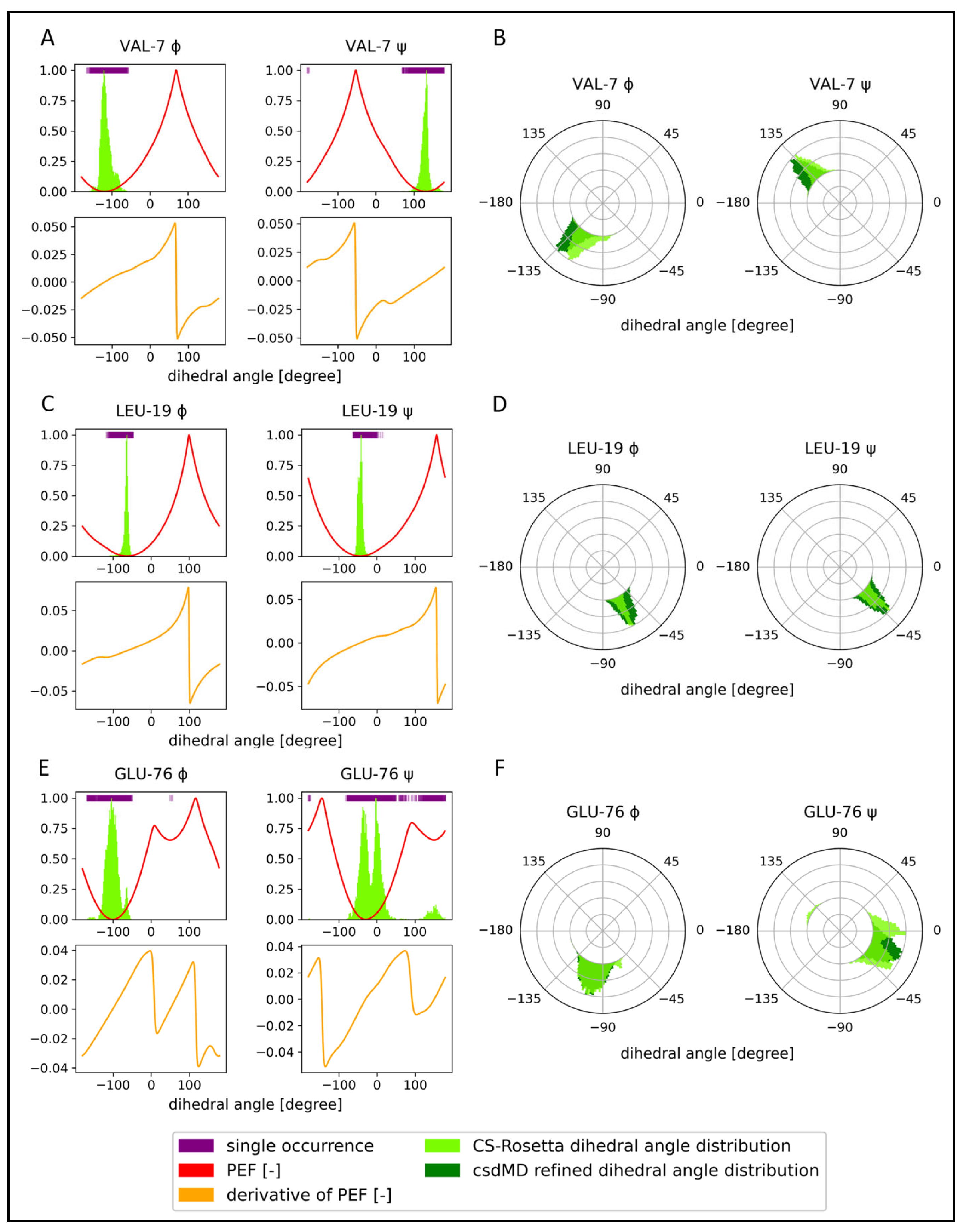
2.2. Chemical-Shift-Driven MD Refinement—Reinsertion of the Nonprotein Components in the Case of the KRAS-G12C/GDP-Mg2+ System
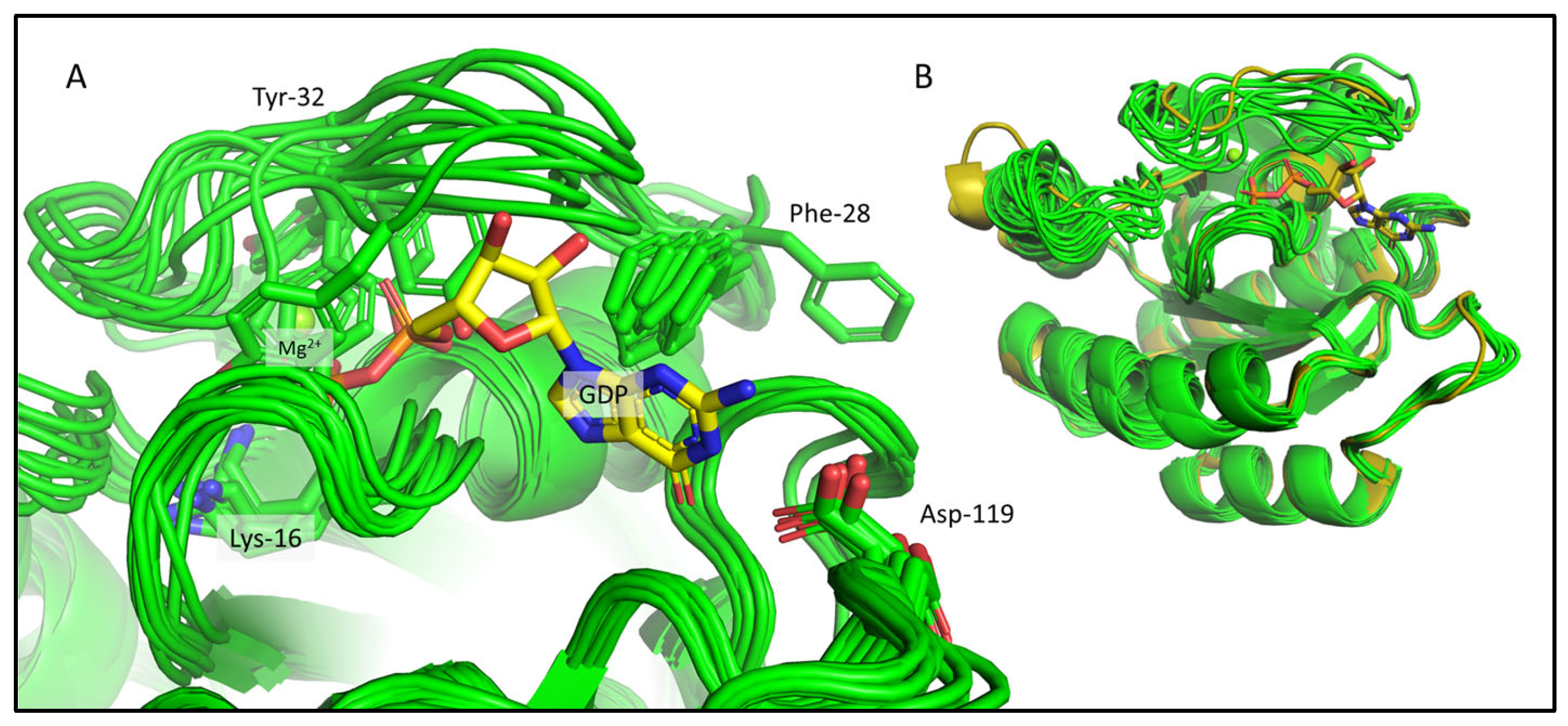
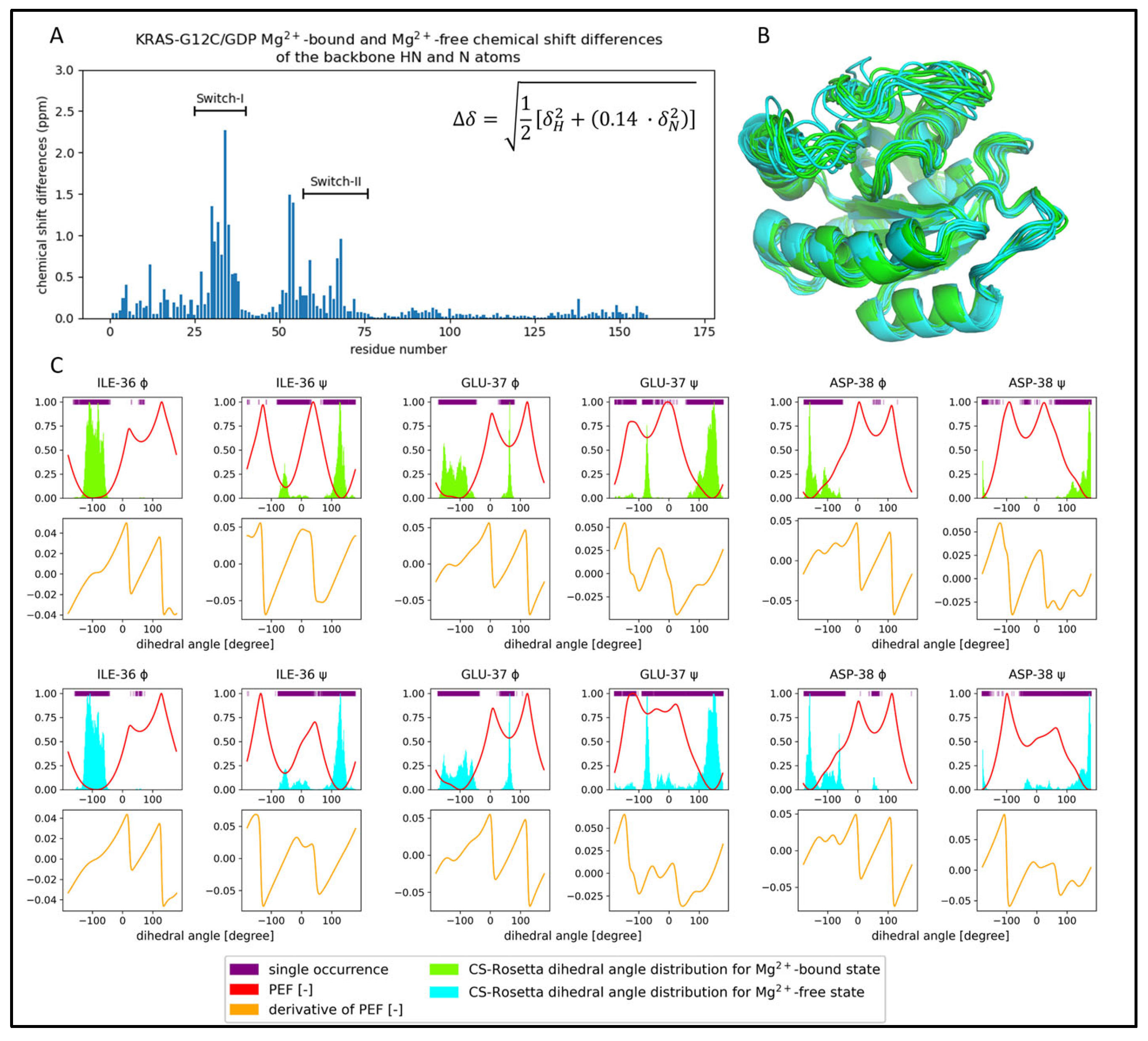
2.3. Comparison of the CS-Rosetta Ensembles of KRAS-G12C/GDP-Mg2+ and the Mg2+-Free KRAS-G12C/GDP System
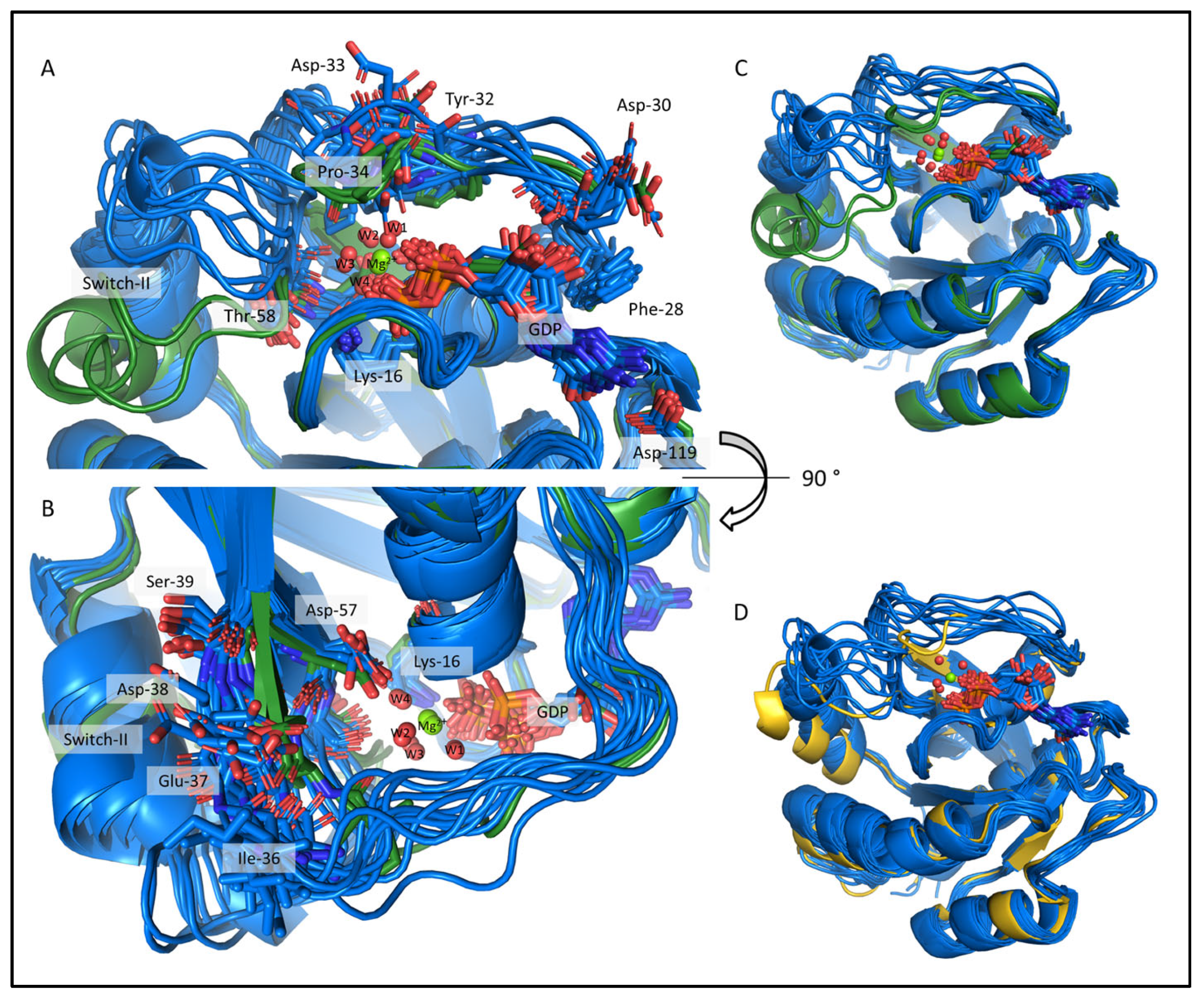
2.4. Comparison of the csdMD Refined Ensembles of KRAS-G12C/GDP-Mg2+ and the Mg2+-Free KRAS-G12C/GDP Systems
2.5. Structure Determination of the Mg2+-Free and GDP-Bound KRAS-G12C by ARTINA
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. NMR Spectroscopy
4.3. CS-Rosetta Structure Determination
4.4. csdMD Model Refinement
4.5. ARTINA Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BMRB | Biological Magnetic Resonance Data Bank |
| csdMD | Chemical-shift-driven MD |
| CS-Rosetta | Chemical-Shift-Rosetta |
| χ1 | Chi-1 dihedral (or torsion) angle |
| GDP | Guanosine diphosphate |
| GEF | Guanosine exchange factor |
| GTP | Guanosine triphosphate |
| MD | Molecular dynamics |
| NMR | Nuclear magnetic resonance |
| NOE | Nuclear Overhauser effect |
| ω | Omega dihedral (or torsion) angle |
| ϕ | Phi dihedral (or torsion) angle |
| PEF | Potential energy function |
| PDB | Protein Data Bank |
| ψ | Psi dihedral (or torsion) angle |
| RMSD | Root-mean-square deviation |
| RMSF | Root-mean-square fluctuation |
| SOS | Son of Sevenless |
References
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 1979, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching AlphaFold models with ligands and cofactors. Nat. Methods 2022, 20, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Guerry, P.; Herrmann, T. Advances in automated NMR protein structure determination. Q. Rev. Biophys. 2011, 44, 257–309. [Google Scholar] [CrossRef]
- Wishart, D.S.; Case, D.A. Use of Chemical Shifts in Macromolecular Structure Determination. Methods Enzymol. 2002, 338, 3–34. [Google Scholar] [CrossRef]
- Markwick, P.R.L.; Malliavin, T.; Nilges, M. Structural Biology by NMR: Structure, Dynamics, and Interactions. PLoS Comput. Biol. 2008, 4, e1000168. [Google Scholar] [CrossRef]
- Cavalli, A.; Salvatella, X.; Dobson, C.M.; Vendruscolo, M. Protein structure determination from NMR chemical shifts. Proc. Natl. Acad. Sci. USA 2007, 104, 9615–9620. [Google Scholar] [CrossRef]
- Shen, Y.; Lange, O.; Delaglio, F.; Rossi, P.; Aramini, J.M.; Liu, G.; Eletsky, A.; Wu, Y.; Singarapu, K.K.; Lemak, A.; et al. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA 2008, 105, 4685–4690. [Google Scholar] [CrossRef]
- Güntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef]
- Guerry, P.; Duong, V.D.; Herrmann, T. CASD-NMR 2: Robust and accurate unsupervised analysis of raw NOESY spectra and protein structure determination with UNIO. J. Biomol. NMR 2015, 62, 473–480. [Google Scholar] [CrossRef]
- Allain, F.; Mareuil, F.; Ménager, H.; Nilges, M.; Bardiaux, B. ARIAweb: A server for automated NMR structure calculation. Nucleic Acids Res. 2020, 48, W41–W47. [Google Scholar] [CrossRef]
- Bermejo, G.A.; Schwieters, C.D. Protein Structure Elucidation from NMR Data with the Program Xplor-NIH. Methods Mol. Biol. 2018, 1688, 311–340. [Google Scholar] [CrossRef]
- Klukowski, P.; Riek, R.; Güntert, P. Rapid protein assignments and structures from raw NMR spectra with the deep learning technique ARTINA. Nat. Commun. 2022, 13, 6151. [Google Scholar] [CrossRef]
- Klukowski, P.; Riek, R.; Güntert, P. NMRtist: An online platform for automated biomolecular NMR spectra analysis. Bioinformatics 2023, 39, btad066. [Google Scholar] [CrossRef]
- Schmidt, E.; Güntert, P. A New Algorithm for Reliable and General NMR Resonance Assignment. J. Am. Chem. Soc. 2012, 134, 12817–12829. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef]
- Kontaxis, G.; Delaglio, F.; Bax, A. Molecular Fragment Replacement Approach to Protein Structure Determination by Chemical Shift and Dipolar Homology Database Mining. Methods Enzym. 2005, 394, 42–78. [Google Scholar] [CrossRef]
- Nerli, S.; McShan, A.C.; Sgourakis, N.G. Chemical shift-based methods in NMR structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 106–107, 1–25. [Google Scholar] [CrossRef]
- Bowers, P.M.; Strauss, C.E.; Baker, D. De novo protein structure determination using sparse NMR data. J. Biomol. NMR 2000, 18, 311–318. [Google Scholar] [CrossRef]
- Nerli, S.; Sgourakis, N.G. CS-ROSETTA. Methods Enzym. 2019, 614, 321–362. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J. Biomol. NMR 2007, 38, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Leman, J.K.; Künze, G. Recent Advances in NMR Protein Structure Prediction with ROSETTA. Int. J. Mol. Sci. 2023, 24, 7835. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Borkakoti, N.; Pravda, L.; Tyzack, J.D.; Thornton, J.M.; Velankar, S. Finding enzyme cofactors in Protein Data Bank. Bioinformatics 2019, 35, 3510–3511. [Google Scholar] [CrossRef]
- Colicelli, J. Human RAS Superfamily Proteins and Related GTPases. Sci. STKE 2004, 2004, re13. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef]
- Kalbitzer, H.R.; Rosnizeck, I.C.; Munte, C.E.; Narayanan, S.P.; Kropf, V.; Spoerner, M. Intrinsic Allosteric Inhibition of Signaling Proteins by Targeting Rare Interaction States Detected by High-Pressure NMR Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 14242–14246. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Pálfy, G.; Menyhárd, D.K.; Perczel, A. Dynamically encoded reactivity of Ras enzymes: Opening new frontiers for drug discovery. Cancer Metastasis Rev. 2020, 39, 1075–1089. [Google Scholar] [CrossRef]
- Zala, D.; Schlattner, U.; Desvignes, T.; Bobe, J.; Roux, A.; Chavrier, P.; Boissan, M. The advantage of channeling nucleotides for very processive functions. F1000Research 2017, 6, 724. [Google Scholar] [CrossRef]
- Killoran, R.C.; Smith, M.J. Conformational resolution of nucleotide cycling and effector interactions for multiple small GTPases determined in parallel. J. Biol. Chem. 2019, 294, 9937–9948. [Google Scholar] [CrossRef]
- RCSB PDB—7KYZ: Solution Structures of Full-Length K-RAS Bound to GDP. Available online: https://www.rcsb.org/structure/7KYZ (accessed on 30 June 2023).
- Menyhárd, D.K.; Pálfy, G.; Orgován, Z.; Vida, I.; Keserű, G.M.; Perczel, A. Structural impact of GTP binding on downstream KRAS signaling. Chem. Sci. 2020, 11, 9272–9289. [Google Scholar] [CrossRef]
- Pálfy, G.; Menyhárd, D.K.; Ákontz-Kiss, H.; Vida, I.; Batta, G.; Tőke, O.; Perczel, A. The Importance of Mg2+-free State in Nucleotide Exchange of Oncogenic K-Ras Mutants. Chem. A Eur. J. 2022, 28, 1449. [Google Scholar] [CrossRef]
- Evangelidis, T.; Nerli, S.; Nováček, J.; Brereton, A.E.; Karplus, P.A.; Dotas, R.R.; Venditti, V.; Sgourakis, N.G.; Tripsianes, K. Automated NMR resonance assignments and structure determination using a minimal set of 4D spectra. Nat. Commun. 2018, 9, 384. [Google Scholar] [CrossRef]
- Sondermann, H.; Soisson, S.M.; Boykevisch, S.; Yang, S.-S.; Bar-Sagi, D.; Kuriyan, J. Structural Analysis of Autoinhibition in the Ras Activator Son of Sevenless. Cell 2004, 119, 393–405. [Google Scholar] [CrossRef]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B.; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Lindert, S.; Meiler, J.; McCammon, J.A. Iterative Molecular Dynamics—Rosetta Protein Structure Refinement Protocol to Improve Model Quality. J. Chem. Theory Comput. 2013, 9, 3843–3847. [Google Scholar] [CrossRef]
- Lindert, S.; McCammon, J.A. Improved cryoEM-guided iterative molecular dynamics-rosetta protein structure refinement protocol for high precision protein structure prediction. J. Chem. Theory Comput. 2015, 11, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Leelananda, S.P.; Lindert, S. Using NMR Chemical Shifts and Cryo-EM Density Restraints in Iterative Rosetta-MD Protein Structure Refinement. J. Chem. Inf. Model. 2020, 60, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Pálfy, G.; Vida, I.; Perczel, A. 1H, 15N backbone assignment and comparative analysis of the wild type and G12C, G12D, G12V mutants of K-Ras bound to GDP at physiological pH. Biomol. NMR Assign. 2020, 14, 1–7. [Google Scholar] [CrossRef]
- Scott, D.W. On optimal and data-based histograms. Biometrika 1979, 66, 605–610. [Google Scholar] [CrossRef]
- Freedman, D.; Diaconis, P. On the histogram as a density estimator: L2 theory. Z. Wahrscheinlichkeitstheorie Verwandte Geb. 1981, 57, 453–476. [Google Scholar] [CrossRef]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef]
- Bauer, P.; Hessand, B.; Lindahl, E. GROMACS, 2022.2 Source code; Zenodo: Geneva, Switzerland, 2022. [CrossRef]
- Aliev, A.E.; Kulke, M.; Khaneja, H.S.; Chudasama, V.; Sheppard, T.D.; Lanigan, R.M. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins Struct. Funct. Bioinform. 2014, 82, 195–215. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Latzer, J.; Case, D.A. Revised AMBER Parameters for Bioorganic Phosphates. J. Chem. Theory Comput. 2012, 8, 4405–4412. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadanecz, M.; Fazekas, Z.; Pálfy, G.; Karancsiné Menyhárd, D.; Perczel, A. NMR-Chemical-Shift-Driven Protocol Reveals the Cofactor-Bound, Complete Structure of Dynamic Intermediates of the Catalytic Cycle of Oncogenic KRAS G12C Protein and the Significance of the Mg2+ Ion. Int. J. Mol. Sci. 2023, 24, 12101. https://doi.org/10.3390/ijms241512101
Gadanecz M, Fazekas Z, Pálfy G, Karancsiné Menyhárd D, Perczel A. NMR-Chemical-Shift-Driven Protocol Reveals the Cofactor-Bound, Complete Structure of Dynamic Intermediates of the Catalytic Cycle of Oncogenic KRAS G12C Protein and the Significance of the Mg2+ Ion. International Journal of Molecular Sciences. 2023; 24(15):12101. https://doi.org/10.3390/ijms241512101
Chicago/Turabian StyleGadanecz, Márton, Zsolt Fazekas, Gyula Pálfy, Dóra Karancsiné Menyhárd, and András Perczel. 2023. "NMR-Chemical-Shift-Driven Protocol Reveals the Cofactor-Bound, Complete Structure of Dynamic Intermediates of the Catalytic Cycle of Oncogenic KRAS G12C Protein and the Significance of the Mg2+ Ion" International Journal of Molecular Sciences 24, no. 15: 12101. https://doi.org/10.3390/ijms241512101
APA StyleGadanecz, M., Fazekas, Z., Pálfy, G., Karancsiné Menyhárd, D., & Perczel, A. (2023). NMR-Chemical-Shift-Driven Protocol Reveals the Cofactor-Bound, Complete Structure of Dynamic Intermediates of the Catalytic Cycle of Oncogenic KRAS G12C Protein and the Significance of the Mg2+ Ion. International Journal of Molecular Sciences, 24(15), 12101. https://doi.org/10.3390/ijms241512101