
Asiaticoside Mitigates Alzheimer’s Disease Pathology by Attenuating Inflammation and Enhancing Synaptic Function
 , and
, and 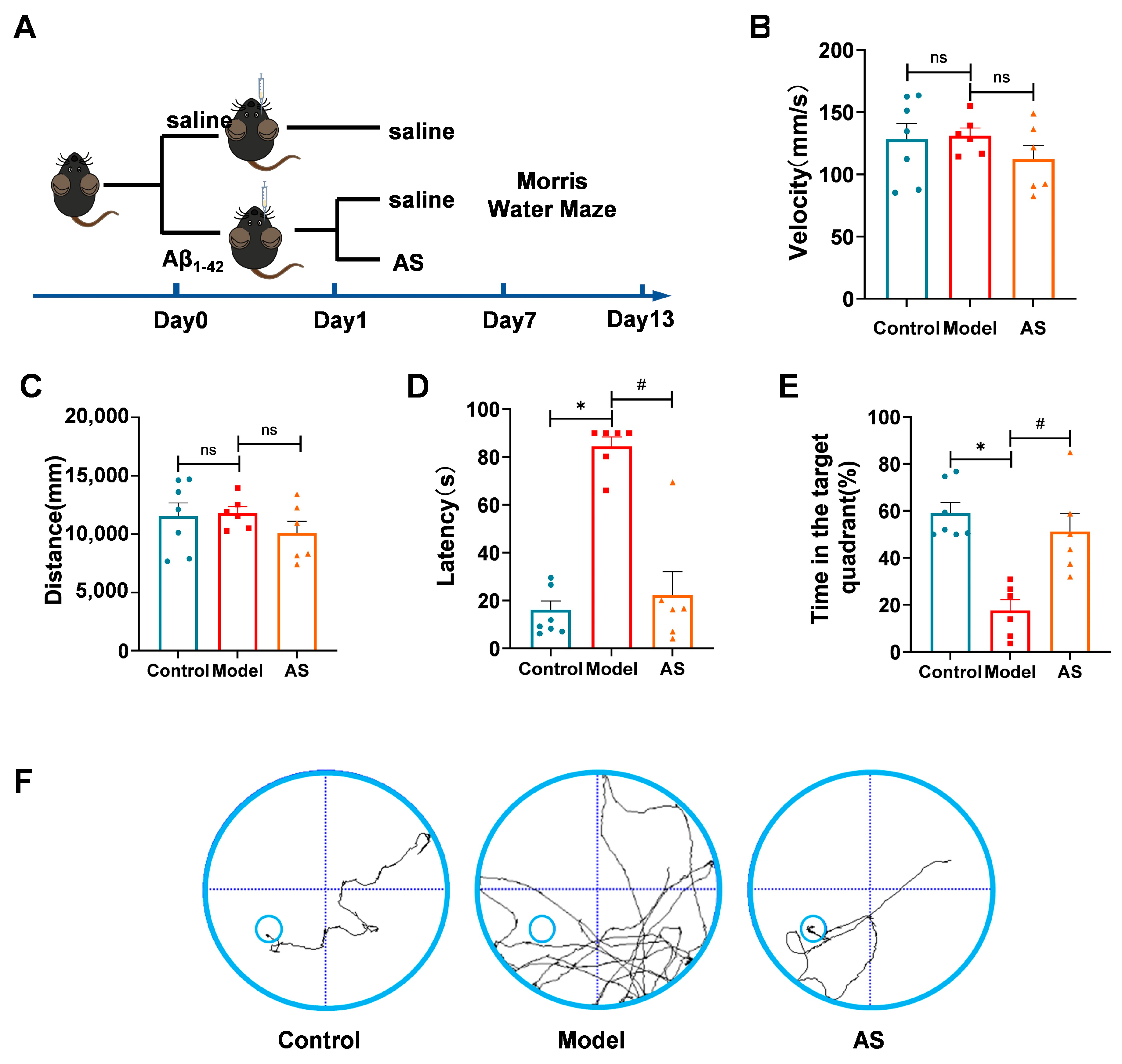{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
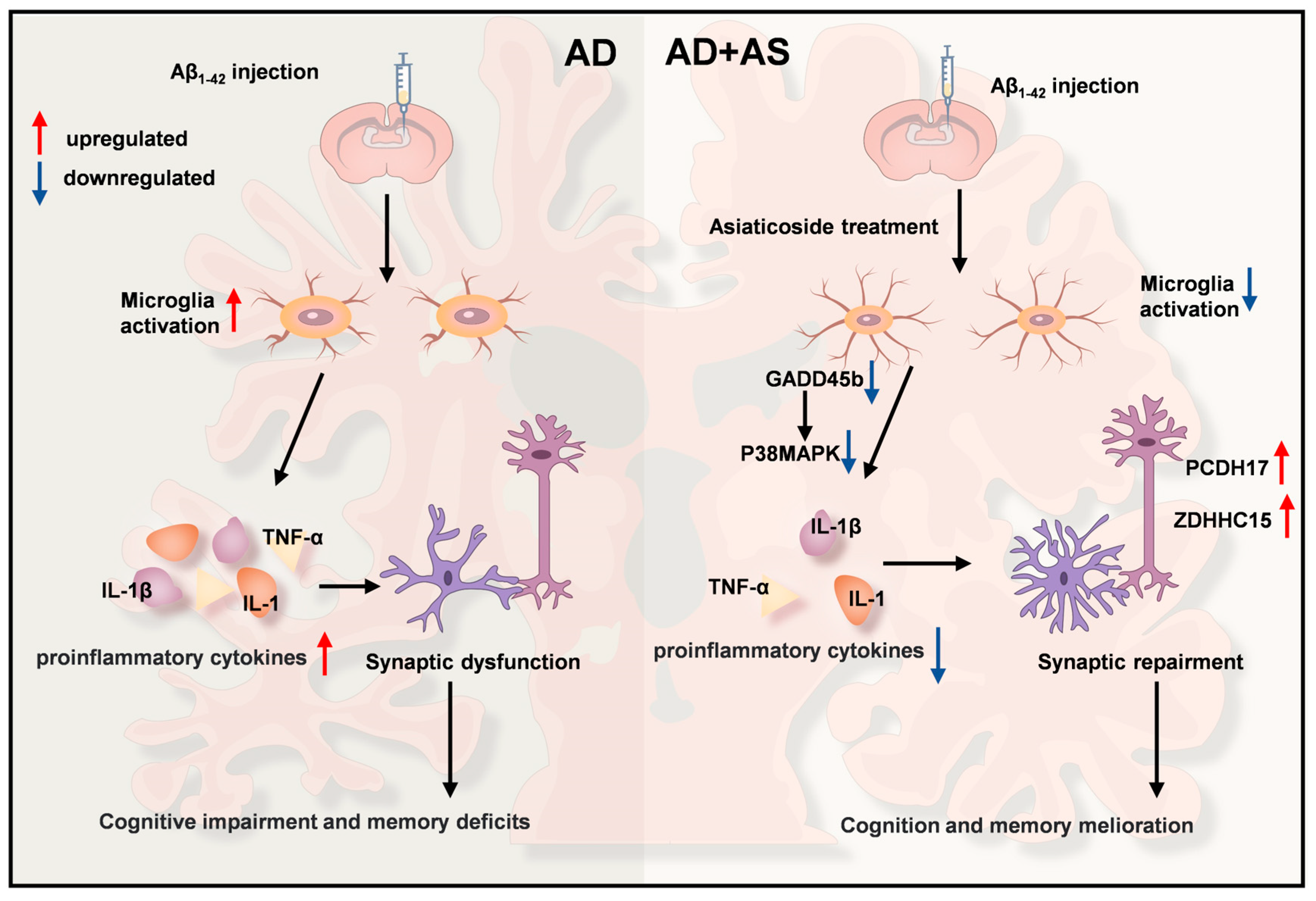
Abstract
1. Introduction
2. Results
2.1. AS Mitigated Learning and Memory Impairments in Aβ1-42-Induced Mice
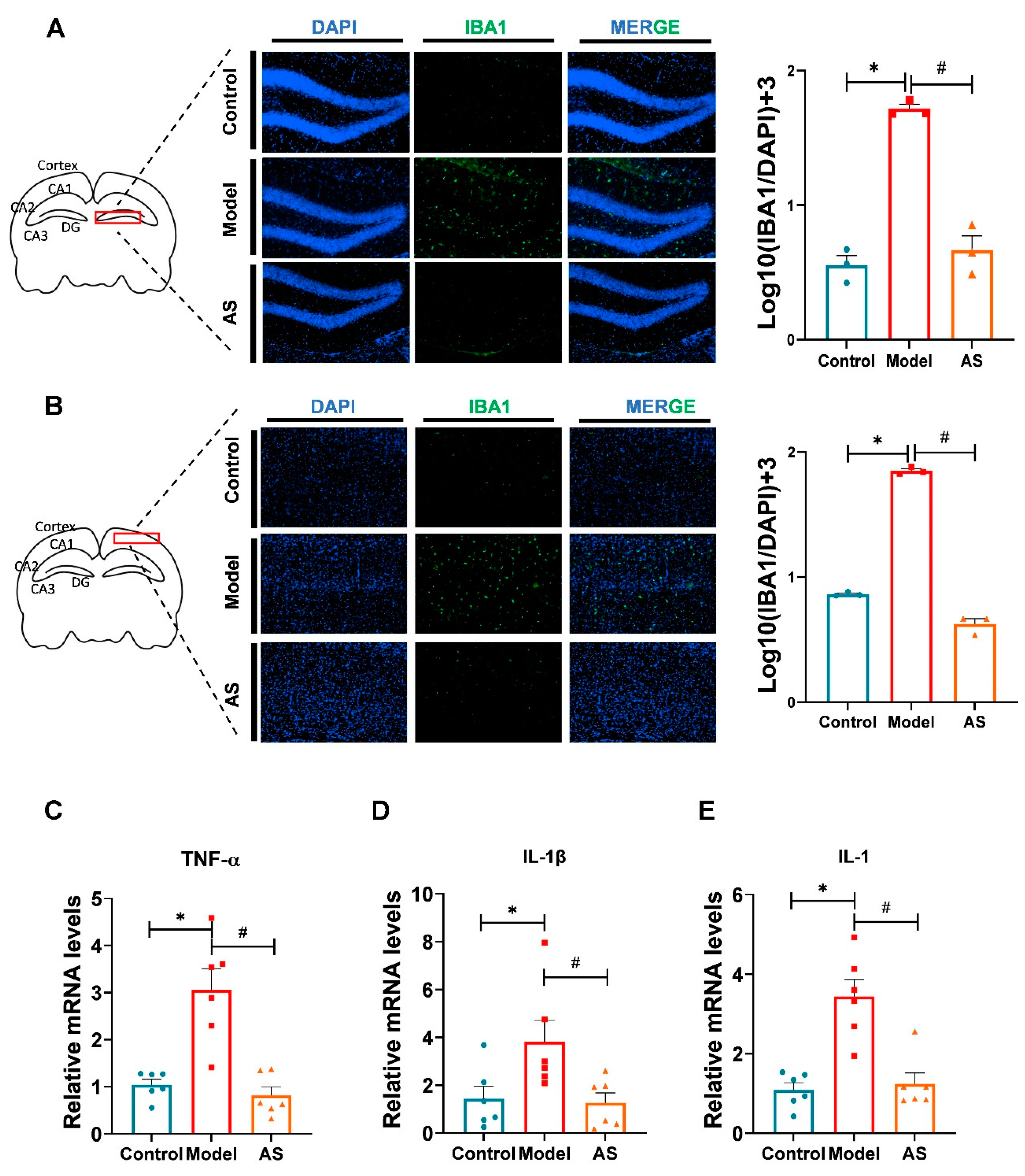
2.2. AS Exerted Anti-Inflammatory Effects in Aβ1-42-Induced Mice
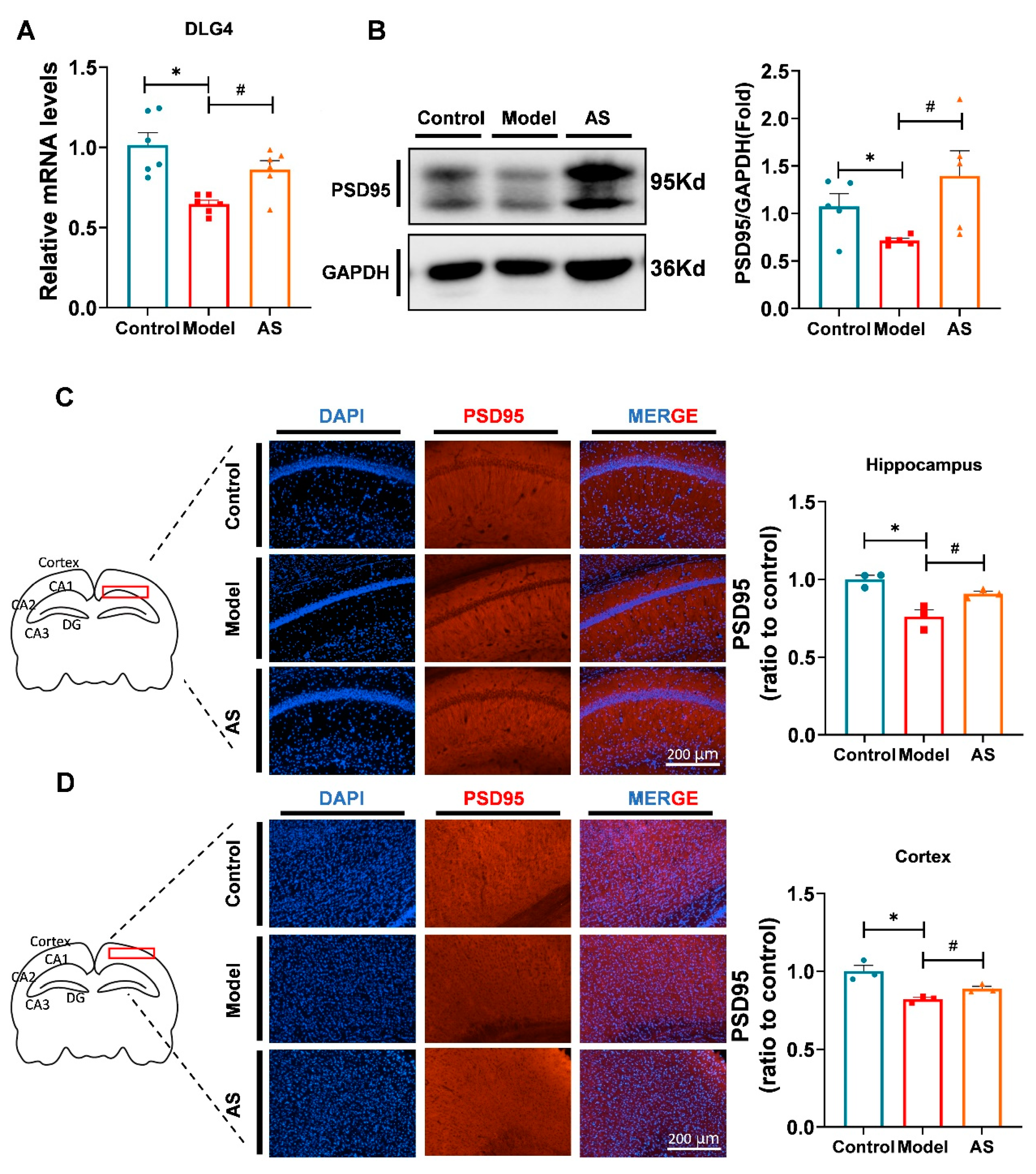
2.3. AS Promoted Synaptic Repairment and Improved Synapse Function in Aβ1-42-Induced Mice
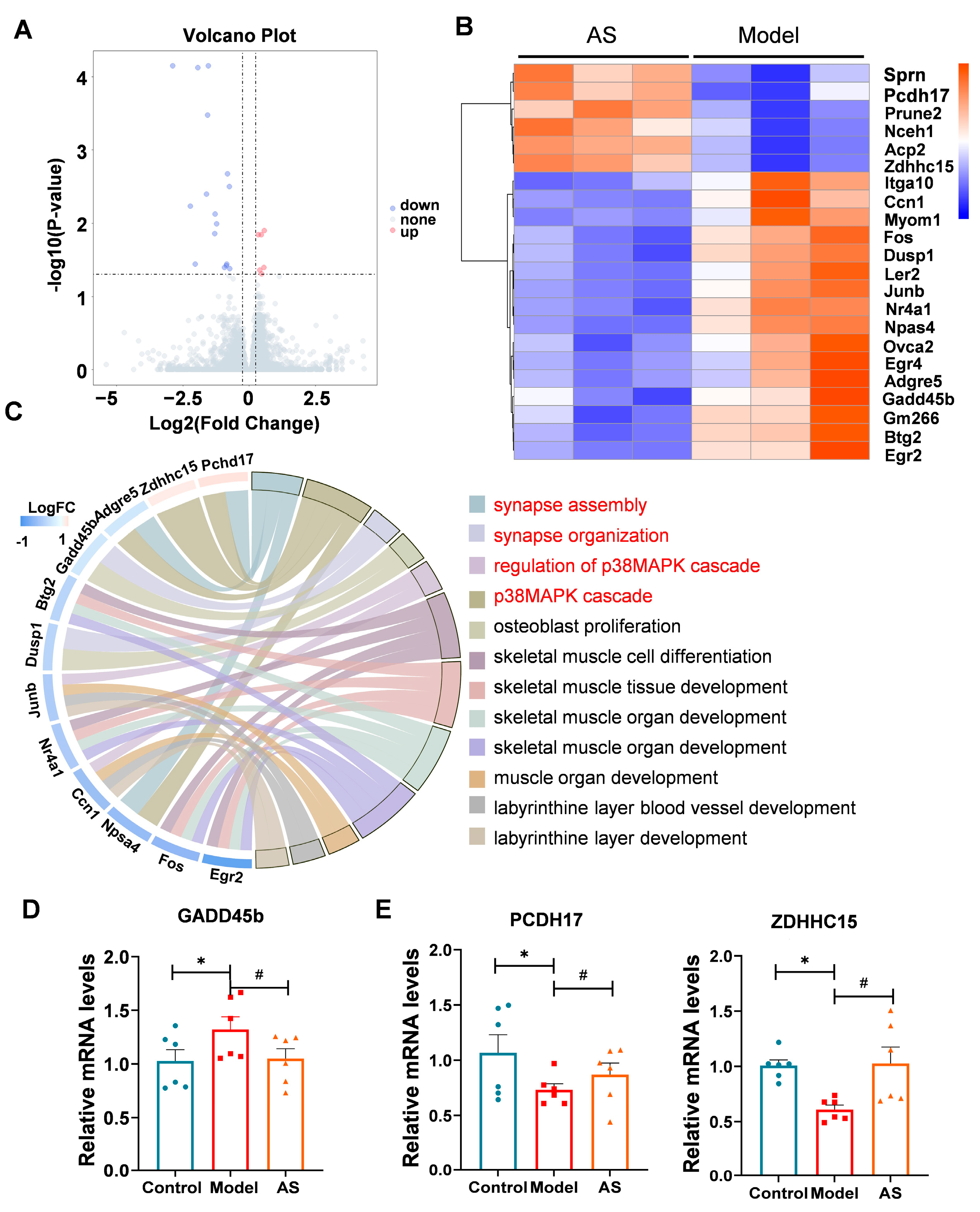
2.4. AS Altered Genes Related to the p38 MAPK Pathway and Synaptic Function
3. Discussion
4. Materials and Methods
4.1. Animals and Manipulations
4.2. Aβ1-42 Injection and Drug Intervention
4.3. Morris Water Maze (MWM)
4.4. Brain Tissue Preparation and Mice Brain Slices
4.5. Immunofluorescence
4.6. RNA Isolation and RNA Sequencing
4.7. RNA-Seq Analysis
4.8. Real-Time PCR
4.9. Western Blotting
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, L.; Grossberg, G.T. Combination therapy for Alzheimer’s disease. Drugs Aging 2011, 28, 539–546. [Google Scholar] [CrossRef]
- Se Thoe, E.; Fauzi, A.; Tang, Y.Q.; Chamyuang, S.; Chia, A.Y.Y. A review on advances of treatment modalities for Alzheimer’s disease. Life Sci. 2021, 276, 119129. [Google Scholar] [CrossRef]
- Liu, L.; Ding, Z.; Yang, Y.; Zhang, Z.; Lu, Q.; Kaplan, D.L. Asiaticoside-laden silk nanofiber hydrogels to regulate inflammation and angiogenesis for scarless skin regeneration. Biomater. Sci. 2021, 9, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Lu, Q.; Ma, Z.; Ding, Y.; Bedarida, T.; Chen, L.; Xie, Z.; Song, P.; Zou, M.H. Circulating miR-103a-3p contributes to angiotensin II-induced renal inflammation and fibrosis via a SNRK/NF-κB/p65 regulatory axis. Nat. Commun. 2019, 10, 2145. [Google Scholar]
- Rao, J.S.; Kellom, M.; Kim, H.W.; Rapoport, S.I.; Reese, E.A. Neuroinflammation and synaptic loss. Neurochem. Res. 2012, 37, 903–910. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- In t’ Veld, B.A.; Ruitenberg, A.; Hofman, A.; Launer, L.J.; van Duijn, C.M.; Stijnen, T.; Breteler, M.M.; Stricker, B.H. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 2001, 345, 1515–1521. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Li, Z.; Zhong, W.; Chen, L.; Liu, S.; Zhang, B.; Zhu, Z.; Li, X. Methylprednisolone alleviates cognitive functions through the regulation of neuroinflammation in Alzheimer’s disease. Front. Immunol. 2023, 14, 1192940. [Google Scholar] [CrossRef]
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J. Alzheimer’s Dis. JAD 2001, 3, 41–48. [Google Scholar] [CrossRef]
- Mohamed, E.A.; Ahmed, H.I.; Zaky, H.S.; Badr, A.M. Sesame oil mitigates memory impairment, oxidative stress, and neurodegeneration in a rat model of Alzheimer’s disease. A pivotal role of NF-κB/p38MAPK/BDNF/PPAR-γ pathways. J. Ethnopharmacol. 2021, 267, 113468. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Fessel, J. Alzheimer’s disease combination treatment. Neurobiol. Aging 2018, 63, 165. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Head, E.; Corrada, M.M.; Kahle-Wrobleski, K.; Kim, R.C.; Sarsoza, F.; Goodus, M.; Kawas, C.H. Synaptic proteins, neuropathology and cognitive status in the oldest-old. Neurobiol. Aging 2009, 30, 1125–1134. [Google Scholar] [CrossRef]
- Chugh, D.; Nilsson, P.; Afjei, S.A.; Bakochi, A.; Ekdahl, C.T. Brain inflammation induces post-synaptic changes during early synapse formation in adult-born hippocampal neurons. Exp. Neurol. 2013, 250, 176–188. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Origlia, N.; Arancio, O.; Domenici, L.; Yan, S.S. MAPK, beta-amyloid and synaptic dysfunction: The role of RAGE. Expert Rev. Neurother. 2009, 9, 1635–1645. [Google Scholar] [CrossRef]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Han, Q.; Lin, Q.; Huang, P.; Chen, M.; Hu, X.; Fu, H.; He, S.; Shen, F.; Zeng, H.; Deng, Y. Microglia-derived IL-1β contributes to axon development disorders and synaptic deficit through p38-MAPK signal pathway in septic neonatal rats. J. Neuroinflamm. 2017, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J. Neuroinflamm. 2019, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.; Lue, L.F. Anti-inflammatory and immune therapy for Alzheimer’s disease: Current status and future directions. Curr. Neuropharmacol. 2007, 5, 232–243. [Google Scholar] [CrossRef]
- Bandopadhyay, S.; Mandal, S.; Ghorai, M.; Jha, N.K.; Kumar, M.; Radha; Ghosh, A.; Proćków, J.; Pérez de la Lastra, J.M.; Dey, A. Therapeutic properties and pharmacological activities of asiaticoside and madecassoside: A review. J. Cell. Mol. Med. 2023, 27, 593–608. [Google Scholar] [CrossRef]
- Song, D.; Jiang, X.; Liu, Y.; Sun, Y.; Cao, S.; Zhang, Z. Asiaticoside Attenuates Cell Growth Inhibition and Apoptosis Induced by Aβ(1-42) via Inhibiting the TLR4/NF-κB Signaling Pathway in Human Brain Microvascular Endothelial Cells. Front. Pharmacol. 2018, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Mook-Jung, I.; Shin, J.E.; Yun, S.H.; Huh, K.; Koh, J.Y.; Park, H.K.; Jew, S.S.; Jung, M.W. Protective effects of asiaticoside derivatives against beta-amyloid neurotoxicity. J. Neurosci. Res. 1999, 58, 417–425. [Google Scholar] [CrossRef]
- Hossain, S.; Hashimoto, M.; Katakura, M.; Al Mamun, A.; Shido, O. Medicinal value of asiaticoside for Alzheimer’s disease as assessed using single-molecule-detection fluorescence correlation spectroscopy, laser-scanning microscopy, transmission electron microscopy, and in silico docking. BMC Complement. Altern. Med. 2015, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, L.; Ruigrok, S.R.; Amelianchik, A.; Ivan, D.; van Dam, A.M.; Lucassen, P.J.; Korosi, A. Early-life stress lastingly alters the neuroinflammatory response to amyloid pathology in an Alzheimer’s disease mouse model. Brain Behav. Immun. 2017, 63, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [CrossRef]
- Chen, Z.; Wan, X.; Hou, Q.; Shi, S.; Wang, L.; Chen, P.; Zhu, X.; Zeng, C.; Qin, W.; Zhou, W.; et al. GADD45B mediates podocyte injury in zebrafish by activating the ROS-GADD45B-p38 pathway. Cell Death Dis. 2016, 7, e2068. [Google Scholar] [CrossRef]
- Hayashi, S.; Inoue, Y.; Kiyonari, H.; Abe, T.; Misaki, K.; Moriguchi, H.; Tanaka, Y.; Takeichi, M. Protocadherin-17 mediates collective axon extension by recruiting actin regulator complexes to interaxonal contacts. Dev. Cell 2014, 30, 673–687. [Google Scholar] [CrossRef]
- Coley, A.A.; Gao, W.J. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 82, 187–194. [Google Scholar] [CrossRef]
- Delrieu, J.; Piau, A.; Caillaud, C.; Voisin, T.; Vellas, B. Managing cognitive dysfunction through the continuum of Alzheimer’s disease: Role of pharmacotherapy. CNS Drugs 2011, 25, 213–226. [Google Scholar] [CrossRef]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W., Jr.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimer’s Dis. JAD 2005, 7, 103–117, discussion 173–180. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, N.J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef]
- Beamer, E.; Corrêa, S.A.L. The p38(MAPK)-MK2 Signaling Axis as a Critical Link Between Inflammation and Synaptic Transmission. Front. Cell Dev. Biol. 2021, 9, 635636. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.M.; Clark, A.R. Dual-specificity phosphatase 1: A critical regulator of innate immune responses. Biochem. Soc. Trans. 2006, 34 Pt 6, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Brondello, J.M.; Pouysségur, J.; McKenzie, F.R. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 1999, 286, 2514–2517. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.S.; Shimell, J.J.; Bamji, S.X. Regulation of dendrite morphology and excitatory synapse formation by zDHHC15. J. Cell Sci. 2019, 132, jcs230052. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Shen, Z.; Anraku, Y.; Kataoka, K.; Chen, X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials 2019, 224, 119491. [Google Scholar] [CrossRef]
- Shah, S.; Dhawan, V.; Holm, R.; Nagarsenker, M.S.; Perrie, Y. Liposomes: Advancements and innovation in the manufacturing process. Adv. Drug Deliv. Rev. 2020, 154–155, 102–122. [Google Scholar] [CrossRef]
- Kayed, R.; Canto, I.; Breydo, L.; Rasool, S.; Lukacsovich, T.; Wu, J.; Albay, R., III; Pensalfini, A.; Yeung, S.; Head, E.; et al. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 2010, 5, 57. [Google Scholar] [CrossRef]
- Lioudyno, M.I.; Broccio, M.; Sokolov, Y.; Rasool, S.; Wu, J.; Alkire, M.T.; Liu, V.; Kozak, J.A.; Dennison, P.R.; Glabe, C.G.; et al. Effect of synthetic aβ peptide oligomers and fluorinated solvents on Kv1.3 channel properties and membrane conductance. PLoS ONE 2012, 7, e35090. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Chen, L.; Li, J.; Sun, Y.; Xu, Y.; Li, Z.; Zhu, Z.; Li, X. Asiaticoside Mitigates Alzheimer’s Disease Pathology by Attenuating Inflammation and Enhancing Synaptic Function. Int. J. Mol. Sci. 2023, 24, 11976. https://doi.org/10.3390/ijms241511976
Liu S, Chen L, Li J, Sun Y, Xu Y, Li Z, Zhu Z, Li X. Asiaticoside Mitigates Alzheimer’s Disease Pathology by Attenuating Inflammation and Enhancing Synaptic Function. International Journal of Molecular Sciences. 2023; 24(15):11976. https://doi.org/10.3390/ijms241511976
Chicago/Turabian StyleLiu, Sai, Long Chen, Jinran Li, Yuan Sun, Yue Xu, Zhaoxing Li, Zheying Zhu, and Xinuo Li. 2023. "Asiaticoside Mitigates Alzheimer’s Disease Pathology by Attenuating Inflammation and Enhancing Synaptic Function" International Journal of Molecular Sciences 24, no. 15: 11976. https://doi.org/10.3390/ijms241511976
APA StyleLiu, S., Chen, L., Li, J., Sun, Y., Xu, Y., Li, Z., Zhu, Z., & Li, X. (2023). Asiaticoside Mitigates Alzheimer’s Disease Pathology by Attenuating Inflammation and Enhancing Synaptic Function. International Journal of Molecular Sciences, 24(15), 11976. https://doi.org/10.3390/ijms241511976