
Autophagy Inhibition via Hydroxychloroquine or 3-Methyladenine Enhances Chemotherapy-Induced Apoptosis in Neuro-Blastoma and Glioblastoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
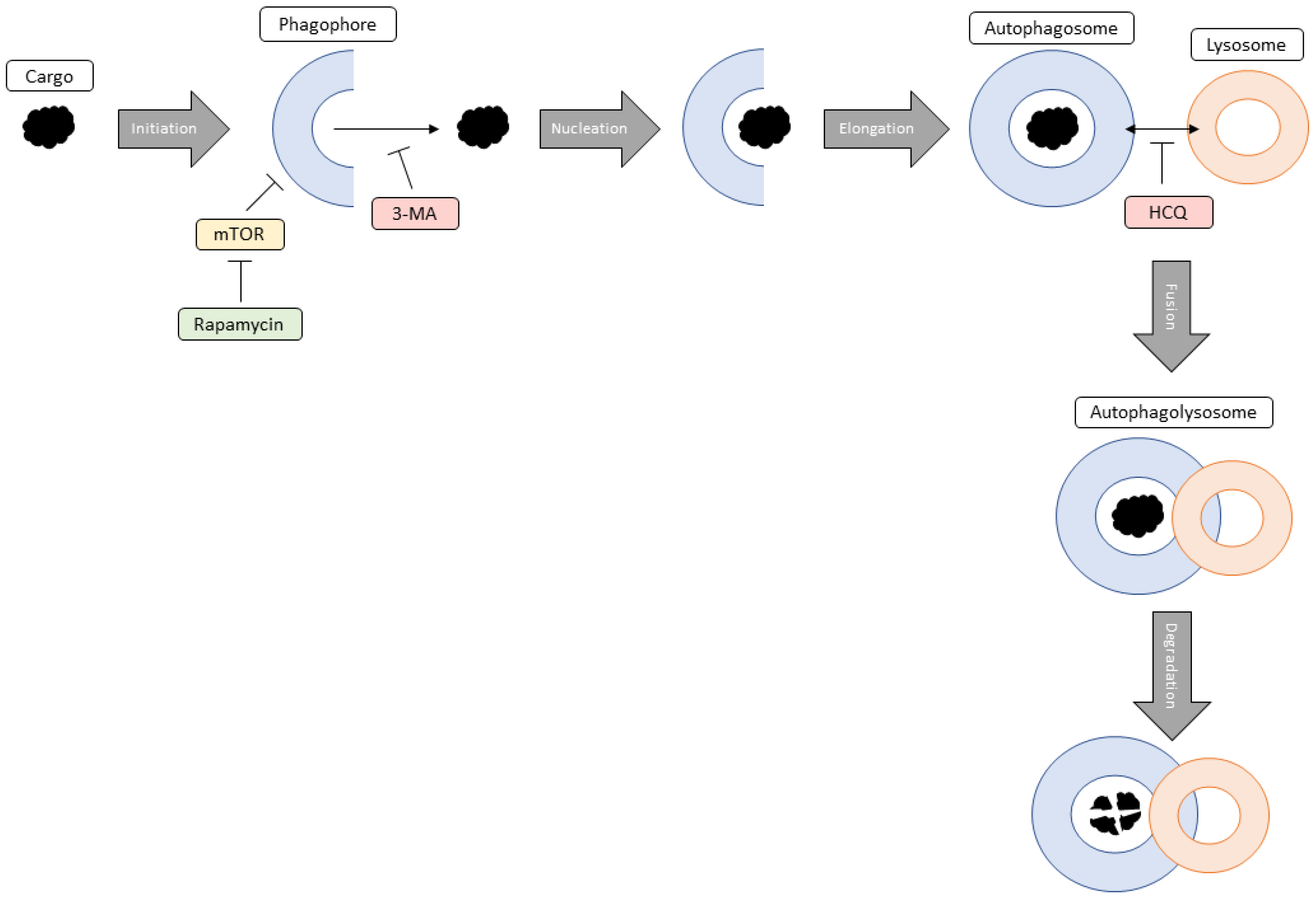
1. Introduction
2. Results
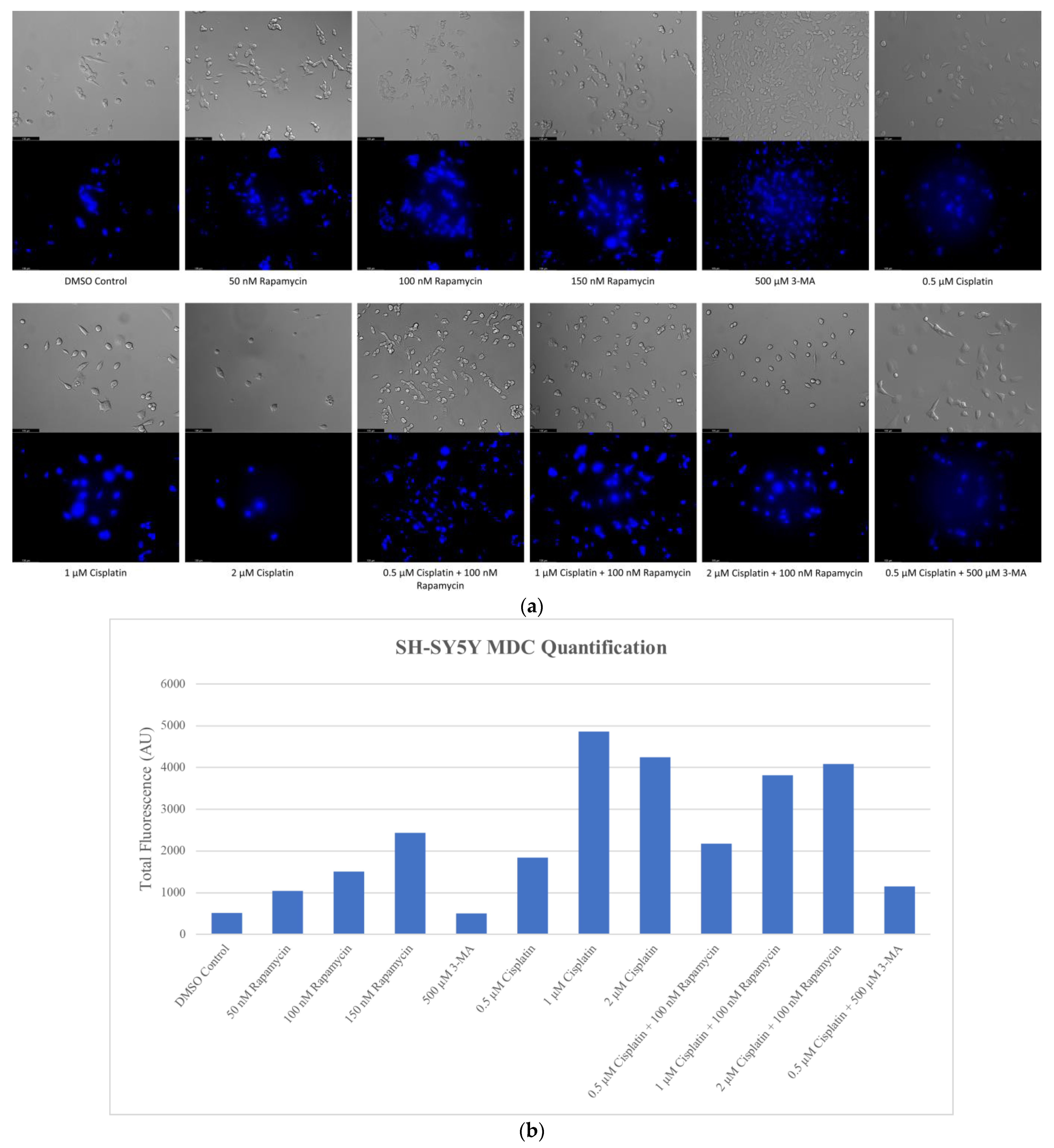
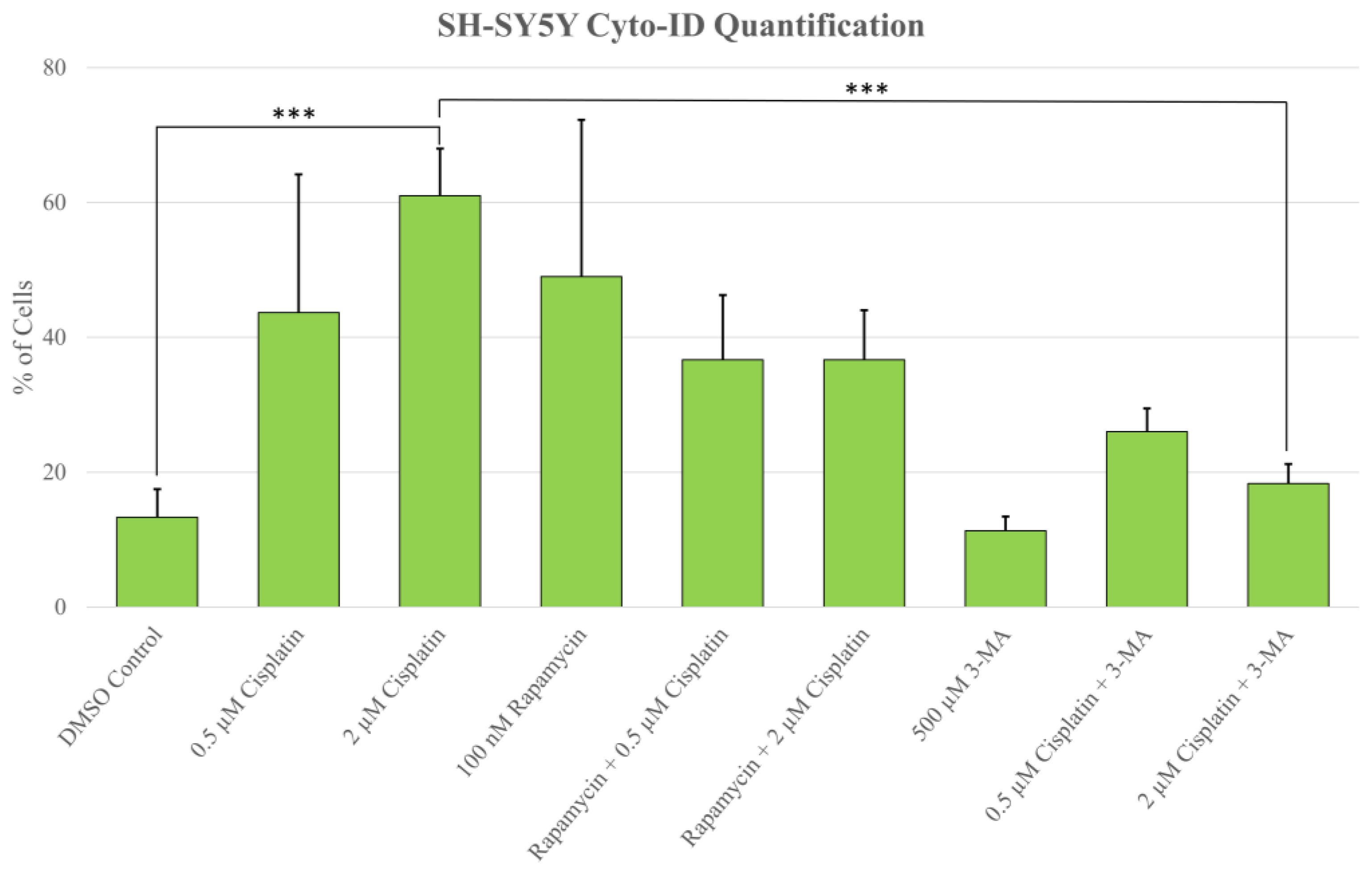
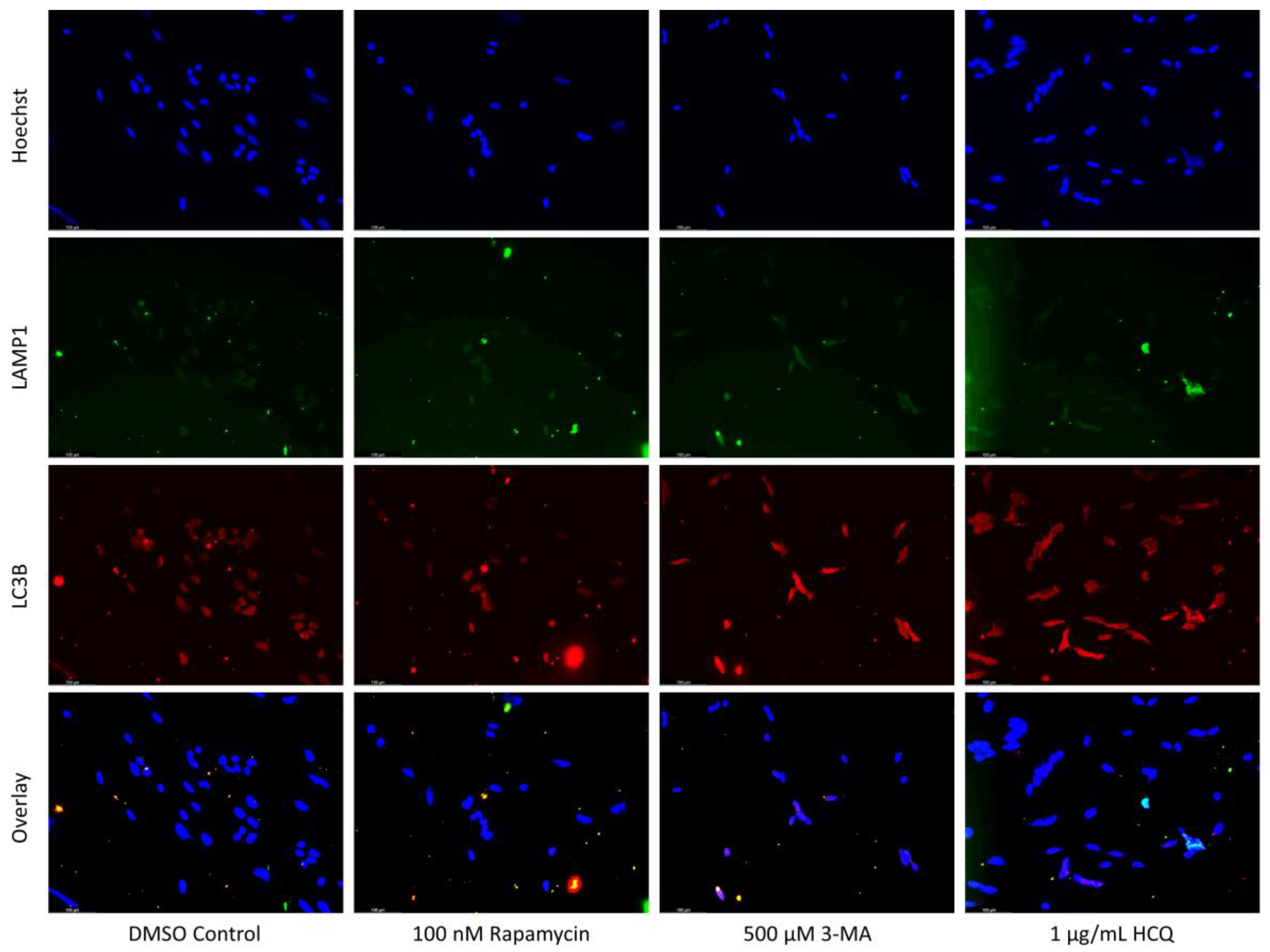
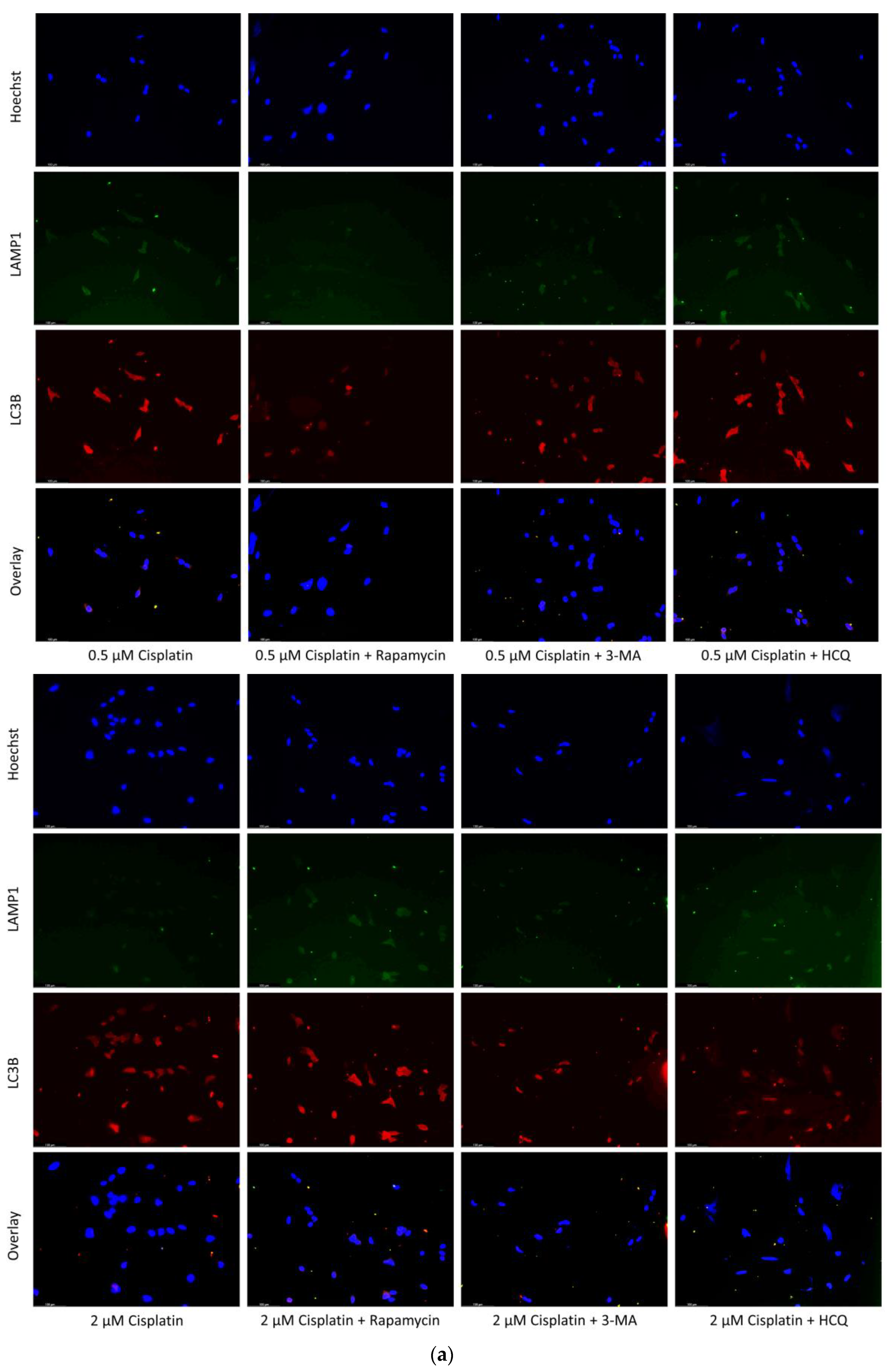
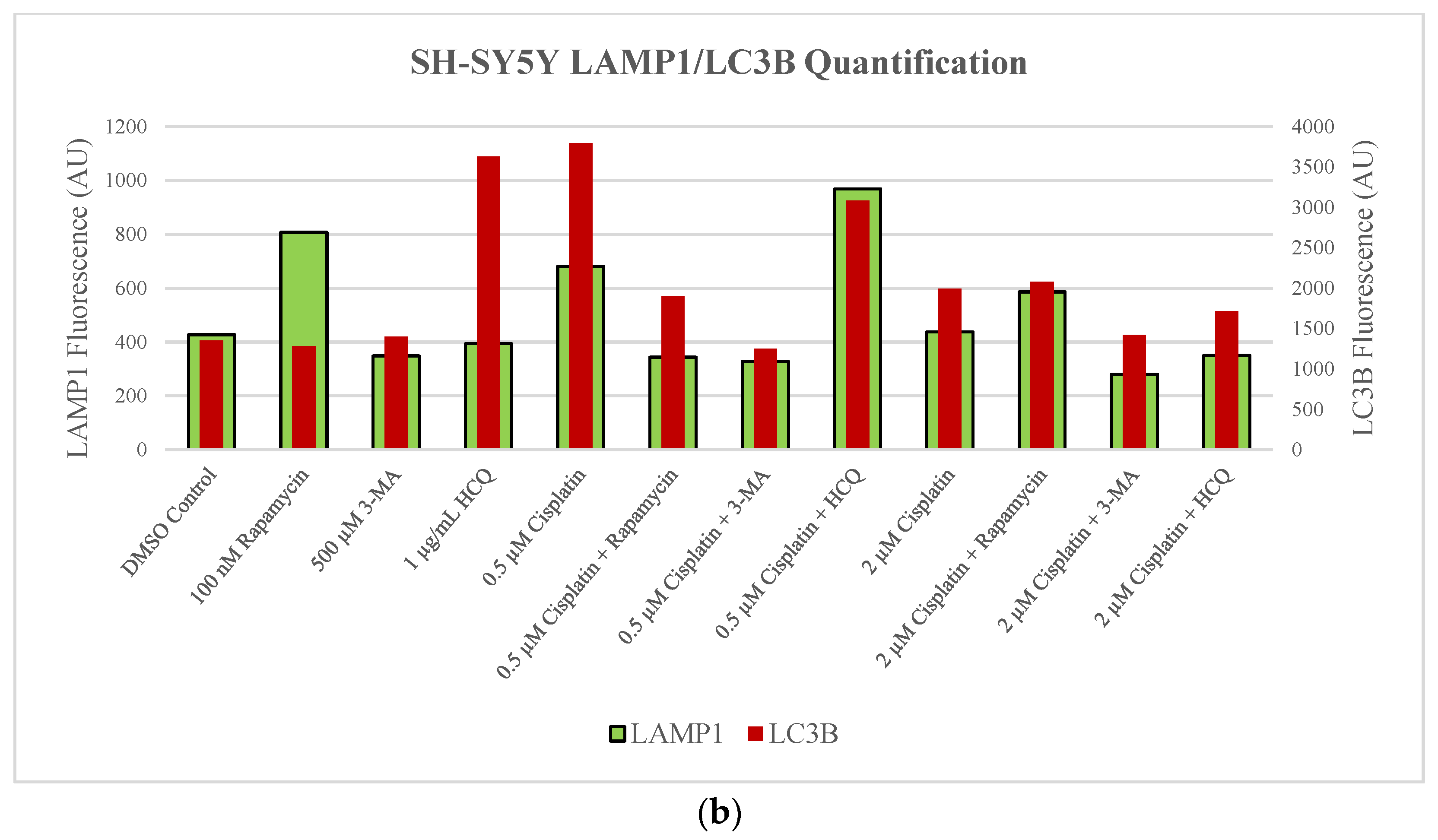
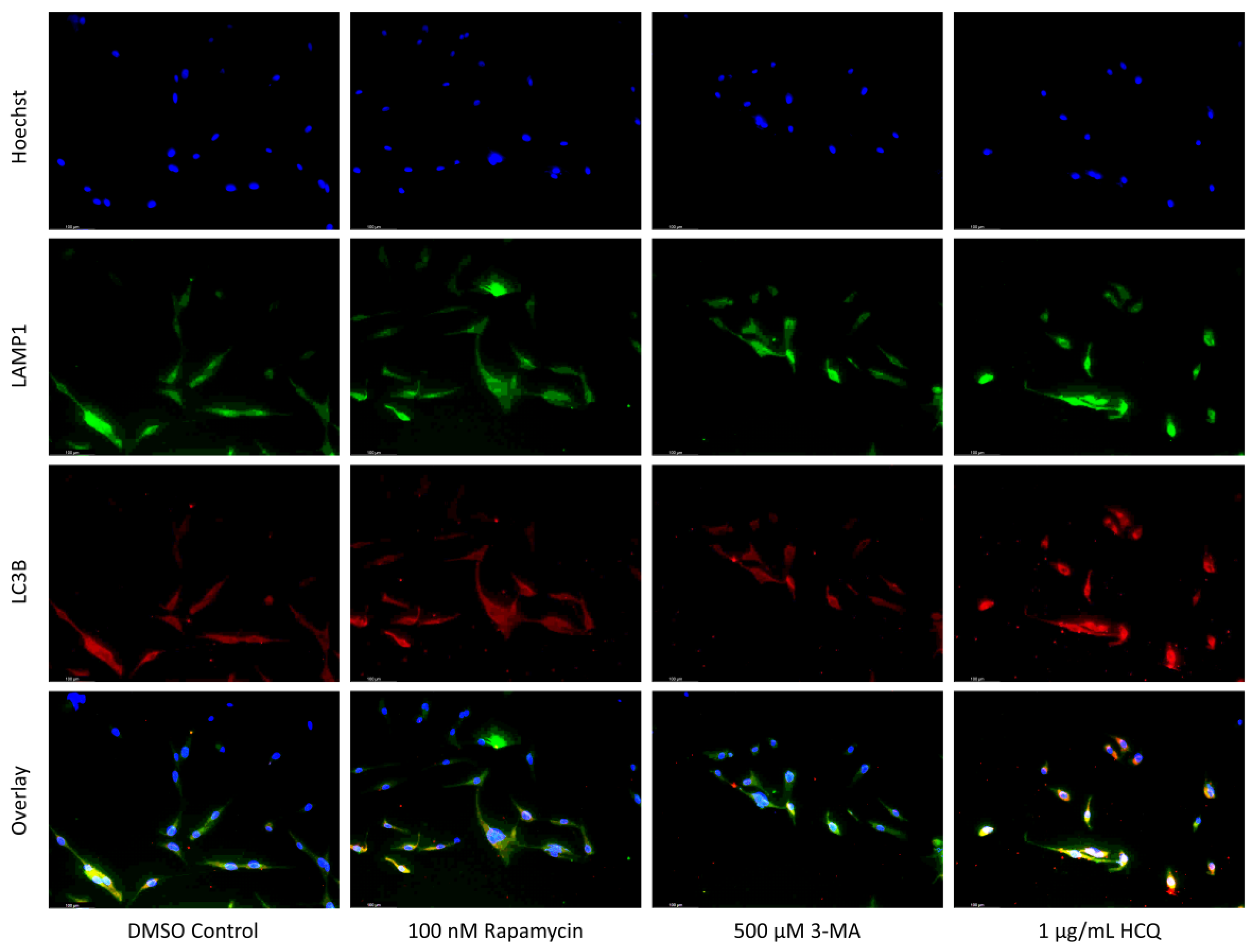
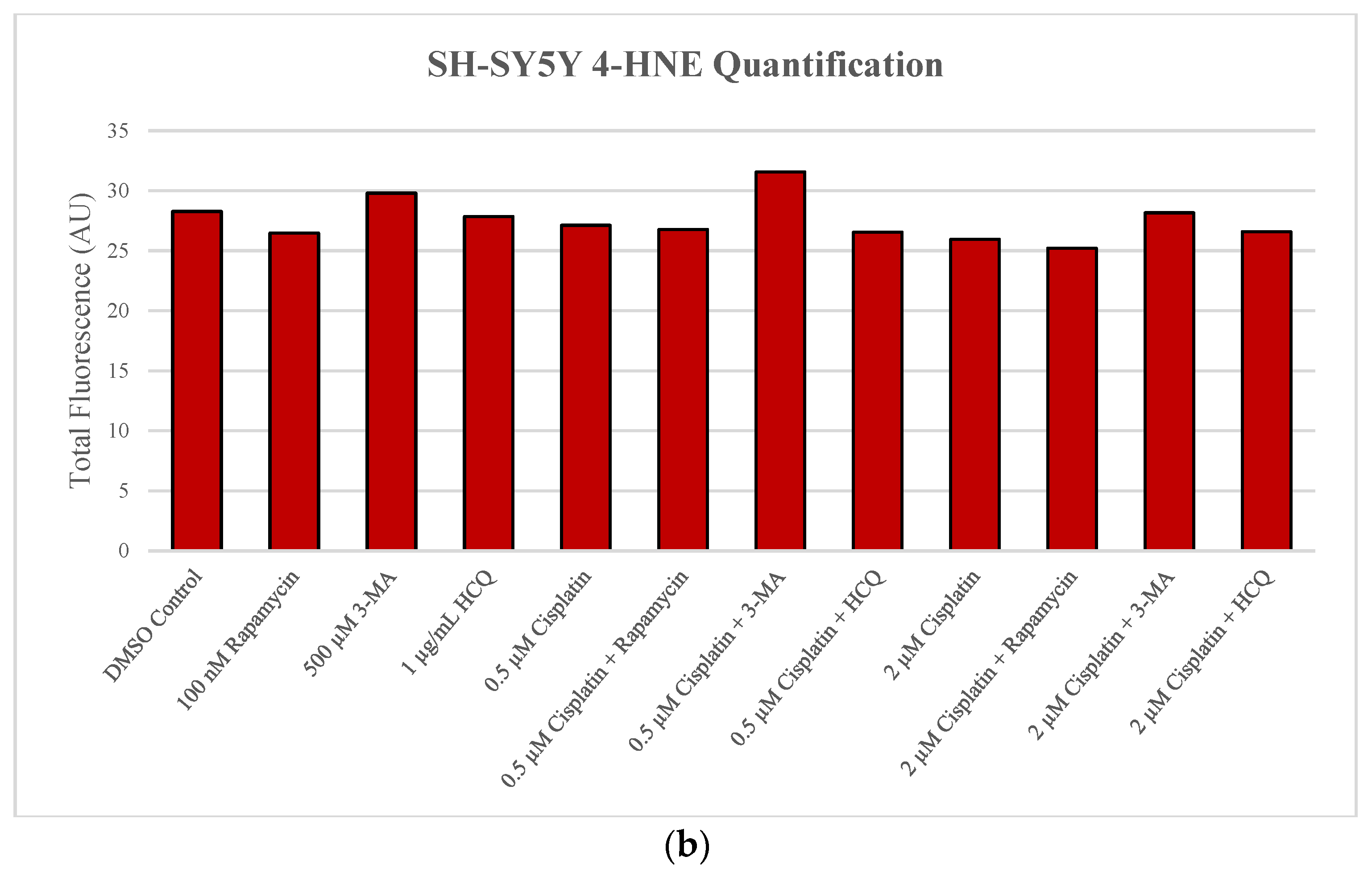
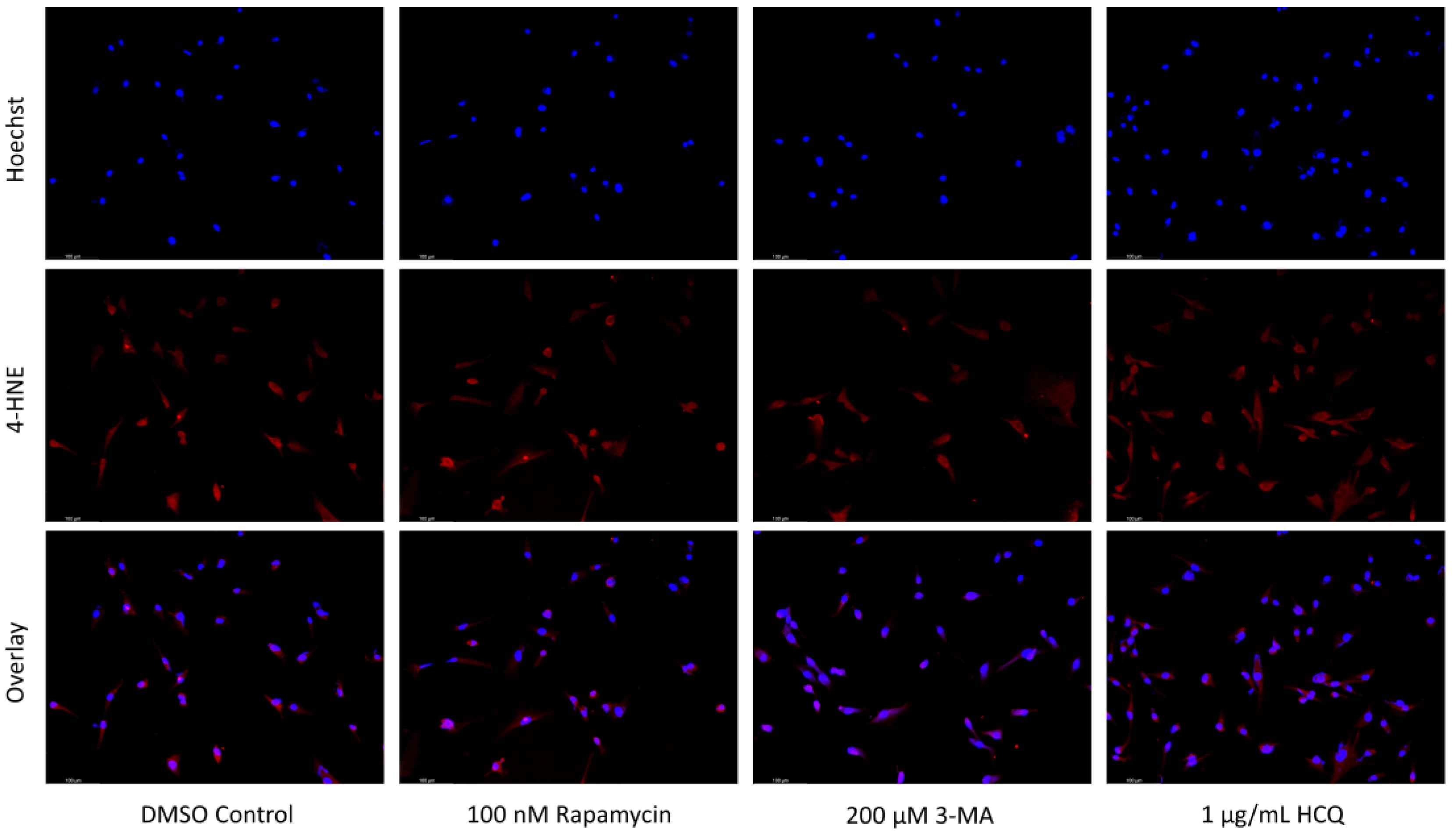
2.1. Cisplatin-Induced Autophagy Is Reduced in SH-SY5Y Neuroblastoma Cells by 3-MA Supplementation
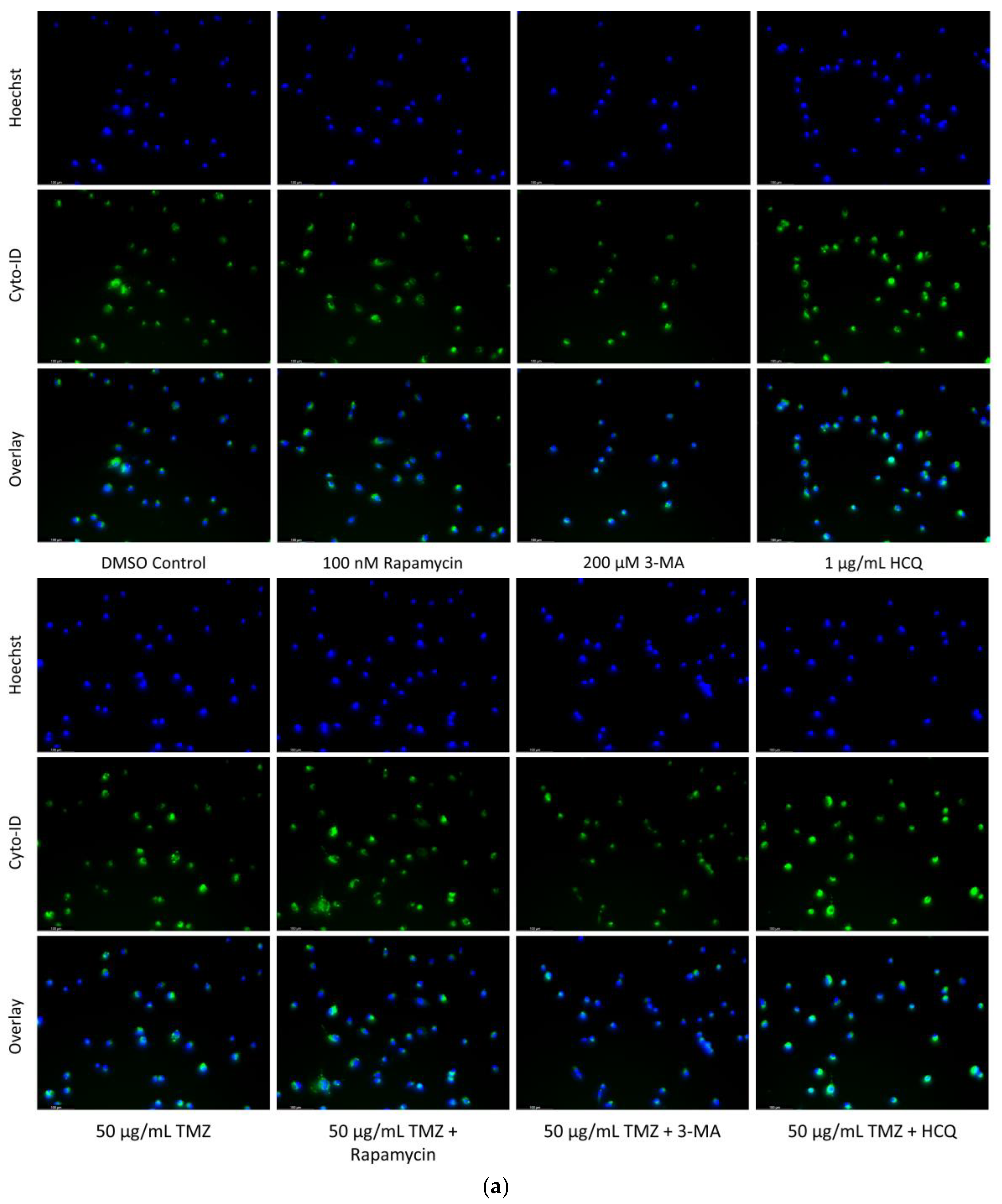
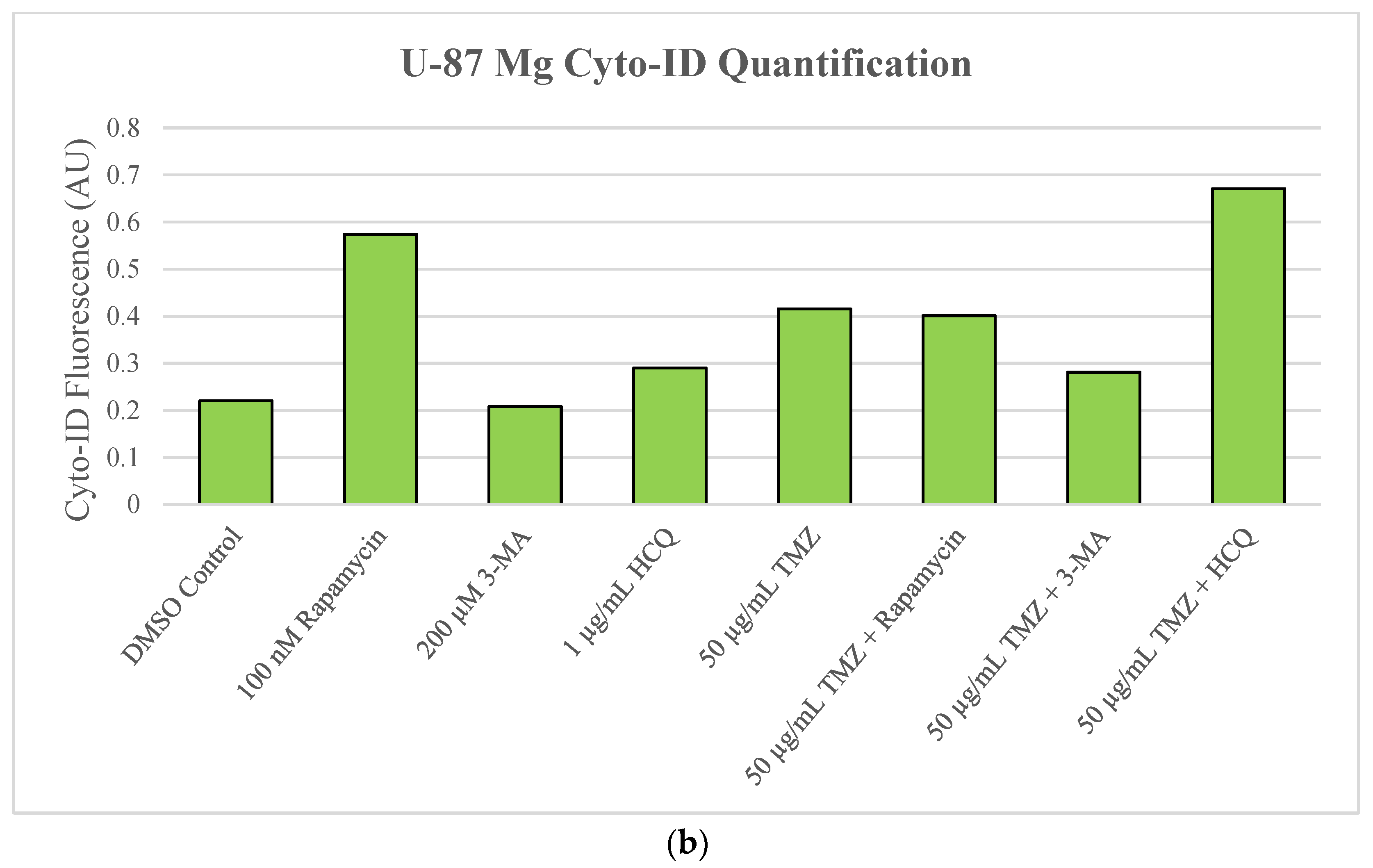
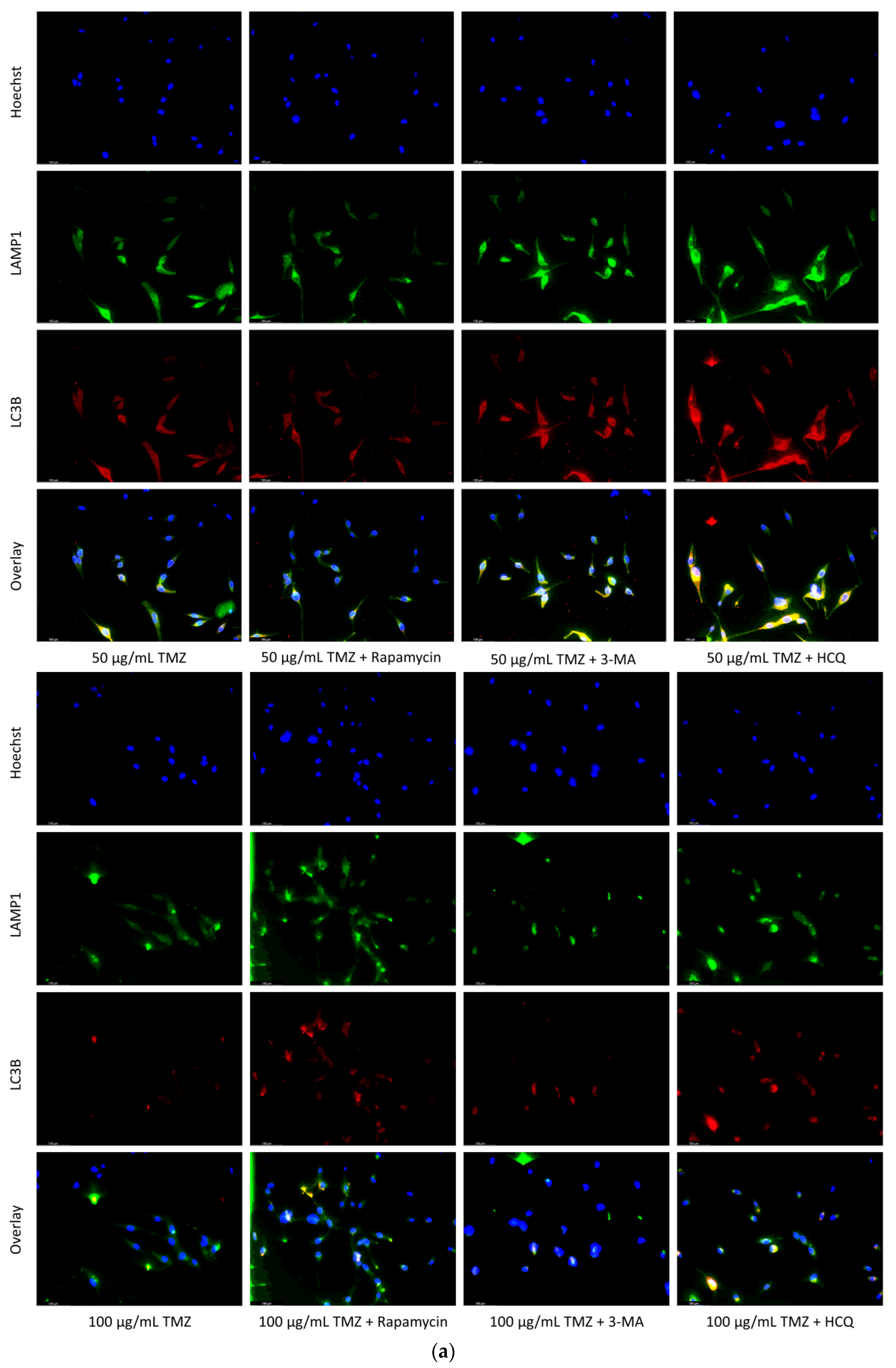
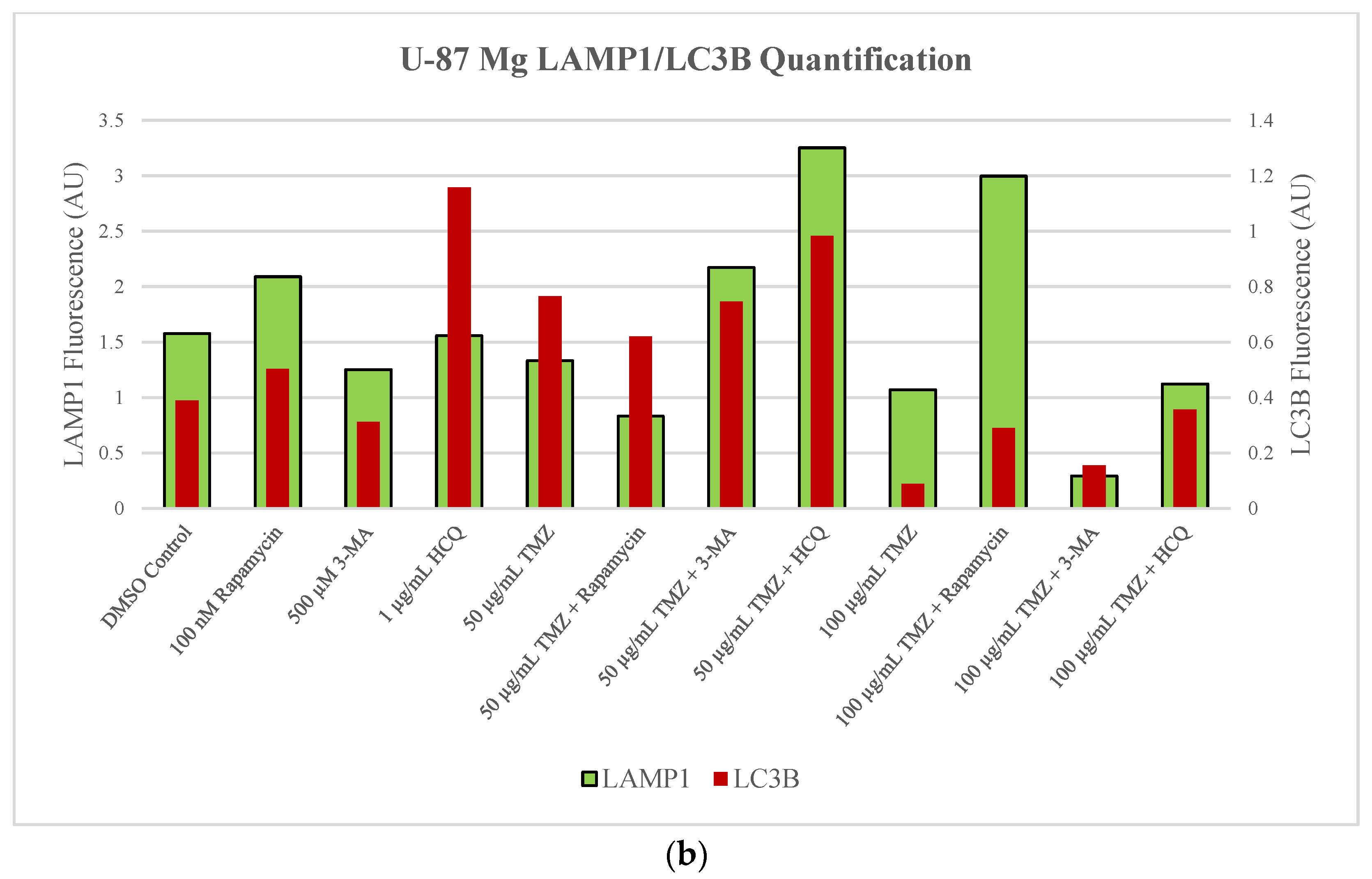
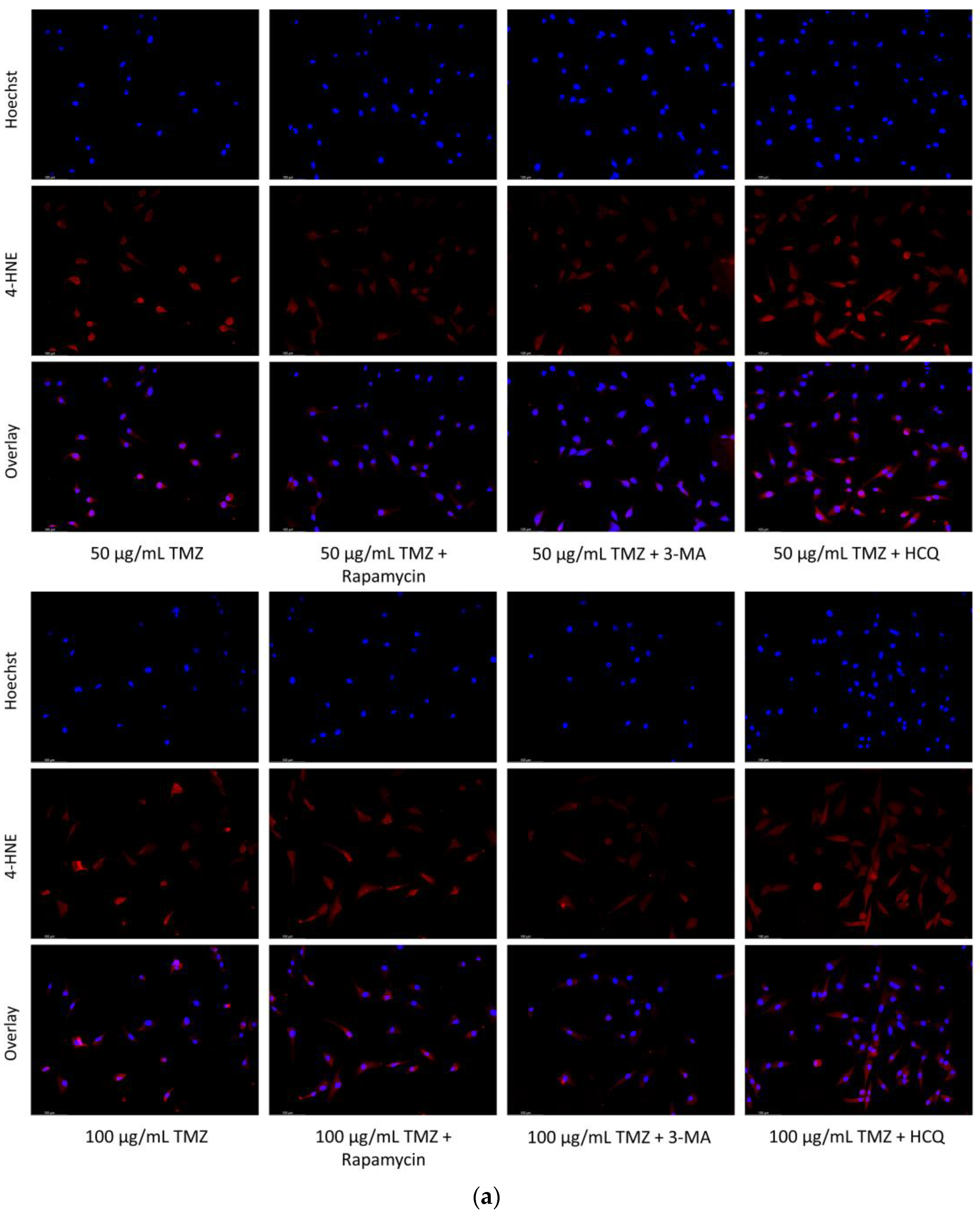
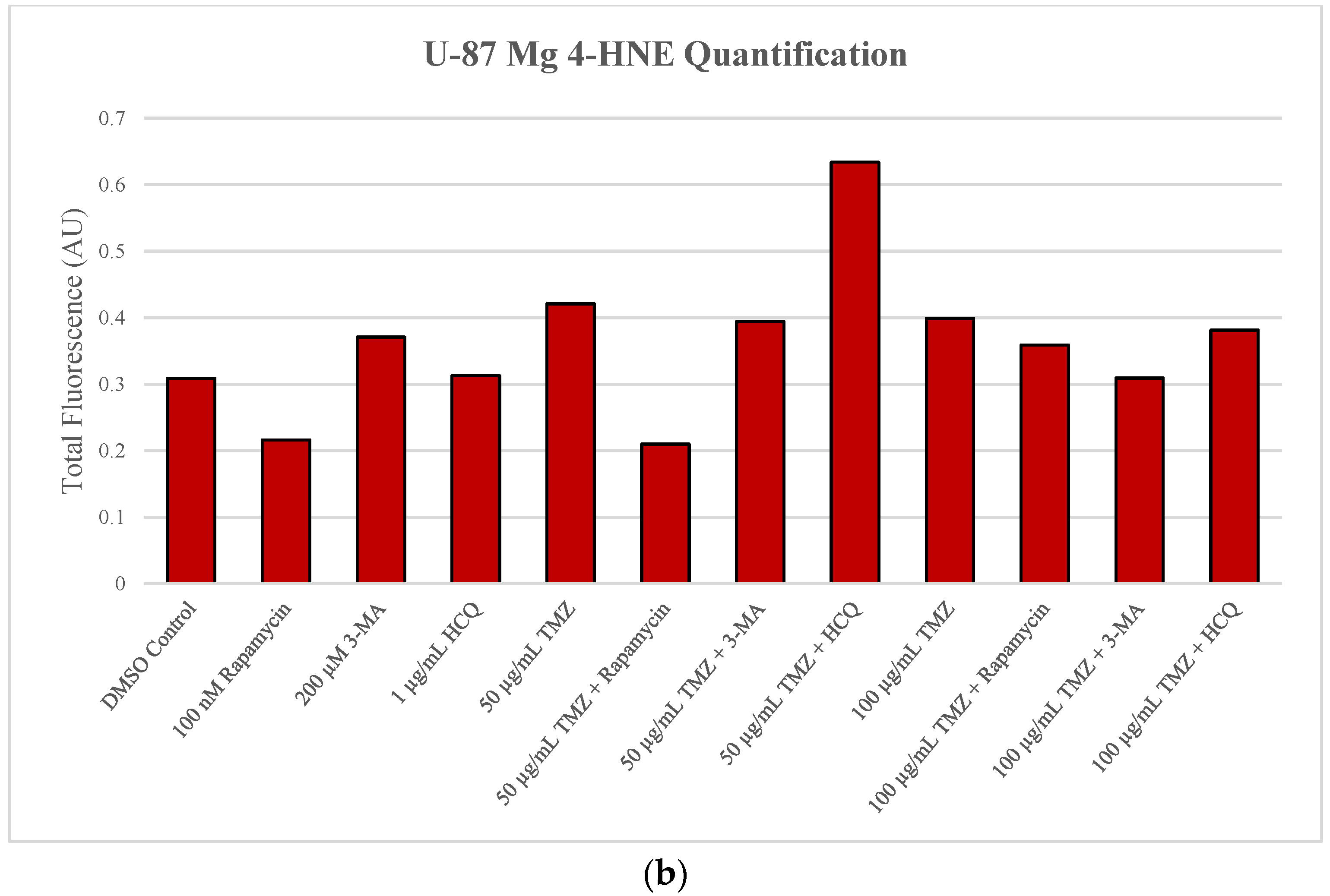
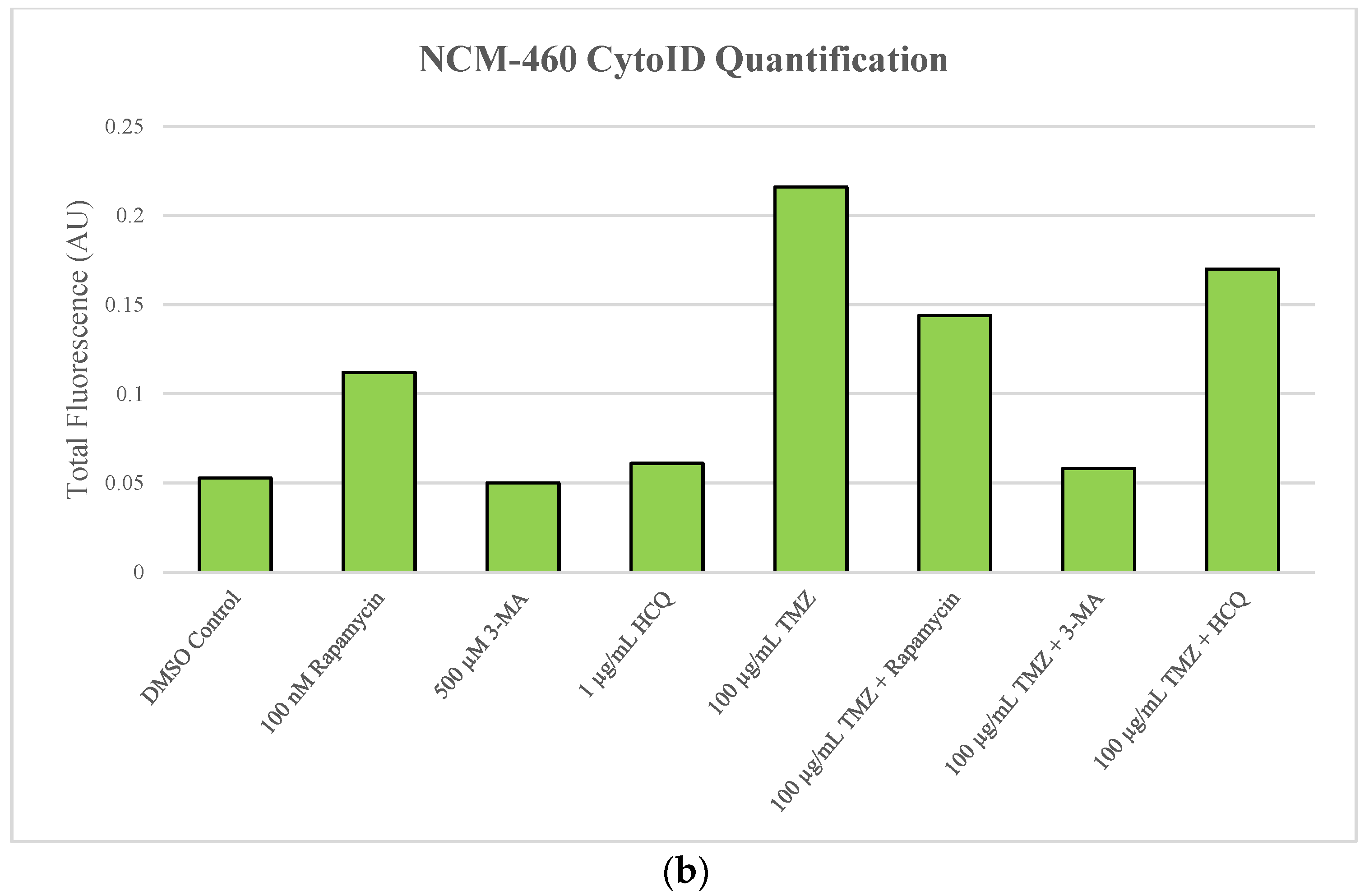
2.2. Autophagic Induction in U-87 Mg Glioblastoma Cells via TMZ Is Decreased by 3-MA or HCQ Supplementation
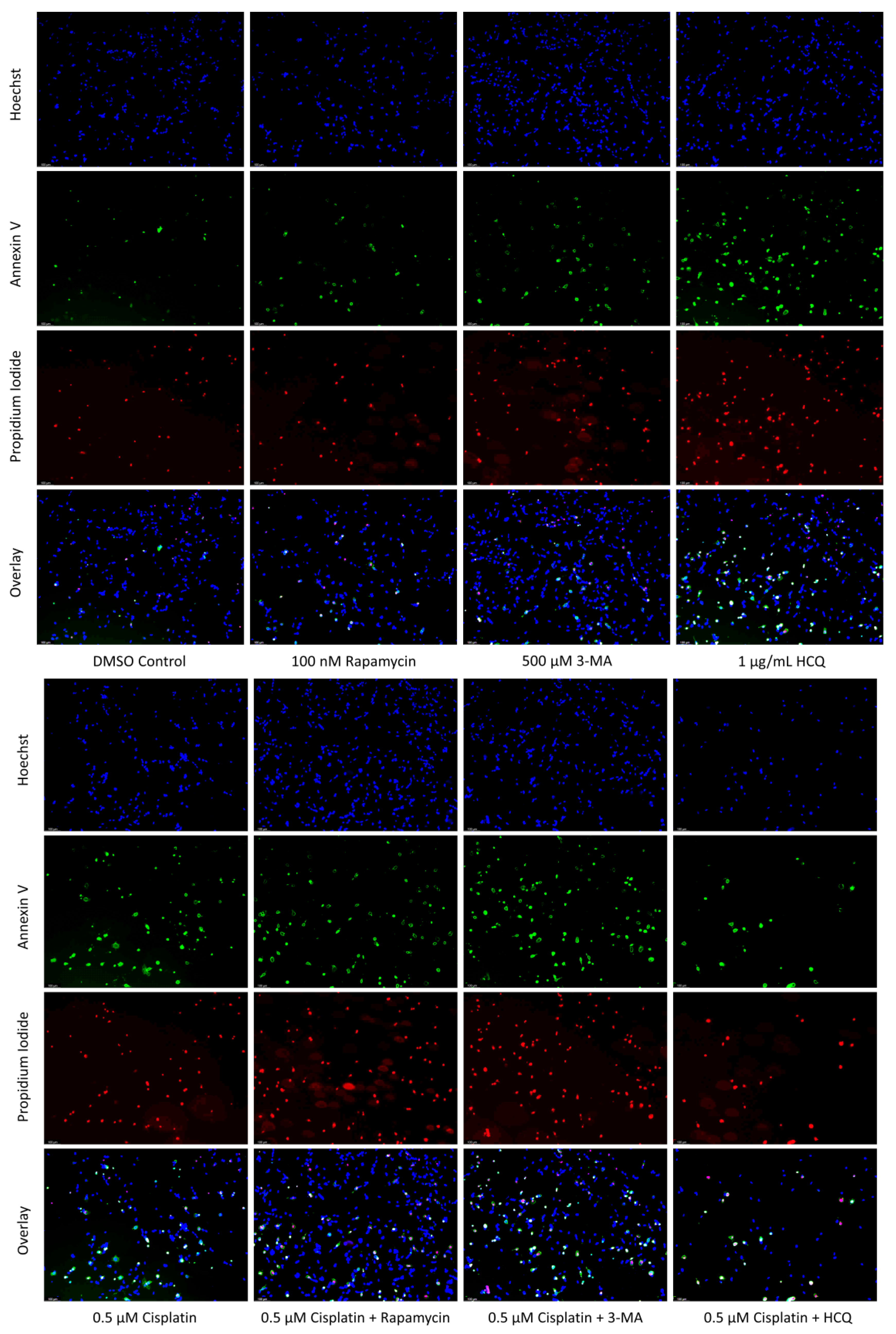
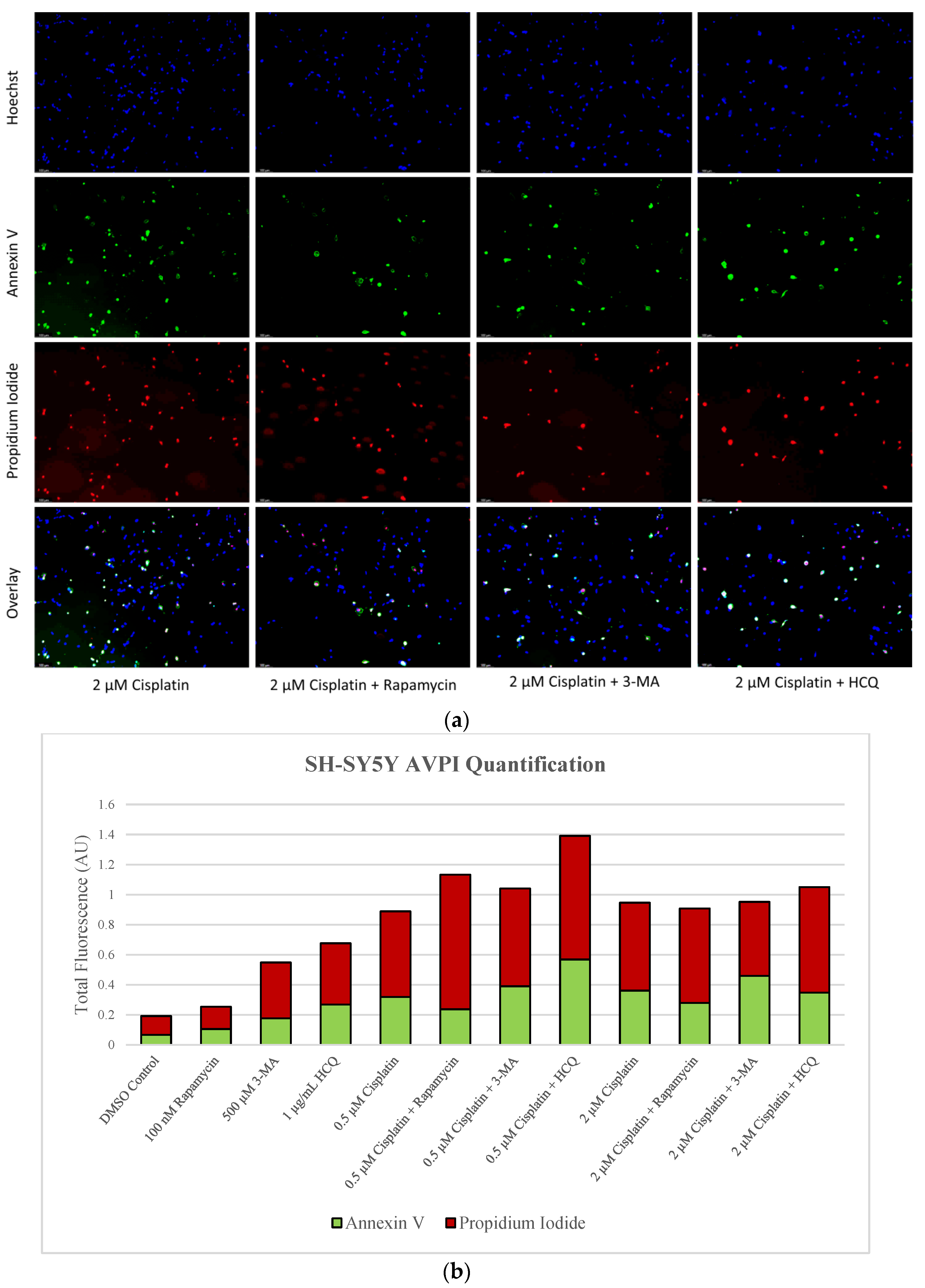
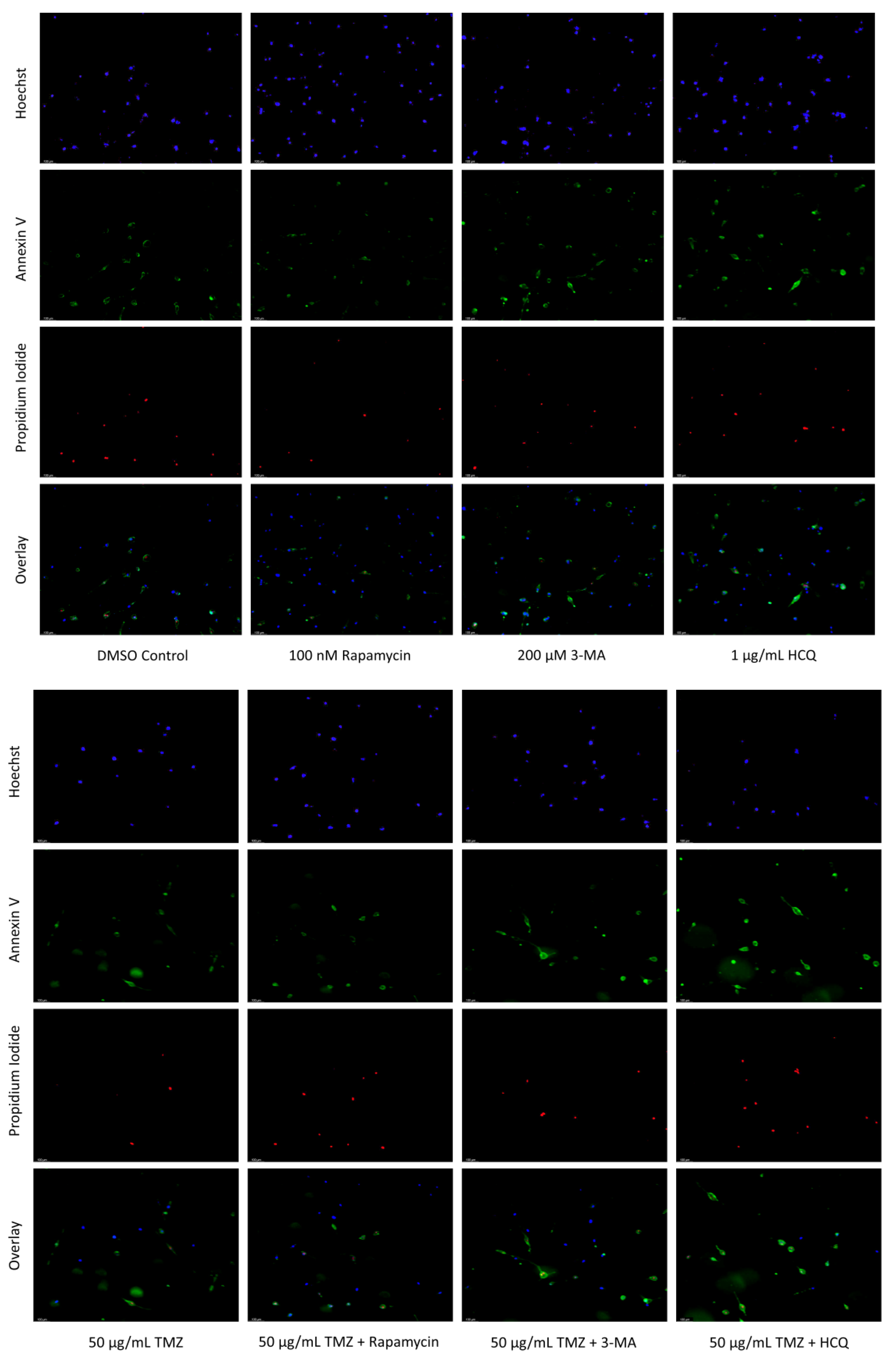
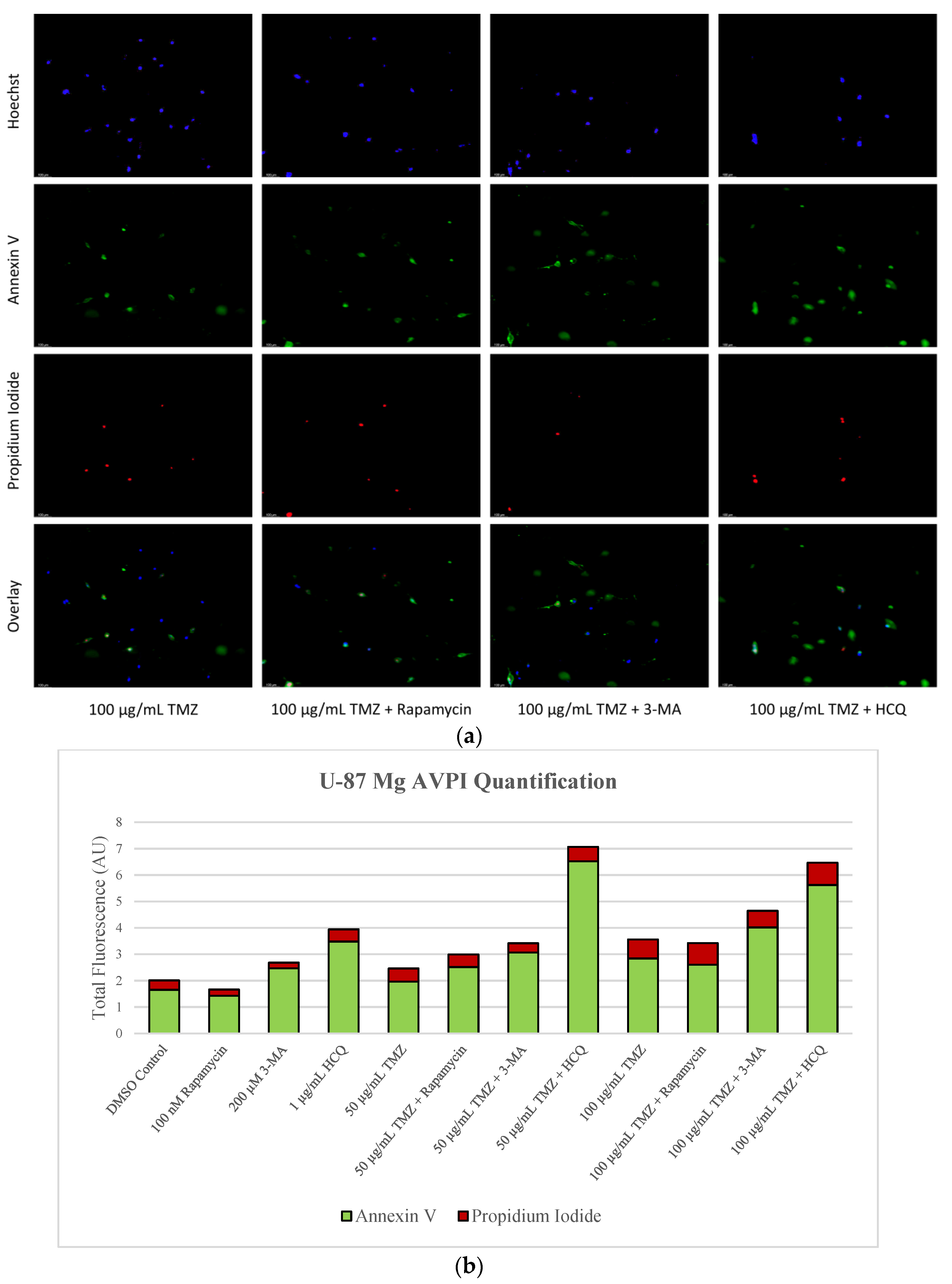
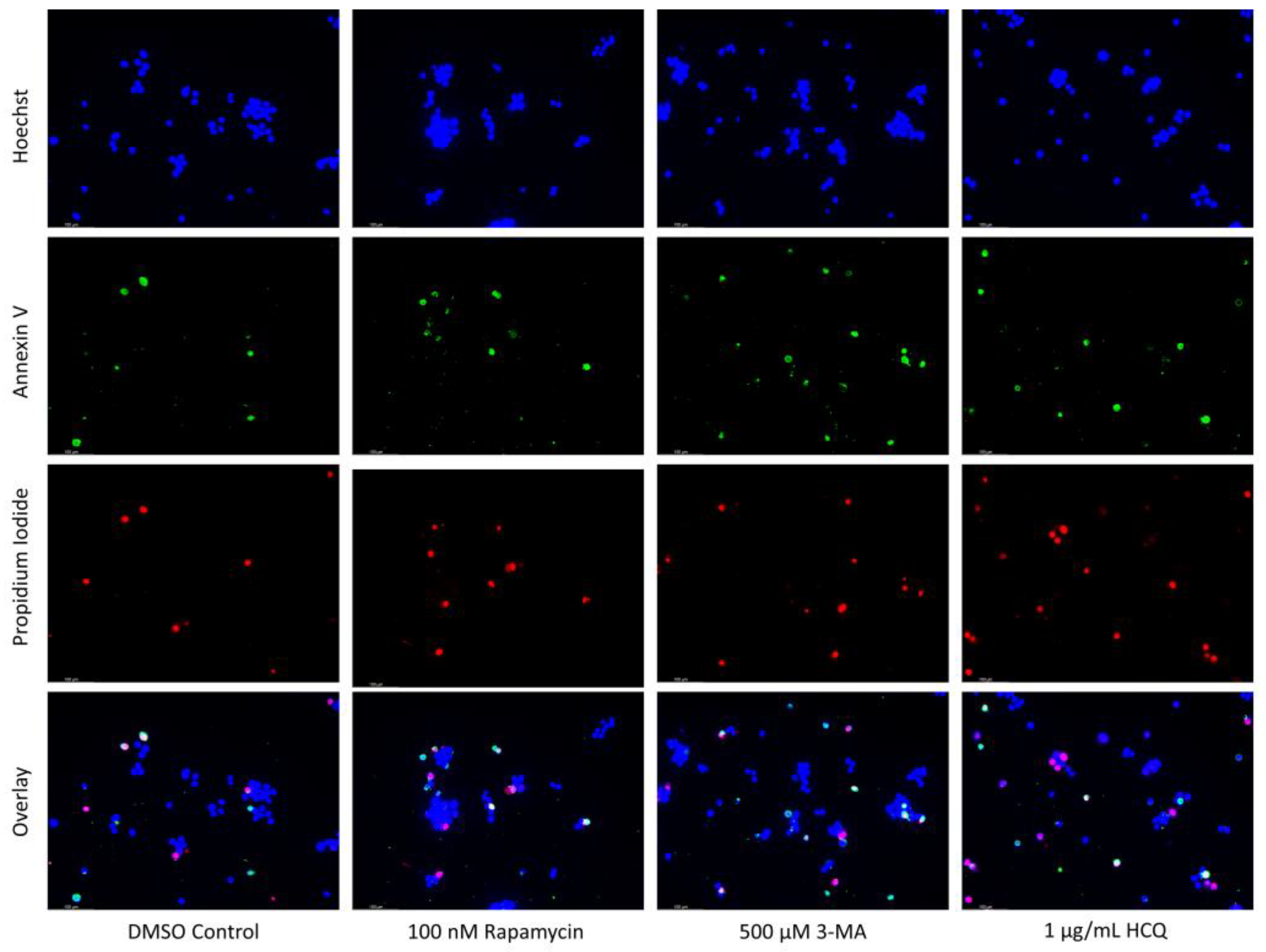
2.3. Autophagy Inhibition via 3-MA or HCQ Enhances Chemotherapy-Induced Apoptosis in Neuroblastoma and Glioblastoma
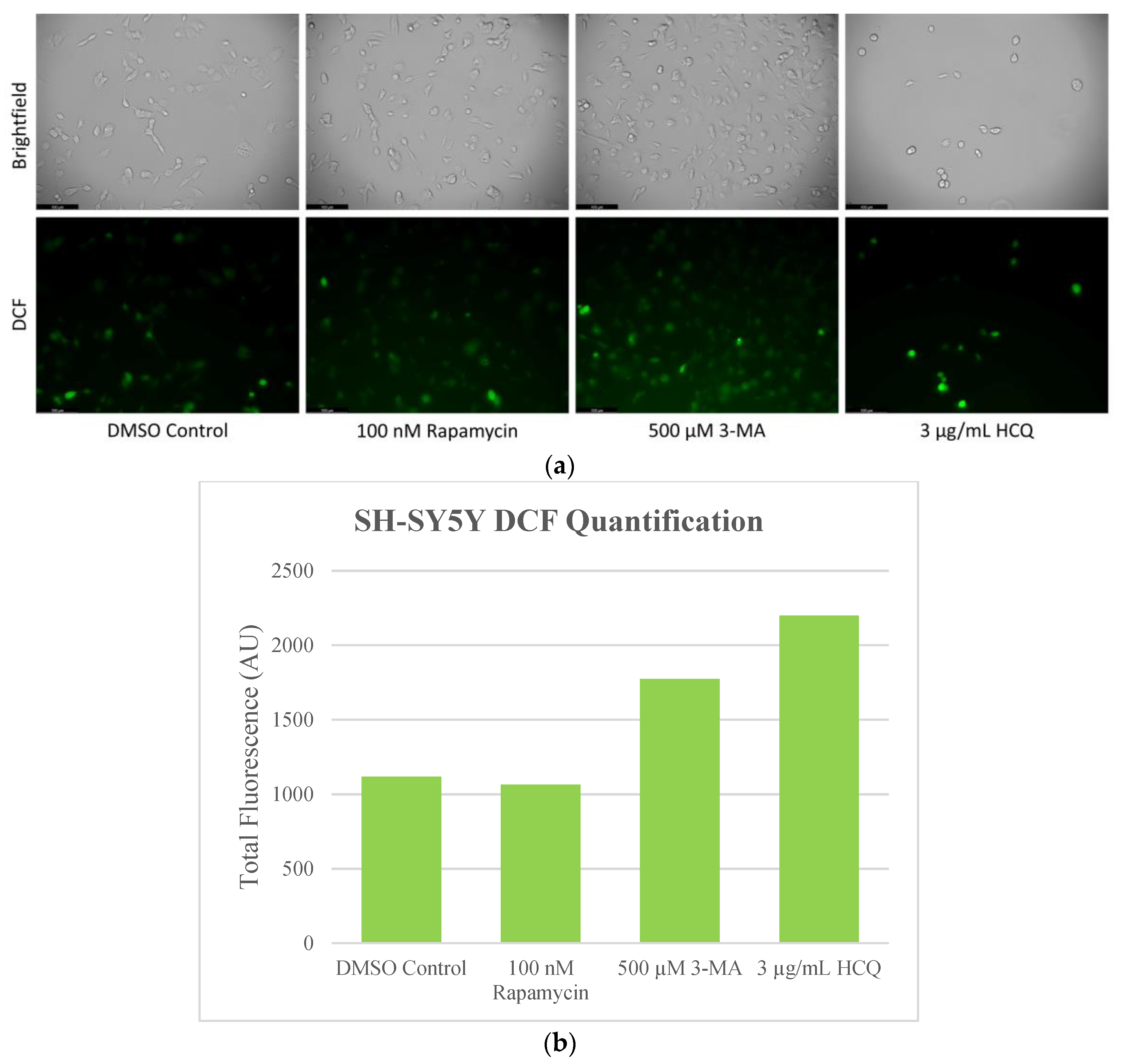
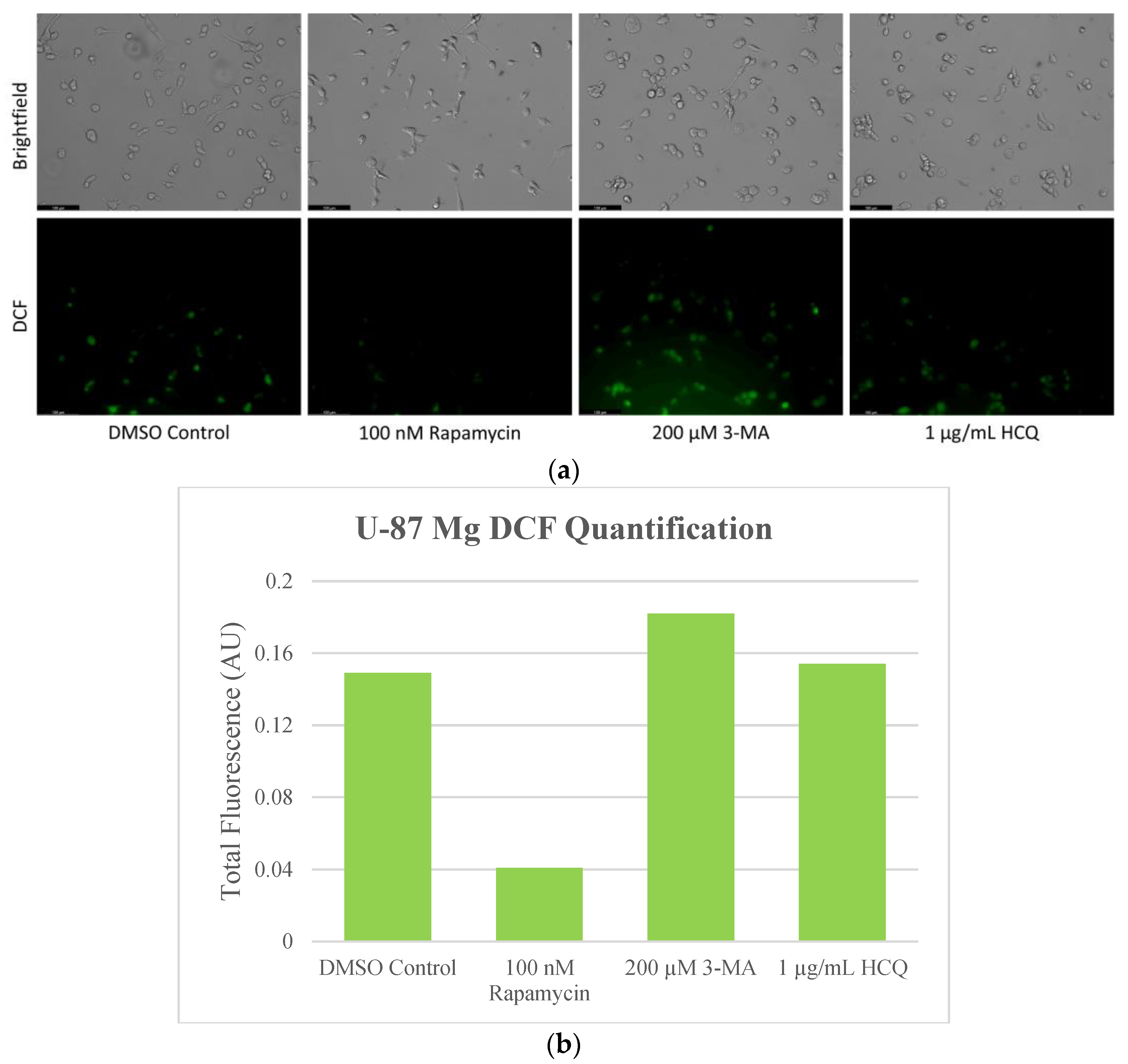
2.4. Autophagy Inhibition Enhances Reactive Oxygen Species (ROS) Production in SH-SY5Y and U-87 Mg Cells
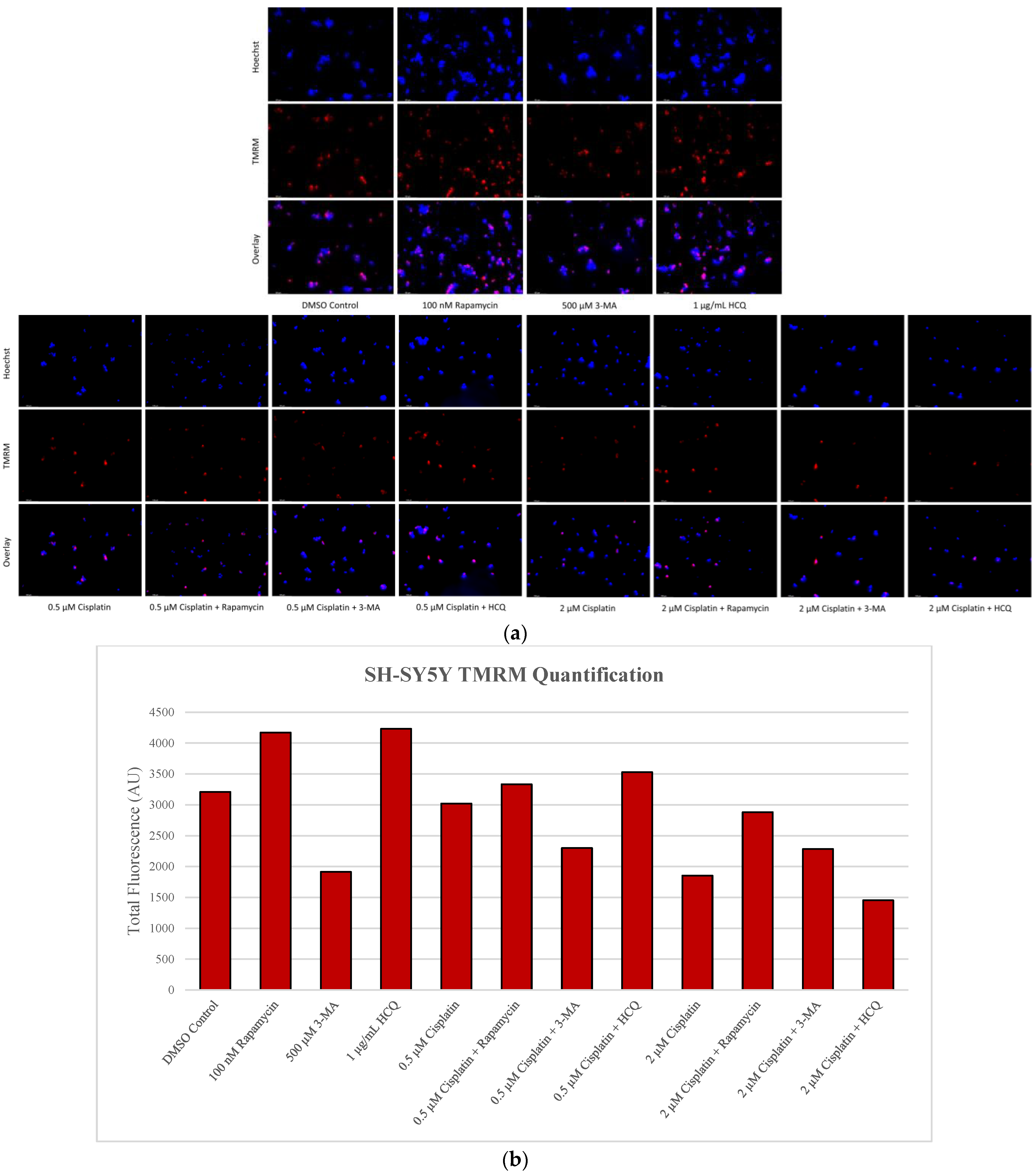
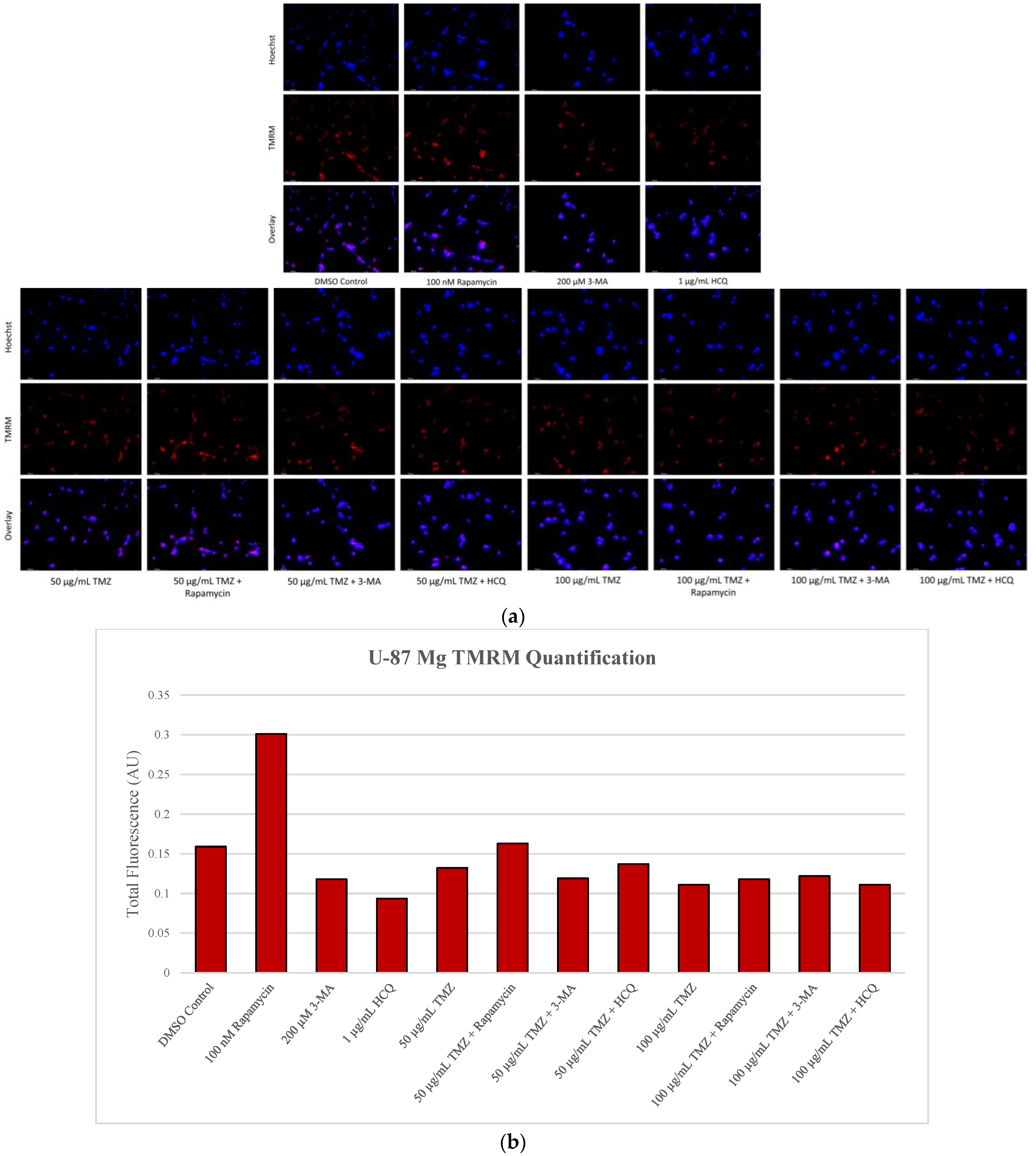
2.5. Rapamycin-Induced Autophagy Activation Increases Mitochondrial Stability While Autophagy Inhibition Reduces Mitochondrial Functionality
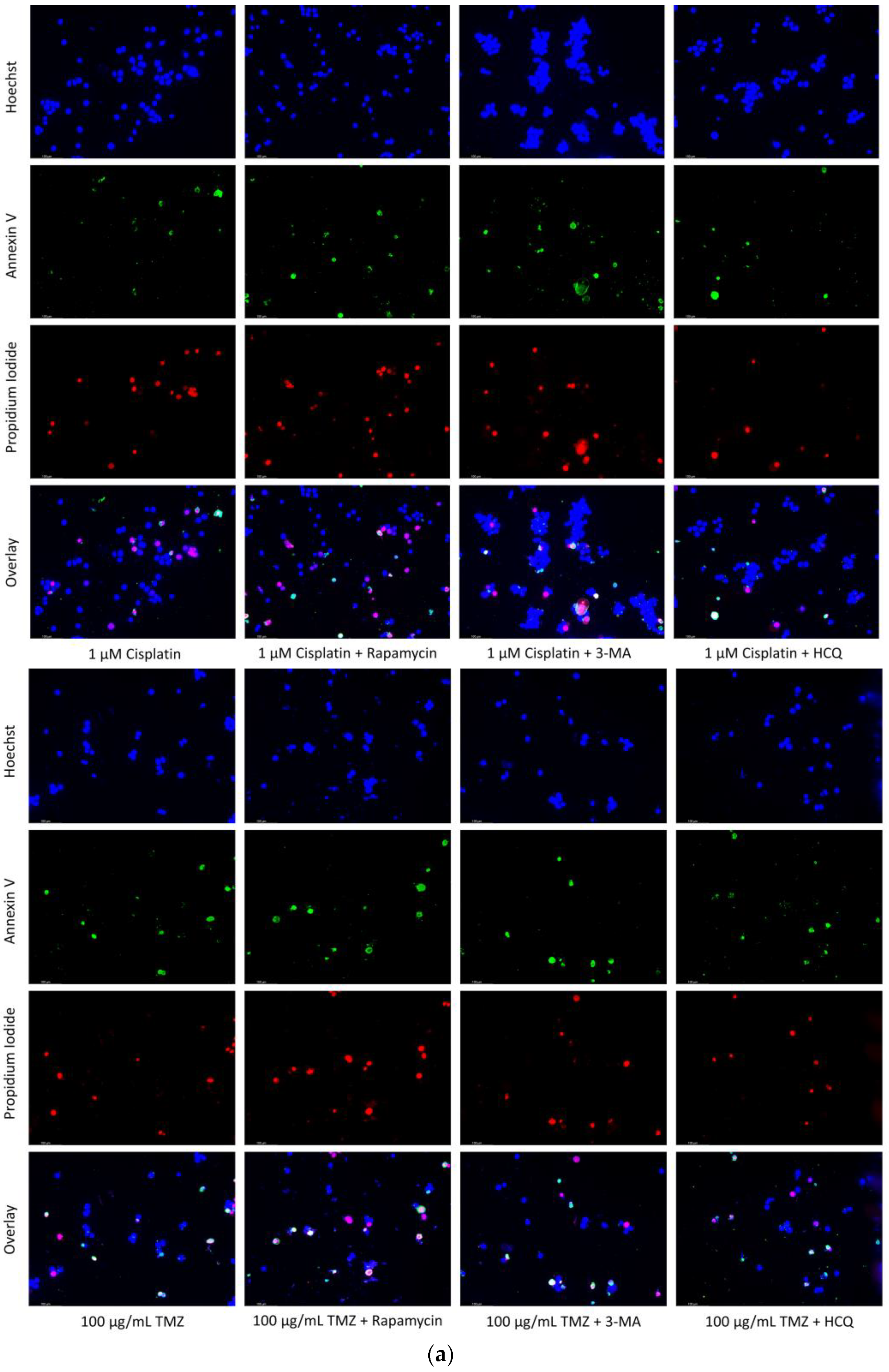
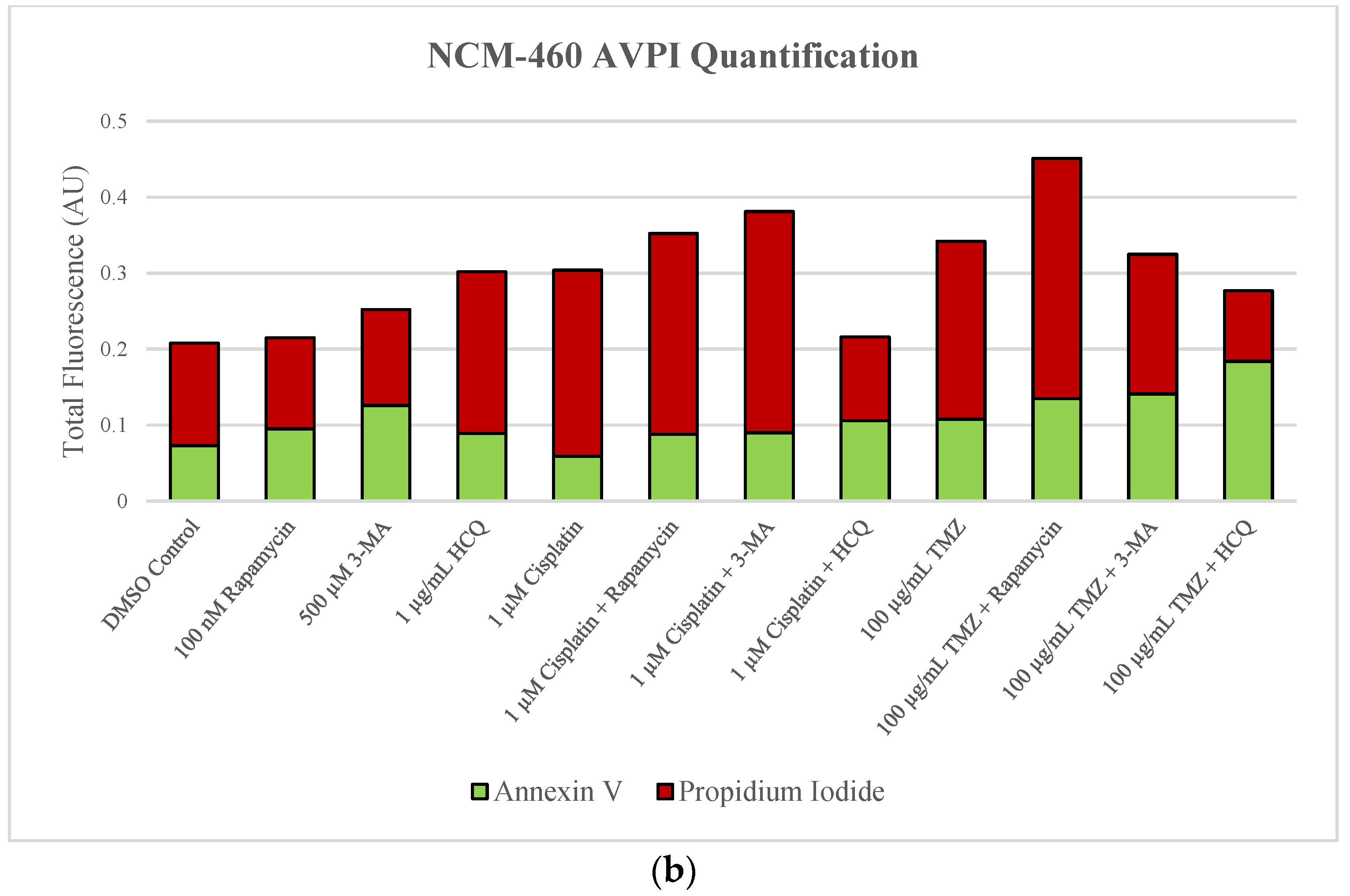
2.6. Supplementation of Standard Chemotherapies with 3-MA or HCQ Has Little Effect on Apoptotic Levels in Normal Healthy Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
- (a)
- U-87 Mg human glioblastoma cells derived from malignant glioma (ATCC, Cat. No. HTB-14, Manassas, VA, USA) were grown and cultured in Eagle’s Minimum Essential Medium with Earle’s salts and nonessential amino acids (Sigma-Aldrich Canada, Mississauga, ON, Canada) supplemented with 10% (v/v) fetal bovine serum (Thermo Scientific, Waltham, MA, USA) and 10 mg/mL gentamicin (Gibco BRL, VWR, Mississauga, ON, Canada). Cells were maintained at 37 °C and 5% CO2.
- (b)
- SH-SY5Y human neuroblastoma cells derived from a metastatic bone tumour (ATCC, Cat. No. CRL-2266, Manassas, VA, USA) were grown and cultured in Dulbecco’s Modified Eagle’s Medium F-12 HAM (Sigma-Aldrich, Mississauga, ON, Canada) supplemented with 10% fetal bovine serum (Thermo Scientific, Waltham, MA, USA) and 10 mg/mL gentamicin (Gibco BRL, VWR, Mississauga, ON, Canada). Cells were maintained at 37 °C and 5% CO2.
- (c)
- NCM-460 human colon epithelial cells (ATCC, Cat. No. CRL-1831, Manassas, VA, USA) were grown and cultured in Dulbecco’s Modified Eagle Medium (Sigma-Aldrich, Mississauga, ON, Canada) supplemented with 10% fetal bovine serum (Thermo Scientific, Waltham, MA, USA) and 10 mg/mL gentamicin (Gibco BRL, VWR, Mississauga, ON, Canada). Cells were maintained at 37 °C and 5% CO2.
- (d)
- NHF2 human skin fibroblasts (Coriell Institute for Medical Research, Cat. No. AG09309, Camden, NJ, USA) were grown and cultured in Eagle’s Minimum Essential Medium with Earle’s salts and nonessential amino acids (Sigma-Aldrich Canada, Mississauga, ON, Canada) supplemented with 10% fetal bovine serum (Thermo Scientific, Waltham, MA, USA) and 10 mg/mL gentamicin (Gibco BRL, VWR, Mississauga, ON, Canada). Cells were maintained at 37 °C and 5% CO2.
4.2. Chemicals and Cell Treatment
- (a)
- Cisplatin (Sigma-Aldrich Canada, Cat. No. PHR1624) was dissolved in 0.9% NaCl to make a 1 mM stock solution. Stock solution was stored at −20 °C until use. SH-SY5Y were treated at doses of 0.5 µM, 1 µM, or 2 µM.
- (b)
- Temozolomide (Ontario Chemicals Inc., Cat. No. T1062) was dissolved in dimethylsulfoxide (DMSO) to make a 20 mg/mL stock solution. Stock solution was stored at −20 °C until use. U-87 Mg were treated at doses of 50 µg/mL or 100 µg/mL.
- (c)
- Rapamycin (Sigma-Aldrich Canada, Cat. No. R8781) was dissolved in DMSO to make a 40 µM stock. Stock solution was stored at −20 °C until use. SH-SY5Y were treated at doses of 50 nM, 100 nM, or 150 nM. U-87 Mg were treated at doses of 100 nM.
- (d)
- 3-Methyladenine (Sigma-Aldrich Canada, Cat. No. M9281) was dissolved in milliQ water to make a 50 mM stock. Stock solution was stored at −20 °C until use. SH-SY5Y were treated at doses of 500 µM. U-87 Mg were treated at doses of 200 µM or 500 µM.
- (e)
- Hydroxychloroquine (Sigma-Aldrich Canada, Cat. No. H0915) was dissolved in DMSO to make a 3 mg/mL stock. Stock solution was stored at −20 °C until use. SH-SY5Y were treated at doses of 1 µg/mL or 3 µg/mL. U-87 Mg were treated at a dose of 1 µg/mL.
4.3. Monodansylcadaverine Staining for Autophagic Vacuoles
4.4. Cyto-ID Staining for Autophagic Flux
4.5. Apoptotic Analysis via Annexin V and Propidium Iodide Staining
4.6. Measurement of Reactive Oxygen Species
4.7. Tetramethylrhodamine, Methyl Ester Staining for Healthy Mitochondria
4.8. Immunofluorescent Staining
4.9. Statistical Testing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241. [Google Scholar] [CrossRef] [PubMed]
- Swift, C.C.; Eklund, M.J.; Kraveka, J.M.; Alazraki, A.L. Updates in Diagnosis, Management, and Treatment of Neuroblastoma. Radiographics 2018, 38, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Colon, N.C.; Chung, D.H. Neuroblastoma. Adv. Pediatr. 2011, 58, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20 (Suppl. S5), S2–S8. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Exon Publications: Brisbane, QLD, Australia, 2017. Available online: http://www.ncbi.nlm.nih.gov/books/NBK470003/ (accessed on 16 May 2022).
- Wirsching, H.G.; Galanis, E.; Weller, M. Chapter 23–Glioblastoma. In Handbook of Clinical Neurology; Berger, M.S., Weller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 134, pp. 381–397. [Google Scholar] [CrossRef]
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, Prognosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2010, 24, 65–86. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Lee, S. Side Effects of Chemotherapy. Canadian Cancer Society. Available online: https://cancer.ca/en/treatments/treatment-types/chemotherapy/side-effects-of-chemotherapy (accessed on 19 May 2022).
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef]
- Dai, S.; Wang, B.; Li, W.; Wang, L.; Song, X.; Guo, C.; Li, Y.; Liu, F.; Zhu, F.; Wang, Q.; et al. Systemic application of 3-methyladenine markedly inhibited atherosclerotic lesion in ApoE−/− mice by modulating autophagy, foam cell formation and immune-negative molecules. Cell Death Dis. 2016, 7, e2498. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. Autophagy modulation for cancer therapy. Cancer Biol. Ther. 2011, 11, 169–176. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Belounis, A.; Nyalendo, C.; Le Gall, R.; Imbriglio, T.V.; Mahma, M.; Teira, P.; Beaunoyer, M.; Cournoyer, S.; Haddad, E.; Vassal, G.; et al. Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer 2016, 16, 891. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef]
- Singh, A.K.; Bissoyi, A.; Kashyap, M.P.; Patra, P.K.; Rizvi, S.I. Autophagy Activation Alleviates Amyloid-β-Induced Oxidative Stress, Apoptosis and Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells. Neurotox. Res. 2017, 32, 351–361. [Google Scholar] [CrossRef]
- Lin, X.; Han, L.; Weng, J.; Wang, K.; Chen, T. Rapamycin inhibits proliferation and induces autophagy in human neuroblastoma cells. Biosci. Rep. 2018, 38, BSR20181822. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.Q.; Toh, X.Y.; Ho, I.A.W.; Sia, K.C.; Newman, J.P.; Yulyana, Y.; Ng, W.-H.; Lai, S.H.; Ho, M.M.F.; Dinesh, N.; et al. Combined treatment of Nimotuzumab and rapamycin is effective against temozolomide-resistant human gliomas regardless of the EGFR mutation status. BMC Cancer 2015, 15, 255. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Cai, M.; Zhang, Y.; Tao, L.; Guo, R. miR-29c-3p inhibits autophagy and cisplatin resistance in ovarian cancer by regulating FOXP1/ATG14 pathway. Cell Cycle 2019, 19, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, Y.; Shen, Y.; Tantai, J. Knockdown of TRIM65 inhibits autophagy and cisplatin resistance in A549/DDP cells by regulating miR-138-5p/ATG7. Cell Death Dis. 2019, 10, 429. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, Z.; Yin, H.; Yang, G. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1α-mediated regulation of apoptosis and autophagy. Theranostics 2019, 9, 1096–1114. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, X.; Zhao, H.; Liu, Y.; Feng, Y.; An, R.; Lv, X.; Li, J.; Chen, B. Down-regulation of OGT promotes cisplatin resistance by inducing autophagy in ovarian cancer. Theranostics 2018, 8, 5200–5212. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Yu, X.; Fan, H.; Jiang, X.; Zheng, W.; Yang, Y.; Jin, M.; Ma, X.; Jiang, W. Apatinib induces apoptosis and autophagy via the PI3K/AKT/mTOR and MAPK/ERK signaling pathways in neuroblastoma. Oncol. Lett. 2020, 20, 52. [Google Scholar] [CrossRef]
- Kommalapati, V.K.; Kumar, D.; Tangutur, A.D. Inhibition of JNJ-26481585-mediated autophagy induces apoptosis via ROS activation and mitochondrial membrane potential disruption in neuroblastoma cells. Mol. Cell. Biochem. 2020, 468, 21–34. [Google Scholar] [CrossRef]
- Wu, X.; Geng, F.; Cheng, X.; Guo, Q.; Zhong, Y.; Cloughesy, T.F.; Yong, W.H.; Chakravarti, A.; Guo, D. Lipid Droplets Maintain Energy Homeostasis and Glioblastoma Growth via Autophagic Release of Stored Fatty Acids. iScience 2020, 23, 101569. [Google Scholar] [CrossRef]
- Josset, E.; Burckel, H.; Noël, G.; Bischoff, P. The mTOR inhibitor RAD001 potentiates autophagic cell death induced by temozolomide in a glioblastoma cell line. Anticancer Res. 2013, 33, 1845–1851. [Google Scholar]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Wang, S.Y.; Wang, X.; Zhang, C.Y. LncRNA SNHG7 enhances chemoresistance in neuroblastoma through cisplatin-induced autophagy by regulating miR-329-3p/MYO10 axis. Eur. Rev. 2020, 24, 3805–3817. [Google Scholar] [CrossRef]
- Yun, E.J.; Kim, S.; Hsieh, J.T.; Baek, S.T. Wnt/β-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef]
- Huang, T.; Wan, X.; Alvarez, A.A.; James, C.D.; Song, X.; Yang, Y.; Sastry, N.; Nakano, I.; Sulman, E.P.; Hu, B.; et al. MIR93 (microRNA-93) regulates tumorigenicity and therapy response of glioblastoma by targeting autophagy. Autophagy 2019, 15, 1100–1111. [Google Scholar] [CrossRef]
- Taylor, M.A.; Das, B.C.; Ray, S.K. Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis 2018, 23, 563–575. [Google Scholar] [CrossRef]
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Ma, D.; Pignanelli, C.; Tarade, D.; Gilbert, T.; Noel, M.; Mansour, F.; Adams, S.; Dowhayko, A.; Stokes, K.; Vshyvenko, S.; et al. Cancer Cell Mitochondria Targeting by Pancratistatin Analogs is Dependent on Functional Complex II and III. Sci. Rep. 2017, 7, 42957. [Google Scholar] [CrossRef]































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wear, D.; Bhagirath, E.; Balachandar, A.; Vegh, C.; Pandey, S. Autophagy Inhibition via Hydroxychloroquine or 3-Methyladenine Enhances Chemotherapy-Induced Apoptosis in Neuro-Blastoma and Glioblastoma. Int. J. Mol. Sci. 2023, 24, 12052. https://doi.org/10.3390/ijms241512052
Wear D, Bhagirath E, Balachandar A, Vegh C, Pandey S. Autophagy Inhibition via Hydroxychloroquine or 3-Methyladenine Enhances Chemotherapy-Induced Apoptosis in Neuro-Blastoma and Glioblastoma. International Journal of Molecular Sciences. 2023; 24(15):12052. https://doi.org/10.3390/ijms241512052
Chicago/Turabian StyleWear, Darcy, Eesha Bhagirath, Arpana Balachandar, Caleb Vegh, and Siyaram Pandey. 2023. "Autophagy Inhibition via Hydroxychloroquine or 3-Methyladenine Enhances Chemotherapy-Induced Apoptosis in Neuro-Blastoma and Glioblastoma" International Journal of Molecular Sciences 24, no. 15: 12052. https://doi.org/10.3390/ijms241512052
APA StyleWear, D., Bhagirath, E., Balachandar, A., Vegh, C., & Pandey, S. (2023). Autophagy Inhibition via Hydroxychloroquine or 3-Methyladenine Enhances Chemotherapy-Induced Apoptosis in Neuro-Blastoma and Glioblastoma. International Journal of Molecular Sciences, 24(15), 12052. https://doi.org/10.3390/ijms241512052