
Targeting KRAS in Colorectal Cancer: A Bench to Bedside Review
 , and
, and
Abstract
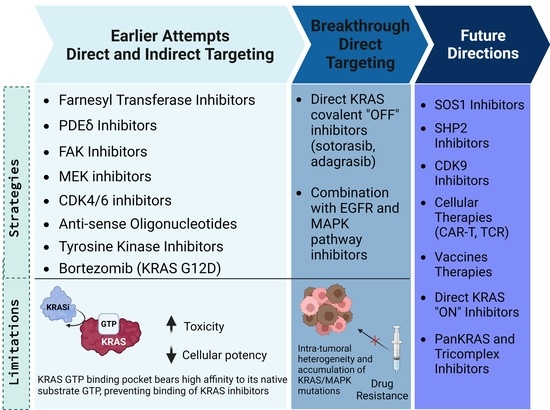
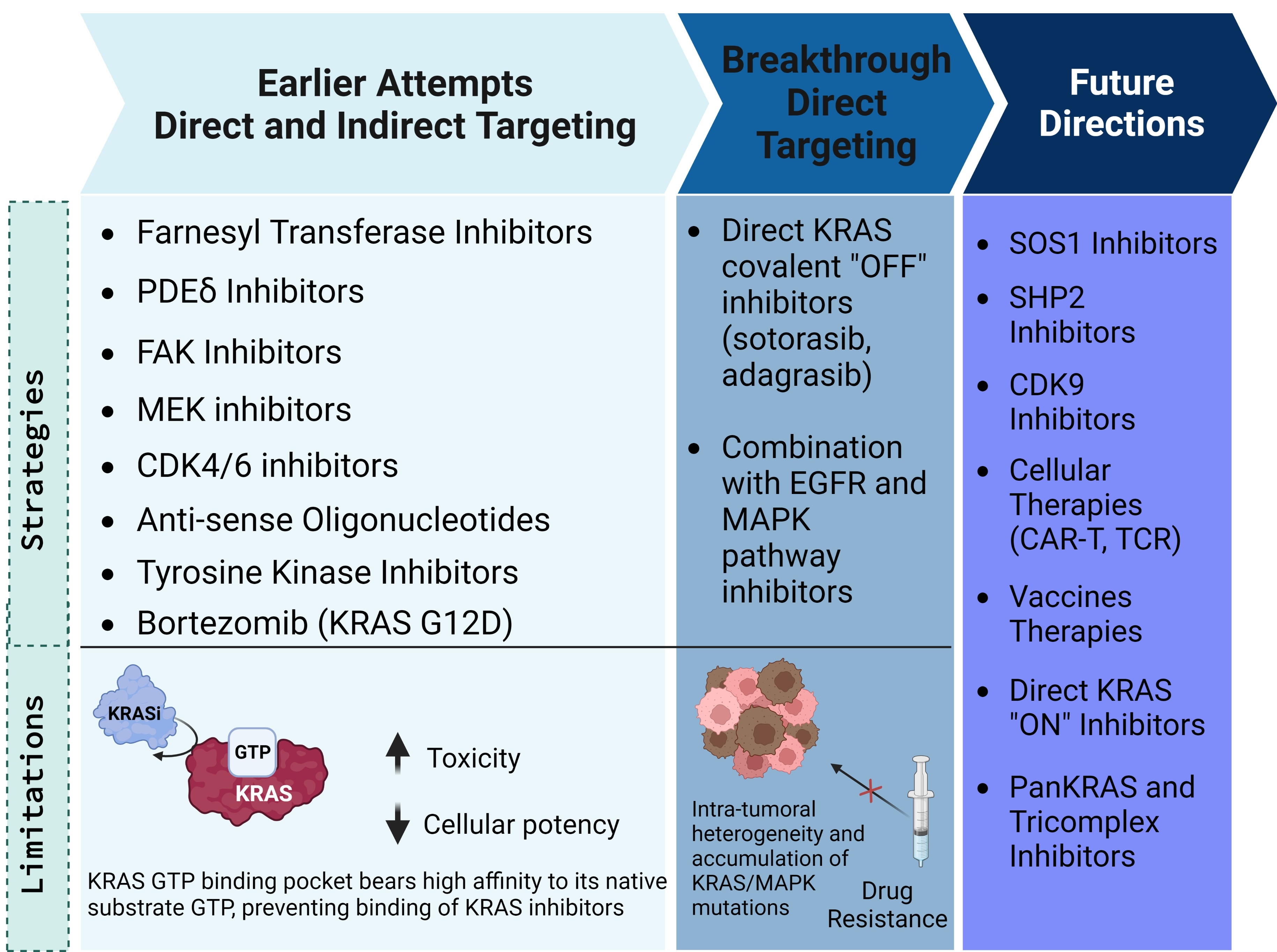{kind=link}
{kind=link}
{kind=link}
1. Introduction
KRAS Molecular Structure, Function, and Mutation
2. Earlier Attempts to Target KRAS-Mutated CRC
3. Direct KRAS Targeting in CRC
3.1. Sotorasib
3.2. Adagrasib
3.3. Comparison between Sotorasib and Adagrasib
3.4. Mechanisms of Resistance to Sotorasib and Adagrasib
4. Future Directions
4.1. Combination with EGFR Inhibitors
4.2. Combination with MEK Inhibitors
4.3. Combination with SOS1 Inhibitors
4.4. Combination with SHP2 Inhibitors
4.5. Newer G12C Inhibitors
4.6. KRAS G12D Targeting
4.7. Tri-Complex and panKRAS Inhibitors
4.8. Other Emerging Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary
| AKT | Protein kinase B (PKB) |
| APC | Adenomatous polyposis coli |
| BRAF | v-Raf murine sarcoma viral oncogene homolog B1 |
| CAR-T | Chimeric antigen receptor (CAR) T-cell |
| CDK | Cyclin-dependent kinase |
| CI | Confidence interval |
| c-MET | c-Mesenchymal-epithelial transition factor |
| CNS | Central nervous system |
| CRC | Colorectal cancer |
| DCC | Deleted in colorectal cancer |
| DCR | Disease control rate |
| DoR | Duration of response |
| EGFR | Epidermal growth factor receptor |
| EMA | European Medicines Agency |
| ERK | Extracellular signal-regulated kinase |
| FAK | Focal adhesion kinase |
| FDA | Food and Drug Administration |
| FGFR | Fibroblast growth factor receptor |
| FOX | Forkhead box |
| FTI | Farnesyl transferase inhibitors |
| GDP | Guanosine diphosphate |
| GRB7 | Growth factor receptor-bound protein 7 |
| GTP | Guanosine triphosphate |
| HLA | Human leukocyte antigens |
| HR | Hazard ratio |
| HRAS | Harvey rat sarcoma viral oncogenes |
| ICIs | Immune checkpoint inhibitors |
| IV | Intravenous |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| KRAS | Kirsten rat sarcoma |
| MAPK | Mitogen-activated protein kinase |
| MEF | Mouse embryonic fibroblast |
| MEK | Mitogen-activated protein kinase |
| mTOR | Mammalian target of rapamycin |
| MYC | Master regulator of cell cycle entry and proliferative metabolism |
| NE | Not evaluable |
| NF-κB | Nuclear factor-kappa B |
| NR | Not reached |
| NRAS | Neuroblastoma rat sarcoma viral oncogenes |
| NSCLC | Non-small-cell lung cancer |
| ORR | Objective response rate |
| OS | Overall survival |
| P+D | Placebo + docetaxel |
| PD-1 | Programmed death 1 |
| PDEδ | Prenyl-binding protein |
| PD-L1 | Programmed death ligand 1 |
| PFS | Progression-free survival |
| PI | Proteasome inhibitor |
| PI3K | Phosphatidylinositol 3-kinase |
| PLCε | Phospholipase Cε |
| RAF | Rapidly accelerated fibrosarcoma |
| RAL | Ras-like protein |
| RalGDS | RAS-related protein Ral |
| RAS | Rat sarcoma |
| RHOA | Ras homolog gene family, member A |
| ROS1 | Ros proto-oncogene 1 |
| RTK | Receptor tyrosine kinase |
| S+D | Selumetinib + docetaxel |
| SHP2 | Protein tyrosine phosphatase 2 |
| S-IIP | Switch-II pocket |
| SOS-1 | Son of sevenless homologue 1 |
| STAT | Signal transducer and activator of transcription |
| TCR | T-cell receptor |
| TEAEs | Treatment emergent adverse events |
| TP53 | Tumor suppressor protein 53 |
| TRAEs | Treatment related adverse events |
| VEGF | Vascular endothelial growth factor |
| WT | Wild-type |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA. Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Sagaert, X.; Vanstapel, A.; Verbeek, S. Tumor Heterogeneity in Colorectal Cancer: What Do We Know So Far? Pathobiology 2018, 85, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Sánchez-de-Diego, C.; Dieste, A.P.; Cerrada, E.; Yoldi, M.J.R. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Clevers, H. Studying cellular heterogeneity and drug sensitivity in colorectal cancer using organoid technology. Curr. Opin. Genet. Dev. 2018, 52, 117–122. [Google Scholar] [CrossRef]
- Fearon, E.F.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Ferreira, A.; Pereira, F.; Reis, C.; Oliveira, M.J.; Sousa, M.J.; Preto, A. Crucial Role of Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation: Therapeutic Implications. Cells 2022, 11, 2183. [Google Scholar] [CrossRef]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Verlaan-de Vries, M.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef]
- Mustachio, L.M.; Chelariu-Raicu, A.; Szekvolgyi, L.; Roszik, J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers 2021, 13, 1204. [Google Scholar] [CrossRef]
- Cheng, W.L.; Feng, P.H.; Lee, K.Y.; Chen, K.Y.; Sun, W.L.; Van Hiep, N.; Luo, C.S.; Wu, S.M. The role of ereg/egfr pathway in tumor progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American society of clinical oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Kong, L.; Wan, Z.; Zhu, F.; Zhong, M.; Lv, Y.; Zhao, P.; Shi, H. Somatic mutation profiling and HER2 status in KRAS-positive Chinese colorectal cancer patients. Sci. Rep. 2019, 9, 16894. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting kras in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57. [Google Scholar] [CrossRef]
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef]
- Dinu, D.; Dobre, M.; Panaitescu, E.; Bîrlă, R.; Iosif, C.; Hoara, P.; Caragui, A.; Boeriu, M.; Constantinoiu, S.; Ardeleanu, C. Prognostic significance of KRAS gene mutations in colorectal cancer-preliminary study. J. Med. Life 2014, 7, 581–587. [Google Scholar]
- Lièvre, A.; Bachet, J.-B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.-F.; Côté, J.-F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; Mccormick, F. The GT Pase superf amily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef]
- Hancock, J.F.; Parton, R.G. Ras plasma membrane signalling platforms. Biochem. J. 2005, 389, 1–11. [Google Scholar] [CrossRef]
- Mao, Z.; Xiao, H.; Shen, P.; Yang, Y.; Xue, J.; Yang, Y.; Shang, Y.; Zhang, L.; Li, X.; Zhang, Y.; et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022, 8, 5. [Google Scholar] [CrossRef]
- Vögler, O.; Barceló, J.M.; Ribas, C.; Escribá, P. V Membrane interactions of G proteins and other related proteins. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 1640–1652. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Braghiroli, M.I.; Chou, J.F.; Hechtman, J.F.; Kemeny, N.; Saltz, L.; Capanu, M.; Yaeger, R. Clinical features and outcomes of patients with colorectal cancers harboring NRAS mutations. Clin. Cancer Res. 2017, 23, 4753–4760. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.E.; Mageean, C.J.; Rusilowicz, E.V.; Wickenden, J.A.; Clague, M.J.; Prior, I.A. Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J. Proteome Res. 2015, 14, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Lou, L.; Song, Z.; Zhang, X.; Xiong, X.F. Targeting mutated GTPase KRAS in tumor therapies. Eur. J. Med. Chem. 2021, 226, 113816. [Google Scholar] [CrossRef]
- Nuevo-Tapioles, C.; Philips, M.R. The role of KRAS splice variants in cancer biology. Front. Cell Dev. Biol. 2022, 10, 1033348. [Google Scholar] [CrossRef]
- Mousavi, N.; Truelsen, S.L.B.; Bernth-Andersen, S.; Koch, A.; Heegaard, S. Mutation of KRAS in colorectal adenocarcinoma in Greenland. APMIS 2022, 130, 493–497. [Google Scholar] [CrossRef]
- Plowman, S.J.; Berry, R.L.; Bader, S.A.; Luo, F.; Arends, M.J.; Harrison, D.J.; Hooper, M.L.; Patek, C.E. K-ras 4A and 4B are co-expressed widely in human tissues, and their ratio is altered in sporadic colorectal cancer. J. Exp. Clin. Cancer Res. 2006, 25, 259–267. [Google Scholar]
- Barceló, C.; Paco, N.; Morell, M.; Alvarez-Moya, B.; Bota-Rabassedas, N.; Jaumot, M.; Vilardell, F.; Capella, G.; Agell, N. Phosphorylation at ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014, 74, 1190–1199. [Google Scholar] [CrossRef]
- Quatela, S.E.; Sung, P.J.; Ahearn, I.M.; Bivona, T.G.; Philips, M.R. Analysis of K-Ras Phosphorylation, Translocation, and Induction of Apoptosis. Methods Enzymol. 2008, 439, 87–102. [Google Scholar]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Bi, Z.; Liu, Y.; Qin, F.; Wei, Y.; Wei, X. Targeting RAS–RAF–MEK–ERK signaling pathway in human cancer: Current status in clinical trials. Genes Dis. 2022, 10, 76–88. [Google Scholar]
- Wang, Y.; Kaiser, C.E.; Frett, B.; Li, H.Y. Targeting mutant KRAS for anticancer therapeutics: A review of novel small molecule modulators. J. Med. Chem. 2013, 56, 5219–5230. [Google Scholar] [CrossRef] [PubMed]
- González-García, A.; Pritchard, C.A.; Paterson, H.F.; Mavria, G.; Stamp, G.; Marshall, C.J. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell 2005, 7, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Hofer, F.; Fields, S.; Schneider, C.; Steven Martin, G. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc. Natl. Acad. Sci. USA 1994, 91, 11089–11093. [Google Scholar] [CrossRef]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Gysin, S.; Salt, M.; Young, A.; McCormick, F. Therapeutic strategies for targeting Ras proteins. Genes Cancer 2011, 2, 359–372. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Hartmann, J.T.; de Braud, F.; Schuch, G.; Zubel, A.; Celik, I.; Schlichting, M.; Koralewski, P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: The OPUS study. Ann. Oncol. 2011, 22, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; John Simes, R.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Socinski, M.A.; Burns, T.F. KRAS mutant lung cancer: Progress thus far on an elusive therapeutic target. Clin. Transl. Med. 2015, 4, 35. [Google Scholar] [CrossRef]
- Parikh, K.; Banna, G.; Liu, S.V.; Friedlaender, A.; Desai, A.; Subbiah, V.; Addeo, A. Drugging KRAS: Current perspectives and state-of-art review. J. Hematol. Oncol. 2022, 15, 152. [Google Scholar] [CrossRef]
- Leung, E.L.-H.; Luo, L.X.; Li, Y.; Liu, Z.-Q.; Li, L.L.; Shi, D.F.; Xie, Y.; Huang, M.; Lu, L.L.; Duan, F.G.; et al. Identification of a new inhibitor of KRAS-PDEδ interaction targeting KRAS mutant nonsmall cell lung cancer. Int. J. Cancer 2019, 145, 1334–1345. [Google Scholar] [CrossRef]
- Appels, N.M.G.M.; Beijnen, J.H.; Schellens, J.H.M. Development of farnesyl transferase inhibitors: A review. Oncologist 2005, 10, 565–578. [Google Scholar] [CrossRef]
- Adjei, A.A.; Mauer, A.; Bruzek, L.; Marks, R.S.; Hillman, S.; Geyer, S.; Hanson, L.J.; Wright, J.J.; Erlichman, C.; Kaufmann, S.H.; et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003, 21, 1760–1766. [Google Scholar] [CrossRef]
- Riely, G.J.; Johnson, M.L.; Medina, C.; Rizvi, N.A.; Miller, V.A.; Kris, M.G.; Pietanza, M.C.; Azzoli, C.G.; Krug, L.M.; Pao, W.; et al. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J. Thorac. Oncol. 2011, 6, 1435–1437. [Google Scholar] [CrossRef]
- Suzuki, M.; Jeng, L.J.B.; Chefo, S.; Wang, Y.; Price, D.; Li, X.; Wang, J.; Li, R.-J.; Ma, L.; Yang, Y.; et al. FDA approval summary for lonafarnib (Zokinvy) for the treatment of Hutchinson-Gilford progeria syndrome and processing-deficient progeroid laminopathies. Genet. Med. 2023, 25, 100335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.L.; Kirschmeier, P.; Carr, D.; James, L.; Bond, R.W.; Wang, L.; Patton, R.; Windsor, W.T.; Syto, R.; Zhang, R.; et al. Characterization of Ha-ras, N-ras, Ki-Ras4A, and Ki-Ras4B as in vitro substrates for farnesyl protein transferase and geranylgeranyl protein transferase type I. J. Biol. Chem. 1997, 272, 10232–10239. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Wang, X.; Li, Y.; Zhou, L.; Lu, X.; Dong, G.; Sheng, C. Discovery of novel KRAS-PDEδ inhibitors with potent activity in patient-derived human pancreatic tumor xenograft models. Acta Pharm. Sin. B 2022, 12, 274–290. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.L.H.; Luo, L.X.; Liu, Z.Q.; Wong, V.K.W.; Lu, L.L.; Xie, Y.; Zhang, N.; Qu, Y.Q.; Fan, X.X.; Li, Y.; et al. Inhibition of KRAS-dependent lung cancer cell growth by deltarasin: Blockage of autophagy increases its cytotoxicity. Cell Death Dis. 2018, 9, 216. [Google Scholar] [CrossRef]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.H.; et al. Small molecule inhibition of the KRAS–PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Papke, B.; Murarka, S.; Vogel, H.A.; Martín-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat. Commun. 2016, 7, 11360. [Google Scholar] [CrossRef]
- Martín-Gago, P.; Fansa, E.K.; Klein, C.H.; Murarka, S.; Janning, P.; Schürmann, M.; Metz, M.; Ismail, S.; Schultz-Fademrecht, C.; Baumann, M.; et al. A PDE6δ-KRas Inhibitor Chemotype with up to Seven H-Bonds and Picomolar Affinity that Prevents Efficient Inhibitor Release by Arl2. Angew. Chem. Int. Ed. Engl. 2017, 56, 2423–2428. [Google Scholar] [CrossRef]
- Uras, I.Z.; Moll, H.P.; Casanova, E. Targeting KRAS Mutant Non-Small-Cell Lung Cancer: Past, Present and Future. Int. J. Mol. Sci. 2020, 21, 4325. [Google Scholar] [CrossRef]
- Konstantinidou, G.; Ramadori, G.; Torti, F.; Kangasniemi, K.; Ramirez, R.E.; Cai, Y.; Behrens, C.; Dellinger, M.T.; Brekken, R.A.; Wistuba, I.I.; et al. RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov. 2013, 3, 444–457. [Google Scholar] [CrossRef]
- Gerber, D.E.; Camidge, D.R.; Morgensztern, D.; Cetnar, J.; Kelly, R.J.; Ramalingam, S.S.; Spigel, D.R.; Jeong, W.; Scaglioni, P.P.; Zhang, S.; et al. Phase 2 Study of the Focal Adhesion Kinase Inhibitor Defactinib (VS-6063) in Previously Treated Advanced KRAS Mutant Non-small Cell Lung Cancer. Lung Cancer 2020, 139, 60–67. [Google Scholar] [CrossRef]
- Capelletto, E.; Bironzo, P.; Denis, L.; Koustenis, A.; Bungaro, M.; Novello, S. Single agent VS-6766 or VS-6766 plus defactinib in KRAS-mutant non-small-cell lung cancer: The RAMP-202 phase II trial. Future Oncol. 2022, 18, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Herbst, R.S.; Wistuba, I.I.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Stewart, D.J.; Hicks, M.E.; Erasmus, J.; Gupta, S.; et al. The BATTLE Trial: Personalizing Therapy for Lung Cancer. Cancer Discov. 2011, 1, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Okazuka, K.; Ishida, T. Proteasome inhibitors for multiple myeloma. Jpn. J. Clin. Oncol. 2018, 48, 785–793. [Google Scholar] [CrossRef]
- Xue, W.; Meylan, E.; Oliver, T.G.; Feldser, D.M.; Winslow, M.M.; Bronson, R.; Jacks, T. Response and resistance to NF-κB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov. 2011, 1, 236–247. [Google Scholar] [CrossRef]
- Drilon, A.; Schoenfeld, A.J.; Arbour, K.C.; Litvak, A.; Ni, A.; Montecalvo, J.; Yu, H.A.; Panora, E.; Ahn, L.; Kennedy, M.; et al. Exceptional responders with invasive mucinous adenocarcinomas: A phase 2 trial of bortezomib in patients with KRAS G12D-mutant lung cancers. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003665. [Google Scholar] [CrossRef]
- Li, T.; Timmins, H.C.; King, T.; Kiernan, M.C.; Goldstein, D.; Park, S.B. Characteristics and risk factors of bortezomib induced peripheral neuropathy: A systematic review of phase III trials. Hematol. Oncol. 2020, 38, 229–243. [Google Scholar] [CrossRef]
- Aghajanian, C.; Soignet, S.; Dizon, D.S.; Pien, C.S.; Adams, J.; Elliott, P.J.; Sabbatini, P.; Miller, V.; Hensley, M.L.; Pezzulli, S.; et al. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin. Cancer Res. 2002, 8, 2505–2511. [Google Scholar]
- Li, S.; Liu, S.; Deng, J.; Akbay, E.A.; Hai, J.; Ambrogio, C.; Zhang, L.; Zhou, F.; Jenkins, R.W.; Adeegbe, D.O.; et al. Assessing Therapeutic Efficacy of MEK Inhibition in a KRASG12C-Driven Mouse Model of Lung Cancers. Clin. Cancer Res. 2018, 24, 4854–4864. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.V.; Logié, A.; Odedra, R.; Heier, A.; Heaton, S.P.; Alferez, D.; Davies, B.R.; Wilkinson, R.W.; Smith, P.D. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br. J. Cancer 2012, 106, 858–866. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Infante, J.R.; Kim, K.B.; Burris, H.A.; Curt, G.; Emeribe, U.; Clemett, D.; Tomkinson, H.K.; Cohen, R.B. A phase I dose-escalation study of selumetinib in combination with docetaxel or dacarbazine in patients with advanced solid tumors. BMC Cancer 2017, 17, 173. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Shaw, A.T.; Pereira, J.R.; Jeannin, G.; Vansteenkiste, J.; Barrios, C.; Franke, F.A.; Grinsted, L.; Zazulina, V.; Smith, P.; et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: A randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013, 14, 38–47. [Google Scholar]
- Jänne, P.A.; Mann, H.; Ghiorghiu, D. Study Design and Rationale for a Randomized, Placebo-Controlled, Double-Blind Study to Assess the Efficacy and Safety of Selumetinib in Combination with Docetaxel as Second-Line Treatment in Patients with KRAS-Mutant Advanced Non-Small Cell Lung Cancer (SELECT-1). Clin. Lung Cancer 2016, 17, e1–e4. [Google Scholar] [PubMed]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Blumenschein, G.R.; Smit, E.F.; Planchard, D.; Kim, D.-W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.-J.; Hanna, N.H.; et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef] [PubMed]
- VanSchaeybroeck, S.; Kalimutho, M.; Dunne, P.D.; Carson, R.; Allen, W.; Jithesh, P.V.; Redmond, K.L.; Sasazuki, T.; Shirasawa, S.; Blayney, J.; et al. ADAM17-Dependent c-MET-STAT3 Signaling Mediates Resistance to MEK Inhibitors in KRAS Mutant Colorectal Cancer. Cell Rep. 2014, 7, 1940–1955. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Luo, D.; Yu, J.; Zhang, M.; Zheng, X.; Xu, G.; Wang, J.; Wang, H.; Xu, Y.; Jiang, K.; et al. Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer. Oncogene 2022, 41, 191–203. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef]
- Ventura, E.; Giordano, A. Cell Cycle. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherland, 2019; ISBN 978-0-12-809633-8. [Google Scholar]
- Piezzo, M.; Chiodini, P.; Riemma, M.; Cocco, S.; Caputo, R.; Cianniello, D.; Di Gioia, G.; Di Lauro, V.; Rella, F.D.; Fusco, G.; et al. Progression-Free Survival and Overall Survival of CDK 4/6 Inhibitors Plus Endocrine Therapy in Metastatic Breast Cancer: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 6400. [Google Scholar]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef]
- Pek, M.; Yatim, S.M.J.M.; Chen, Y.; Li, J.; Gong, M.; Jiang, X.; Zhang, F.; Zheng, J.; Wu, X.; Yu, Q. Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene 2017, 36, 4975–4986. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.W.; Mazieres, J.; Barlesi, F.; Dragnev, K.H.; Koczywas, M.; Göskel, T.; Cortot, A.B.; Girard, N.; Wesseler, C.; Bischoff, H.; et al. A Randomized Phase III Study of Abemaciclib Versus Erlotinib in Patients with Stage IV Non-small Cell Lung Cancer With a Detectable KRAS Mutation Who Failed Prior Platinum-Based Therapy: JUNIPER. Front. Oncol. 2020, 10, 578756. [Google Scholar] [PubMed]
- Sorokin, A.V.; Marie, P.K.; Bitner, L.; Syed, M.; Woods, M.; Manyam, G.; Kwong, L.N.; Johnson, B.; Morris, V.K.; Jones, P.; et al. Targeting RAS Mutant Colorectal Cancer with Dual Inhibition of MEK and CDK4/6. Cancer Res. 2022, 82, 3335–3344. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Zemla, T.J.; Ciombor, K.K.; McRee, A.J.; Akce, M.; Dakhil, S.R.; Jaszewski, B.L.; Ou, F.-S.; Bekaii-Saab, T.S.; Kopetz, S. A randomized phase II trial of MEK and CDK4/6 inhibitors vesus tipiracil/trifluridine (TAS-102) in metastatic KRAS/NRAS mutant (mut) colorectal cancer (CRC). J. Clin. Oncol. 2022, 40, 116. [Google Scholar] [CrossRef]
- Rodrigues, R.; Duarte, D.; Vale, N. Drug Repurposing in Cancer Therapy: Influence of Patient’s Genetic Background in Breast Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 4280. [Google Scholar] [CrossRef] [PubMed]
- Mertens, S.; Huismans, M.A.; Verissimo, C.S.; Ponsioen, B.; Overmeer, R.; Proost, N.; van Tellingen, O.; van de Ven, M.; Begthel, H.; Boj, S.F.; et al. Drug-repurposing screen on patient-derived organoids identifies therapy-induced vulnerability in KRAS-mutant colon cancer. Cell Rep. 2023, 42, 112324. [Google Scholar] [CrossRef] [PubMed]
- Srisongkram, T.; Weerapreeyakul, N. Drug Repurposing against KRAS Mutant G12C: A Machine Learning, Molecular Docking, and Molecular Dynamics Study. Int. J. Mol. Sci. 2023, 24, 669. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.L.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRASG12C inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef]
- Goebel, L.; Müller, M.P.; Goody, R.S.; Rauh, D. KRasG12C inhibitors in clinical trials: A short historical perspective. RSC Med. Chem. 2020, 11, 760. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargi, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Hunter, J.C.; Gurbani, D.; Ficarro, S.B.; Carrasco, M.A.; Lim, S.M.; Choi, H.G.; Xie, T.; Marto, J.A.; Chen, Z.; Gray, N.S.; et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA 2014, 111, 8895–8900. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Nagashima, T.; Nishizono, Y.; Kuramoto, K.; Mori, K.; Homboh, K.; Yuri, M.; Shimazaki, M. Characterisation of a novel KRAS G12C inhibitor ASP2453 that shows potent anti-tumour activity in KRAS G12C-mutated preclinical models. Br. J. Cancer 2022, 126, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Sotorasib: First Approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- de Langen, A.J.; Johnson, M.L.; Mazieres, J.; Dingemans, A.-M.C.; Mountzios, G.; Pless, M.; Wolf, J.; Schuler, M.; Lena, H.; Skoulidis, F.; et al. Sotorasib versus docetaxel for previously treated non-small-cell lung cancer with KRASG12C mutation: A randomised, open-label, phase 3 trial. Lancet 2023, 401, 733–746. [Google Scholar] [PubMed]
- Fakih, M.G.; Kopetz, S.; Kuboki, Y.; Kim, T.W.; Munster, P.N.; Krauss, J.C.; Falchook, G.S.; Han, S.-W.; Heinemann, V.; Muro, K.; et al. Sotorasib for previously treated colorectal cancers with KRASG12C mutation (CodeBreaK100): A prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022, 23, 115–124. [Google Scholar] [CrossRef]
- Ji, J.; Wang, C.; Fakih, M. Targeting KRASG12C-Mutated Advanced Colorectal Cancer: Research and Clinical Developments. Onco Targets Ther. 2022, 15, 747–756. [Google Scholar] [CrossRef]
- Kuboki, Y.; Yaeger, R.; Fakih, M.; Strickler, J.H.; Masuishi, T.; Kim, E.J.-H.; Bestvina, C.M.; Langer, C.J.; Krauss, J.C.; Puri, S.; et al. 45MO Sotorasib in combination with panitumumab in refractory KRAS G12C-mutated colorectal cancer: Safety and efficacy for phase Ib full expansion cohort. Ann. Oncol. 2022, 33, S1445–S1446. [Google Scholar] [CrossRef]
- Dhillon, S. Adagrasib: First Approval. Drugs 2023, 83, 275–285. [Google Scholar] [CrossRef]
- Sabari, J.K.; Velcheti, V.; Shimizu, K.; Strickland, M.R.; Heist, R.S.; Singh, M.; Nayyar, N.; Giobbie-Hurder, A.; Digumarthy, S.R.; Gainor, J.F.; et al. Activity of Adagrasib (MRTX849) in Brain Metastases: Preclinical Models and Clinical Data from Patients with KRASG12C-Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 3318–3328. [Google Scholar] [CrossRef]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Klempner, S.J.; Weiss, J.; Pelster, M.; Spira, A.; Barve, M.; Ou, S.-H.I.; Leal, T.A.; Bekaii-Saab, T.; Christensen, J.G.; Kheoh, T.; et al. LBA24 KRYSTAL-1: Updated efficacy and safety of adagrasib (MRTX849) with or without cetuximab in patients with advanced colorectal cancer (CRC) harboring a KRASG12C mutation. Ann. Oncol. 2022, 33, S1391. [Google Scholar] [CrossRef]
- Corral de la Fuente, E.; Olmedo Garcia, M.E.; Gomez Rueda, A.; Lage, Y.; Garrido, P. Targeting KRAS in Non-Small Cell Lung Cancer. Front. Oncol. 2022, 11, 792635. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Velcheti, V.; Price, T.J.; Hong, D.S.; Fakih, M.; Kim, D.-W.; Falchook, G.S.; Delord, J.-P.; Dy, G.K.; Ramalingam, S.S.; et al. Largest evaluation of acquired resistance to sotorasib in KRAS p.G12C-mutated non–small cell lung cancer (NSCLC) and colorectal cancer (CRC): Plasma biomarker analysis of CodeBreaK100. J. Clin. Oncol. 2022, 40, 102. [Google Scholar]
- Akhave, N.S.; Biter, A.B.; Hong, D.S. Mechanisms of resistance to KRASG12C-targeted therapy. Cancer Discov. 2021, 11, 1345–1352. [Google Scholar] [CrossRef]
- Ning, W.; Marti, T.M.; Dorn, P.; Peng, R.-W. Non-genetic adaptive resistance to KRASG12C inhibition: EMT is not the only culprit. Front. Oncol. 2022, 12, 1004669. [Google Scholar]
- Zhao, Y.; Murciano-Goroff, Y.R.; Xue, J.Y.; Ang, A.; Lucas, J.; Mai, T.T.; Da Cruz Paula, A.F.; Saiki, A.Y.; Mohn, D.; Achanta, P.; et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021, 599, 679–683. [Google Scholar] [CrossRef]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ignatius Ou, S.-H.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef]
- Knickelbein, K.; Tong, J.; Chen, D.; Wang, Y.J.; Misale, S.; Bardelli, A.; Yu, J.; Zhang, L. Restoring PUMA induction overcomes KRAS-mediated resistance to anti-EGFR antibodies in colorectal cancer. Oncogene 2018, 37, 4599–4610. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Lai, L.P.; Brel, V.; Sharma, K.; Frappier, J.; Le-Henanf, N.; Vivet, B.; Muzet, N.; Schell, E.; Morales, R.; Rooney, E.; et al. Sensitivity of Oncogenic KRAS-Expressing Cells to CDK9 Inhibition. SLAS Discov. 2021, 26, 922–932. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. Egfr blockade reverts resistance to krasg12c inhibition in colorectal cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; de la Cruz, F.F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical pathway inhibition overcomes adaptive feedback resistance to KrasG12C inhibition. Clin. Cancer Res. 2020, 26, 1617–1643. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Fakih, M.; Strickler, J.; Govindan, R.; Li, B.T.; Goldberg, S.; Gandara, D.; Burns, T.; Barve, M.; Shu, C.; et al. Abstract P05-01: A phase 1b study evaluating the safety and efficacy of sotorasib, a KRASG12C inhibitor, in combination with trametinib, a MEK inhibitor, in KRAS p.G12C-Mutated Solid Tumors. Mol. Cancer Ther. 2021, 20, P05-01. [Google Scholar] [CrossRef]
- Hong, D.S.; Yaeger, R.; Kuboki, Y.; Masuishi, T.; Barve, M.A.; Falchook, G.S.; Govindan, R.; Sohal, D.; Kasi, P.M.; Burns, T.F.; et al. A phase 1b study of sotorasib, a specific and irreversible KRASG12C inhibitor, in combination with other anticancer therapies in advanced colorectal cancer (CRC) and other solid tumors (CodeBreaK 101). J. Clin. Oncol. 2022, 40, TPS214. [Google Scholar] [CrossRef]
- Coma, S.; Chowdhury, S.; Pachter, J.A. Abstract 1263: Dual RAF/MEK inhibitor VS-6766 enhances antitumor efficacy of KRAS-G12C inhibitors through a vertical pathway inhibition strategy. Cancer Res. 2021, 81, 1263. [Google Scholar] [CrossRef]
- Luo, G.; Wang, B.; Hou, Q.; Wu, X. Development of Son of Sevenless Homologue 1 (SOS1) Modulators To Treat Cancers by Regulating RAS Signaling. J. Med. Chem. 2023, 66, 4324–4341. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef]
- Thatikonda, V.; Lu, H.; Jurado, S.; Kostyrko, K.; Bristow, C.A.; Bosch, K.; Feng, N.; Gao, S.; Gerlach, D.; Gmachl, M.; et al. Combined KRASG12C and SOS1 inhibition enhances and extends the anti-tumor response in KRASG12C-driven cancers by addressing intrinsic and acquired resistance. bioRxiv 2023. [Google Scholar] [CrossRef]
- Mohi, M.G.; Neel, B.G. The role of Shp2 (PTPN11) in cancer. Curr. Opin. Genet. Dev. 2007, 17, 23–30. [Google Scholar] [CrossRef]
- Marasco, M.; Berteotti, A.; Weyershaeuser, J.; Thorausch, N.; Sikorska, J.; Krausze, J.; Brandt, H.J.; Kirkpatrick, J.; Rios, P.; Schamel, W.W.; et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci. Adv. 2020, 6, eaay4458. [Google Scholar] [CrossRef]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK inhibitors in Multiple Cancer Models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Long, Y.-Q. Recent advances in the discovery of protein tyrosine phosphatase SHP2 inhibitors. RSC Med. Chem. 2023, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Weng, J.; Fan, X.; Wang, E.; Zhu, Q.; Tao, L.; Han, Z.; Wang, Z.; Niu, H.; Jiang, Y.; et al. Abstract 932: Discovery of D-1553, a novel and selective KRas-G12C Inhibitor with potent anti-tumor activity in a broad spectrum of tumor cell lines and xenograft models. Cancer Res. 2021, 81, 932. [Google Scholar] [CrossRef]
- Frank, K.J.; Mulero-Sánchez, A.; Berninger, A.; Ruiz-Cañas, L.; Bosma, A.; Görgülü, K.; Wu, N.; Diakopoulos, K.N.; Kaya-Aksoy, E.; Ruess, D.A.; et al. Extensive preclinical validation of combined RMC-4550 and LY3214996 supports clinical investigation for KRAS mutant pancreatic cancer. Cell Reports. Med. 2022, 3, 100815. [Google Scholar] [CrossRef]
- Wong, G.S.; Zhou, J.; Liu, J.B.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R. CT028—A first-in-human phase 1 study of LY3537982, a highly selective and potent KRAS G12C inhibitor in patients with KRAS G12C-mutant advanced solid tumors. In Proceedings of the AACR 2023, Orlando, FL, USA, 14–19 April 2023. [Google Scholar]
- Peng, S.-B.; Si, C.; Zhang, Y.; Van Horn, R.D.; Lin, X.; Gong, X.; Huber, L.; Donoho, G.; Curtis, C.; Strelow, J.M.; et al. Abstract 1259: Preclinical characterization of LY3537982, a novel, highly selective and potent KRAS-G12C inhibitor. Cancer Res. 2021, 81, 1259. [Google Scholar] [CrossRef]
- Sacher, A.; Patel, M.R.; Miller, W.H.; Desai, J.; Garralda, E.; Bowyer, S.; Kim, T.W.; De Miguel, M.; Falcon, A.; Krebs, M.G.; et al. OA03.04 Phase I A Study to Evaluate GDC-6036 Monotherapy in Patients with Non-small Cell Lung Cancer (NSCLC) with KRAS G12C Mutation. J. Thorac. Oncol. 2022, 17, S8–S9. [Google Scholar] [CrossRef]
- Desai, J. 362P-Phase Ia study to evaluate GDC-6036 monotherapy in patients with colorectal cancer (CRC) with KRAS G12C mutation. In Proceedings of the ESMO Congress 2022, Paris, France, 9–13 September 2022. [Google Scholar]
- Desai, J.; Han, S.-W.; Lee, J.-S.; Shacham-Shmueli, E.; Massarelli, E.; Cervantes, A.; Garralda, E.; Falcon, A.; Miller, W.H.; Gort, E.; et al. Abstract CT029: Phase Ib study of GDC-6036 in combination with cetuximab in patients with colorectal cancer (CRC) with KRAS G12C mutation. Cancer Res. 2023, 83, CT029. [Google Scholar] [CrossRef]
- Koulouridi, A.; Karagianni, M.; Messaritakis, I.; Sfakianaki, M.; Voutsina, A.; Trypaki, M.; Bachlitzanaki, M.; Koustas, E.; Karamouzis, M.V.; Ntavatzikos, A.; et al. Prognostic Value of KRAS Mutations in Colorectal Cancer Patients. Cancers 2022, 14, 3320. [Google Scholar] [CrossRef]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Tajiknia, V.; El-Deiry, W.S.; Schwermann, M.; Huntington, K.; Zhou, L.; Srinivasan, P. Combination of 5-FU plus KRAS G12D inhibitor MRTX1133 against human colorectal and pancreatic cancer cells and the affects on inhibition of pERK and immune-stimulatory cytokine patterns in in KRAS G12D and KRAS G12V tumor cells. J. Clin. Oncol. 2023, 41, e16301. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R. Glimmers of hope for targeting oncogenic KRAS-G12D. Cancer Gene Ther. 2022, 30, 391–393. [Google Scholar]
- Kim, D.; Xue, J.Y.; Lito, P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell 2020, 183, 850–859. [Google Scholar] [CrossRef]
- Shu, C.-L.; Liu, Y.-L. The Path to Personalized Treatment in KRAS-Mutant Non-Small Cell Lung Cancer: A Review of Targeted Therapies and Immunotherapy. Cancer Manag. Res. 2022, 14, 3485–3492. [Google Scholar] [PubMed]
- Nichols, R.J.; Yang, Y.C.; Cregg, J.; Schulze, C.J.; Wang, Z.; Dua, R.; Jiang, J.; Garrenton, L.S.; Nasholm, N.; Bermingham, A.; et al. Abstract 3595: RMC-6291, a next-generation tri-complex KRASG12C(ON) inhibitor, outperforms KRASG12C(OFF) inhibitors in preclinical models of KRASG12C cancers. Cancer Res. 2022, 82, 3595. [Google Scholar]
- Koltun, E.S.; Rice, M.A.; Gustafson, W.C.; Wilds, D.; Jiang, J.; Lee, B.J.; Wang, Z.; Chang, S.; Flagella, M.; Mu, Y.; et al. Abstract 3597: Direct targeting of KRASG12X mutant cancers with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Cancer Res. 2022, 82, 3597. [Google Scholar] [CrossRef]
- Keeton, A.B.; Ward, A.; Chen, X.; Valiyaveettil, J.; Zhu, B.; Ramirez-Alcantara, V.; Maxuitenko, Y.; Berry, K.; Mattox, T.E.; Boyd, M.R.; et al. Abstract 2707: A novel RAS inhibitor, MCI-062, inhibits colon tumor growth in vivo and activates antitumor immunity. Cancer Res. 2019, 79, 2707. [Google Scholar]
- Kim, D.; Herdeis, L.; Rudolph, D.; Zhao, Y.; Böttcher, J.; Vides, A.; Ayala-Santos, C.I.; Pourfarjam, Y.; Cuevas-Navarro, A.; Xue, J.Y.; et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 2023, 619, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Asimgil, H.; Ertetik, U.; Çevik, N.C.; Ekizce, M.; Doğruöz, A.; Gökalp, M.; Arık-Sever, E.; Istvanffy, R.; Friess, H.; Ceyhan, G.O.; et al. Targeting the undruggable oncogenic KRAS: The dawn of hope. JCI Insight 2023, 7, e153688. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef]
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.-P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.L.; Cole, D.; Kenefeck, R.; OKelly, I.; Lepore, M.; Jakobsen, B.K. Novel TCR-based biologics: Mobilising T cells to warm ‘cold’ tumours. Cancer Treat. Rev. 2019, 77, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, M.K.; Bakka, A.; Breivik, J.; Saeterdal, I.; Solheim, B.G.; Søreide, O.; Thorsby, E.; Gaudernack, G. Vaccination with mutant ras peptides and induction of T-cell responsiveness in pancreatic carcinoma patients carrying the corresponding RAS mutation. Lancet 1995, 346, 1399–1400. [Google Scholar] [CrossRef]
- Palmer, D.H.; Valle, J.W.; Ma, Y.T.; Faluyi, O.; Neoptolemos, J.P.; Jensen Gjertsen, T.; Iversen, B.; Amund Eriksen, J.; Møller, A.-S.; Aksnes, A.-K.; et al. TG01/GM-CSF and adjuvant gemcitabine in patients with resected RAS-mutant adenocarcinoma of the pancreas (CT TG01-01): A single-arm, phase 1/2 trial. Br. J. Cancer 2020, 122, 971–977. [Google Scholar] [PubMed]
- Bear, A.S.; Rech, A.J.; Richman, L.P.; O’Hara, M.H.; Linette, G.P.; Carreno, B.M.; Vonderheide, R.H. Abstract B04: Identification of T-cell receptors targeting mutant KRAS in pancreatic cancer. Cancer Res. 2019, 79, B04. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bteich, F.; Mohammadi, M.; Li, T.; Bhat, M.A.; Sofianidi, A.; Wei, N.; Kuang, C. Targeting KRAS in Colorectal Cancer: A Bench to Bedside Review. Int. J. Mol. Sci. 2023, 24, 12030. https://doi.org/10.3390/ijms241512030
Bteich F, Mohammadi M, Li T, Bhat MA, Sofianidi A, Wei N, Kuang C. Targeting KRAS in Colorectal Cancer: A Bench to Bedside Review. International Journal of Molecular Sciences. 2023; 24(15):12030. https://doi.org/10.3390/ijms241512030
Chicago/Turabian StyleBteich, Fernand, Mahshid Mohammadi, Terence Li, Muzaffer Ahmed Bhat, Amalia Sofianidi, Ning Wei, and Chaoyuan Kuang. 2023. "Targeting KRAS in Colorectal Cancer: A Bench to Bedside Review" International Journal of Molecular Sciences 24, no. 15: 12030. https://doi.org/10.3390/ijms241512030
APA StyleBteich, F., Mohammadi, M., Li, T., Bhat, M. A., Sofianidi, A., Wei, N., & Kuang, C. (2023). Targeting KRAS in Colorectal Cancer: A Bench to Bedside Review. International Journal of Molecular Sciences, 24(15), 12030. https://doi.org/10.3390/ijms241512030