
Surveying the Metabolic and Dysfunctional Profiles of T Cells and NK Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome


Abstract
1. Introduction
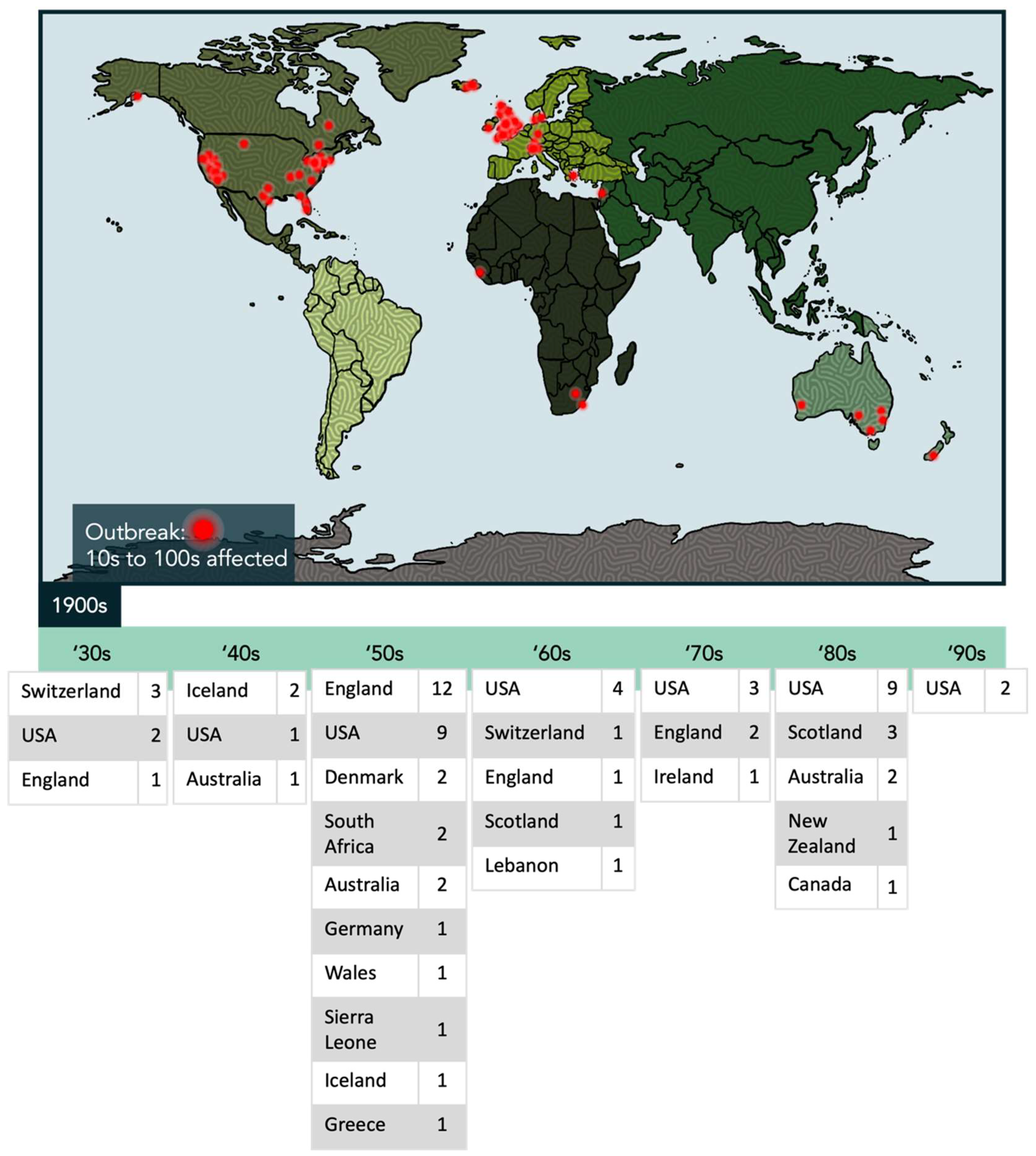
1.1. An Overview of ME/CFS
1.2. Implicated Viral Infections in ME/CFS
2. Metabolic Dynamics in T and NK Cell Populations
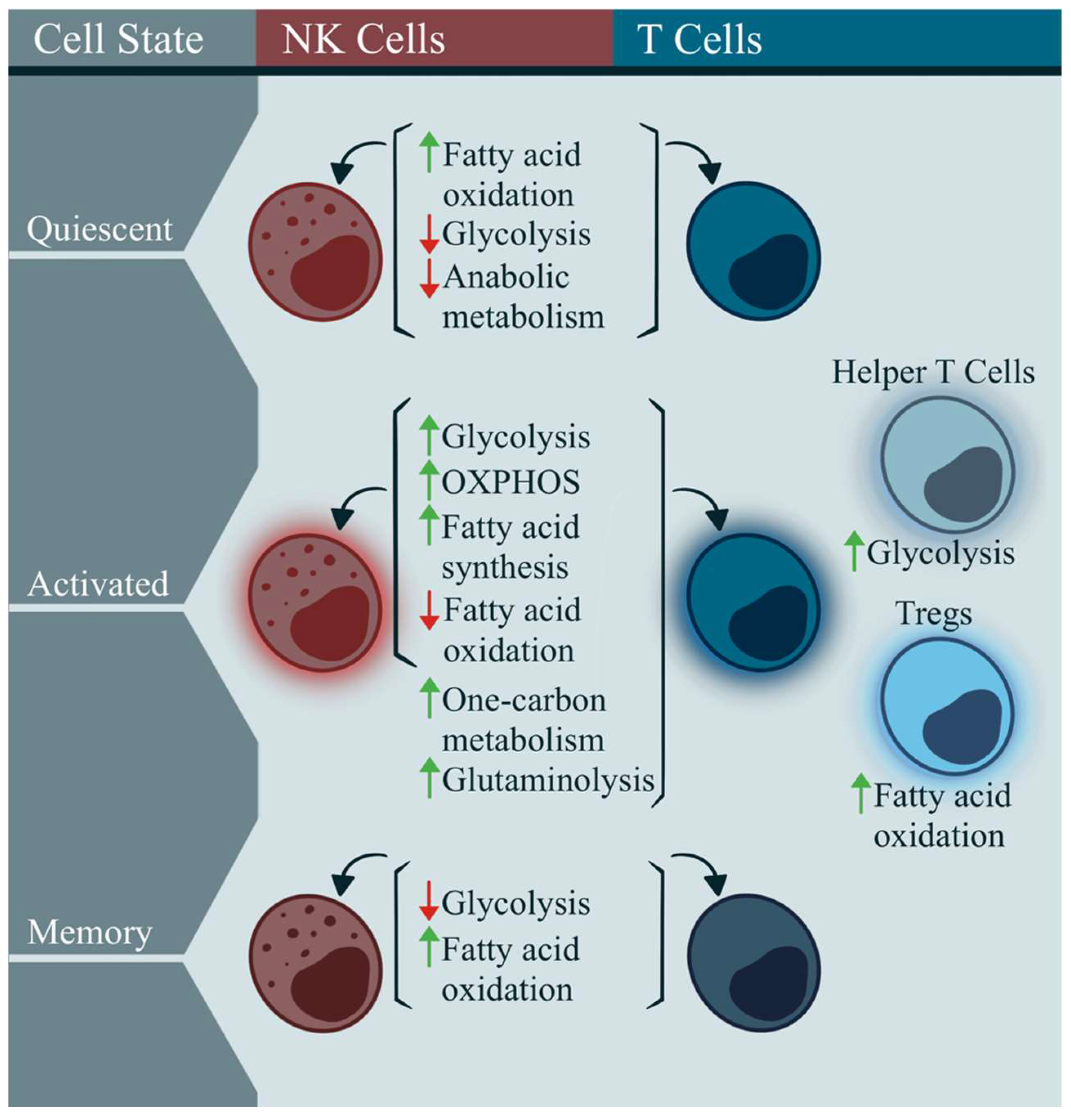
2.1. Immunometabolism in Classic T and NK Cells
2.2. Immunometabolism in ME/CFS
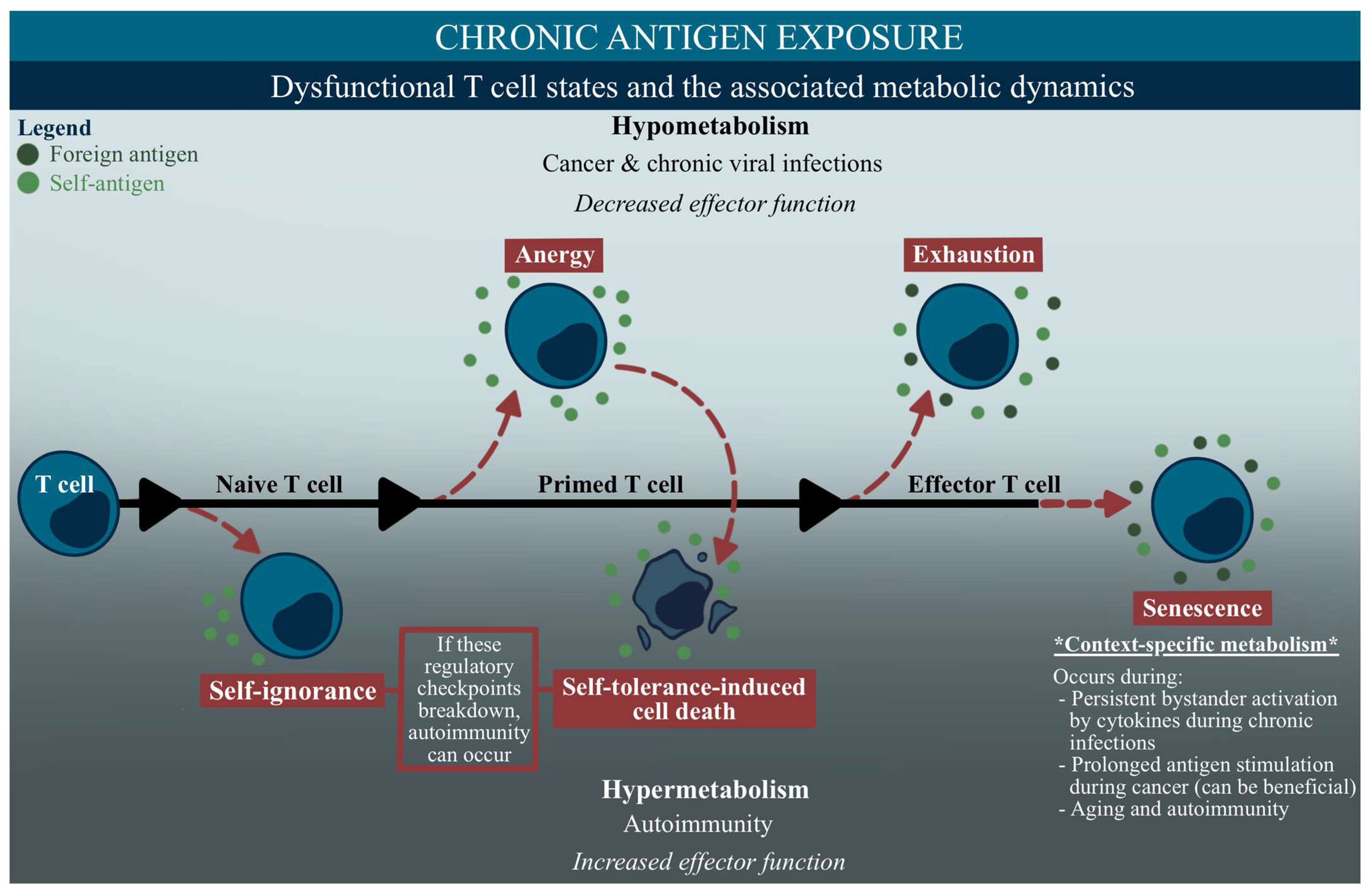
2.3. Immunometabolism in Dysfunctional Immune Cell States
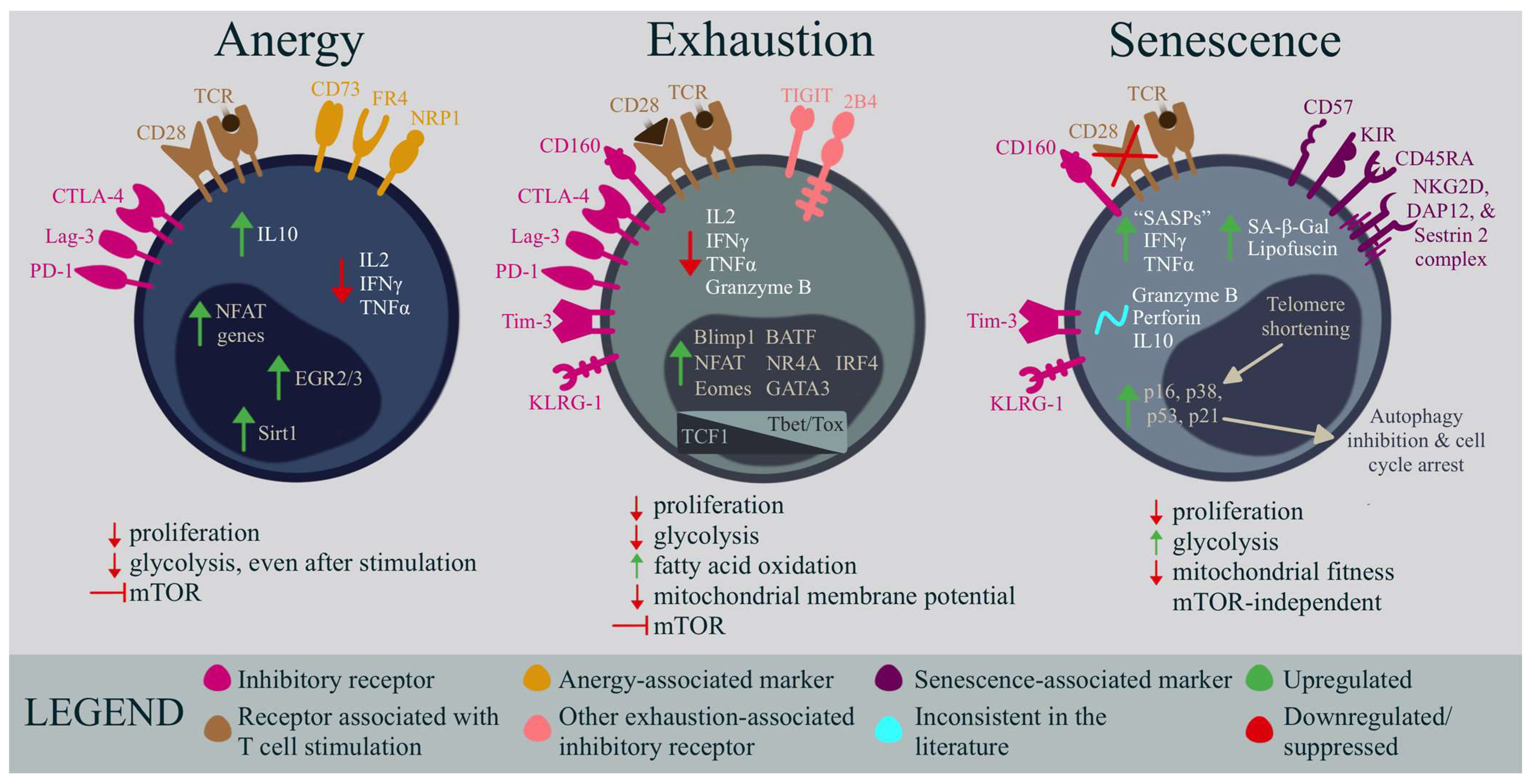
3. Anergy
Comparative Analysis of Anergy and ME/CFS T Cells
4. Exhaustion
Comparative Analysis of Exhaustion and ME/CFS T Cells
5. Senescence
Comparative Analysis of Senescence and ME/CFS T Cells
6. Comparative Analysis of Dysfunctional NK Cell States and ME/CFS NK Cells
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanson, M.R.; Germain, A. Letter to the Editor of Metabolites. Metabolites 2020, 10, 216. [Google Scholar] [CrossRef]
- Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome; Board on the Health of Select Populations; Institute of Medicine. The National Academies Collection: Reports funded by National Institutes of Health. In Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness; National Academies Press: Washington, DC, USA, 2015. [Google Scholar]
- Chu, L.; Valencia, I.J.; Garvert, D.W.; Montoya, J.G. Onset patterns and course of myalgic encephalomyelitis/chronic fatigue syndrome. Front. Pediatr. 2019, 7, 12. [Google Scholar] [CrossRef]
- Jason, L.A.; Porter, N.; Brown, M.; Anderson, V.; Brown, A.; Hunnell, J.; Lerch, A. CFS: A Review of Epidemiology and Natural History Studies. Bull. IACFS/ME 2009, 17, 88–106. [Google Scholar] [PubMed]
- Jason, L.A.; Sunnquist, M.; Brown, A.; Evans, M.; Vernon, S.D.; Furst, J.; Simonis, V. Examining case definition criteria for chronic fatigue syndrome and myalgic encephalomyelitis. Fatigue 2014, 2, 40–56. [Google Scholar] [CrossRef]
- FDA. The voice of the patient: Chronic fatigue syndrome and myalgic encephalomyelitis. In A Series of Reports from the U.S. Food and Drug Administration’s Patient-Focused Drug Development Initiative; Center for Drug Evaluation and Research (CDER): Bethesda, MD, USA, 2013. [Google Scholar]
- O’Neal, A.J.; Hanson, M.R. The Enterovirus Theory of Disease Etiology in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Critical Review. Front. Med. 2021, 8, 688486. [Google Scholar] [CrossRef]
- Hyde, B.M. Understanding Myalgic Encephalomyelitis; Nightingale Press: Ottawa, ON, Canada, 2020; Volume 315. [Google Scholar]
- Bansal, A.S.; Kraneveld, A.D.; Oltra, E.; Carding, S. What Causes ME/CFS: The Role of the Dysfunctional Immune System and Viral Infections. J. Immunol. Allergy 2022, 3, 1–14. [Google Scholar] [CrossRef]
- Chia, J.K. The role of enterovirus in chronic fatigue syndrome. J. Clin. Pathol. 2005, 58, 1126–1132. [Google Scholar] [CrossRef]
- Broderick, G.; Katz, B.Z.; Fernandes, H.; Fletcher, M.A.; Klimas, N.; Smith, F.A.; O’Gorman, M.R.; Vernon, S.D.; Taylor, R. Cytokine expression profiles of immune imbalance in post-mononucleosis chronic fatigue. J. Transl. Med. 2012, 10, 191. [Google Scholar] [CrossRef]
- Buchwald, D.; Cheney, P.R.; Peterson, D.L.; Henry, B.; Wormsley, S.B.; Geiger, A.; Ablashi, D.V.; Salahuddin, S.Z.; Saxinger, C.; Biddle, R.; et al. A chronic illness characterized by fatigue, neurologic and immunologic disorders, and active human herpesvirus type 6 infection. Ann. Intern. Med. 1992, 116, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.M.; Beqaj, S.H.; Deeter, R.G.; Fitzgerald, J.T. Valacyclovir treatment in Epstein-Barr virus subset chronic fatigue syndrome: Thirty-six months follow-up. In Vivo 2007, 21, 707–713. [Google Scholar]
- Chia, J.; Chia, A.; Voeller, M.; Lee, T.; Chang, R. Acute enterovirus infection followed by myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and viral persistence. J. Clin. Pathol. 2010, 63, 165–168. [Google Scholar] [CrossRef]
- Chia, J.K.; Chia, A.Y. Chronic fatigue syndrome is associated with chronic enterovirus infection of the stomach. J. Clin. Pathol. 2008, 61, 43–48. [Google Scholar] [CrossRef]
- Rasa-Dzelzkaleja, S.; Krumina, A.; Capenko, S.; Nora-Krukle, Z.; Gravelsina, S.; Vilmane, A.; Ievina, L.; Shoenfeld, Y.; Murovska, M. The persistent viral infections in the development and severity of myalgic encephalomyelitis/chronic fatigue syndrome. J. Transl. Med. 2023, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, E.I.; Macal, M.; Lewis, G.M.; Harker, J.A. Innate and Adaptive Immune Regulation during Chronic Viral Infections. Annu. Rev. Virol. 2015, 2, 573–597. [Google Scholar] [CrossRef]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.C.P.; et al. Myalgic encephalomyelitis/chronic fatigue syndrome: Clinical working case definition, diagnostic and treatment protocols. J. Chronic Fatigue Syndr. 2003, 11, 7–115. [Google Scholar] [CrossRef]
- Parish, J.G. Reference Index of Papers Published on Epidemics of ME 1934-80; ME Research UK: Perth, UK, 1980. [Google Scholar]
- Hyde, B.M.; Goldstein, J.; Levine, P.H. The Clinical and Scientific Basis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome; Nightingale Research Foundation: Ottawa, ON, Canada, 1992. [Google Scholar]
- Monro, J.A.; Puri, B.K. A Molecular Neurobiological Approach to Understanding the Aetiology of Chronic Fatigue Syndrome (Myalgic Encephalomyelitis or Systemic Exertion Intolerance Disease) with Treatment Implications. Mol. Neurobiol. 2018, 55, 7377–7388. [Google Scholar] [CrossRef] [PubMed]
- Kendell, R.E. The psychiatric sequelae of benign myalgic encephalomyelitis. Br. J. Psychiatry 1967, 113, 833–840. [Google Scholar] [CrossRef]
- Medical Staff of the Royal Free Hospital. An outbreak of encephalomyelitis in the Royal Free Hospital Group, London, in 1955. Br. Med. J. 1957, 2, 895–904. [Google Scholar]
- Fukuda, K.; Straus, S.E.; Hickie, I.; Sharpe, M.C.; Dobbins, J.G.; Komaroff, A. The chronic fatigue syndrome: A comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann. Intern. Med. 1994, 121, 953–959. [Google Scholar] [CrossRef]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Laboratory Diagnosis of Virus Diseases. Fenner White’s Med. Virol. 2017, 11, 135–154. [Google Scholar]
- Louten, J. Detection and Diagnosis of Viral Infections. Essent. Hum. Virol. 2016, 111–132. [Google Scholar] [CrossRef]
- Ruiz-Pablos, M.; Paiva, B.; Montero-Mateo, R.; Garcia, N.; Zabaleta, A. Epstein-Barr Virus and the Origin of Myalgic Encephalomyelitis or Chronic Fatigue Syndrome. Front. Immunol. 2021, 12, 4637. [Google Scholar] [CrossRef]
- Mozhgani, S.H.; Rajabi, F.; Qurbani, M.; Erfani, Y.; Yaslianifard, S.; Moosavi, A.; Pourrostami, K.; Baradaran Bagheri, A.; Soleimani, A.; Behzadian, F.; et al. Human Herpesvirus 6 Infection and Risk of Chronic Fatigue Syndrome: A Systematic Review and Meta-Analysis. Intervirology 2022, 65, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.V.; Cox, B.; Lafuse, W.P.; Ariza, M.E. Epstein-Barr Virus dUTPase Induces Neuroinflammatory Mediators: Implications for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Clin. Ther. 2019, 41, 848–863. [Google Scholar] [CrossRef]
- Shikova, E.; Reshkova, V.; Kumanova, A.; Raleva, S.; Alexandrova, D.; Capo, N.; Murovska, M. Cytomegalovirus, Epstein-Barr virus, and human herpesvirus-6 infections in patients with myalgic encephalomyelitis/chronic fatigue syndrome. J. Med. Virol. 2020, 92, 3682–3688. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.R. Epstein-Barr Virus Induced Gene-2 Upregulation Identifies a Particular Subtype of Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. Front. Pediatr. 2019, 7, 59. [Google Scholar] [CrossRef]
- Loebel, M.; Strohschein, K.; Giannini, C.; Koelsch, U.; Bauer, S.; Doebis, C.; Thomas, S.; Unterwalder, N.; von Baehr, V.; Reinke, P.; et al. Deficient EBV-specific B- and T-cell response in patients with chronic fatigue syndrome. PLoS ONE 2014, 9, e85387. [Google Scholar] [CrossRef]
- Yalcin, S.; Kuratsune, H.; Yamaguchi, K.; Kitani, T.; Yamanishi, K. Prevalence of human herpesvirus 6 variants A and B in patients with chronic fatigue syndrome. Microbiol. Immunol. 1994, 38, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Cuende, J.I.; Civeira, P.; Diez, N.; Prieto, J. High prevalence without reactivation of herpes virus 6 in subjects with chronic fatigue syndrome. Med. Interna 1997, 14, 441–444. [Google Scholar]
- Komaroff, A.L. Is human herpesvirus-6 a trigger for chronic fatigue syndrome? J. Clin. Virol. 2006, 37 (Suppl. S1), S39–S46. [Google Scholar] [CrossRef]
- Nicolson, G.L.; Nasralla, M.Y.; Meirleir, K.D.; Gan, R.; Haier, J. Evidence for Bacterial (Mycoplasma, Chlamydia) and Viral (HHV-6) Co-Infections in Chronic Fatigue Syndrome Patients. J. Chronic Fatigue Syndr. 2003, 11, 7–19. [Google Scholar] [CrossRef]
- Ablashi, D.V.; Eastman, H.B.; Owen, C.B.; Roman, M.M.; Friedman, J.; Zabriskie, J.B.; Peterson, D.L.; Pearson, G.R.; Whitman, J.E. Frequent HHV-6 reactivation in multiple sclerosis (MS) and chronic fatigue syndrome (CFS) patients. J. Clin. Virol. 2000, 16, 179–191. [Google Scholar] [CrossRef]
- Kasimir, F.; Toomey, D.; Liu, Z.; Kaiping, A.C.; Ariza, M.E.; Prusty, B.K. Tissue specific signature of HHV-6 infection in ME/CFS. Front. Mol. Biosci. 2022, 9, 1369. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, L.; Bowles, N.E.; Lane, R.J.M.; Dubowitz, V.; Archard, L.C. Persistence of enteroviral RNA in chronic fatigue syndrome is associated with the abnormal production of equal amounts of positive and negative strands of enteroviral RNA. J. Gen. Virol. 1990, 71, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Gow, J.W.; Behan, W.M.; Clements, G.B.; Woodall, C.; Riding, M.; Behan, P.O. Enteroviral RNA sequences detected by polymerase chain reaction in muscle of patients with postviral fatigue syndrome. BMJ 1991, 302, 692–696. [Google Scholar] [CrossRef]
- Yousef, G.E.; Bell, E.J.; Mann, G.F.; Murugesan, V.; Smith, D.G.; McCartney, R.A.; Mowbray, J.F. Chronic enterovirus infection in patients with postviral fatigue syndrome. Lancet 1988, 1, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.K.; Chia, A.Y.; Wang, D.; El-Habbal, R. Functional dyspepsia and chronic gastritis associated with enteroviruses. Open J. Gastroenterol. 2015, 5, 21–27. [Google Scholar] [CrossRef]
- Montoya, J.G.; Kogelnik, A.M.; Bhangoo, M.; Lunn, M.R.; Flamand, L.; Merrihew, L.E.; Watt, T.; Kubo, J.T.; Paik, J.; Desai, M. Randomized clinical trial to evaluate the efficacy and safety of valganciclovir in a subset of patients with chronic fatigue syndrome. J. Med. Virol. 2013, 85, 2101–2109. [Google Scholar] [CrossRef]
- Diaz-Mitoma, F.; Turgonyi, E.; Kumar, A.; Lim, W.; Larocque, L.; Hyde, B.M. Clinical Improvement in Chronic Fatigue Syndrome Is Associated with Enhanced Natural Killer Cell-Mediated Cytotoxicity: The Results of a Pilot Study with Isoprinosine®. J. Chronic Fatigue Syndr. 2003, 11, 71–95. [Google Scholar] [CrossRef]
- Kerr, J.R.; Cunniffe, V.S.; Kelleher, P.; Bernstein, R.M.; Bruce, I.N. Successful Intravenous Immunoglobulin Therapy in 3 Cases of Parvovirus B19-Associated Chronic Fatigue Syndrome. Clin. Infect. Dis. 2003, 36, e100–e106. [Google Scholar] [CrossRef]
- Lloyd, A.; Hickie, I.; Wakefield, D.; Boughton, C.; Dwyer, J. A double-blind, placebo-controlled trial of intravenous immunoglobulin therapy in patients with chronic fatigue syndrome. Am. J. Med. 1990, 89, 561–568. [Google Scholar] [CrossRef]
- Peterson, P.K.; Shepard, J.; Macres, M.; Schenck, C.; Crosson, J.; Rechtman, D.; Lurie, N. A controlled trial of intravenous immunoglobulin G in chronic fatigue syndrome. Am. J. Med. 1990, 89, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Rowe, K.S. Double-blind randomized controlled trial to assess the efficacy of intravenous gammaglobulin for the management of chronic fatigue syndrome in adolescents. J. Psychiatr. Res. 1997, 31, 133–147. [Google Scholar] [CrossRef]
- Qanneta, R. Long COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome: Similarities and differences of two peas in a pod. Reum. Clin. 2022, 18, 626–628. [Google Scholar] [CrossRef]
- Xu, W.; Cao, Y.; Wu, L. No Causal Effects Detected in COVID-19 and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Two Sample Mendelian Randomization Study. Int. J. Environ. Res. Public Health 2023, 20, 2437. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.L.; Weitzer, D.J. Long COVID and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)—A Systemic Review and Comparison of Clinical Presentation and Symptomatology. Medicina 2021, 57, 418. [Google Scholar] [CrossRef]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef]
- Nunes, J.M.; Kruger, A.; Proal, A.; Kell, D.B.; Pretorius, E. The Occurrence of Hyperactivated Platelets and Fibrinaloid Microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Pharmaceuticals 2022, 15, 931. [Google Scholar]
- Gao, P.; Liu, J.; Liu, M. Effect of COVID-19 Vaccines on Reducing the Risk of Long COVID in the Real World: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 19, 12422. [Google Scholar] [CrossRef]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naive to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Hua, X.; Thompson, C.B. Quiescent T cells: Actively maintaining inactivity. Nat. Immunol. 2001, 2, 1097–1098. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Gardiner, C.M. NK Cell Metabolism. J. Leukoc. Biol. 2019, 105, 1235–1242. [Google Scholar] [CrossRef]
- Gardiner, C.M.; Finlay, D.K. What Fuels Natural Killers? Metabolism and NK Cell Responses. Front. Immunol. 2017, 8, 367. [Google Scholar] [CrossRef]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef]
- Keating, S.E.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Keane, C.; Brennan, K.; Finlay, D.K.; Gardiner, C.M. Metabolic Reprogramming Supports IFN-gamma Production by CD56bright NK Cells. J. Immunol. 2016, 196, 2552–2560. [Google Scholar] [CrossRef]
- Keppel, M.P.; Cooper, M.A. Assessment of NK Cell Metabolism. Methods Mol. Biol. 2016, 1441, 27–42. [Google Scholar] [CrossRef]
- Finlay, D.K. Metabolic regulation of natural killer cells. Biochem. Soc. Trans. 2015, 43, 758–762. [Google Scholar] [CrossRef]
- Troen, A.M.; Mitchell, B.; Sorensen, B.; Wener, M.H.; Johnston, A.; Wood, B.; Selhub, J.; McTiernan, A.; Yasui, Y.; Oral, E.; et al. Unmetabolized folic acid in plasma is associated with reduced natural killer cell cytotoxicity among postmenopausal women. J. Nutr. 2006, 136, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, S.M.; Singh, K.; Ritchie, T.M.; Aguiar, J.A.; Fan, I.Y.; Portillo, A.L.; Rojas, E.A.; Vahedi, F.; El-Sayes, A.; Xing, S.; et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 2021, 33, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Ashkar, A.A. What Defines NK Cell Functional Fate: Phenotype or Metabolism? Front. Immunol. 2019, 10, 1414. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-Cell Metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Yang, K.; Li, Y.; Shaw, T.I.; Wang, Y.; Blanco, D.B.; Wang, X.; Cho, J.H.; Wang, H.; Rankin, S.; et al. Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 2017, 46, 488–503. [Google Scholar] [CrossRef]
- Ron-Harel, N.; Santos, D.; Ghergurovich, J.M.; Sage, P.T.; Reddy, A.; Lovitch, S.B.; Dephoure, N.; Satterstrom, F.K.; Sheffer, M.; Spinelli, J.B.; et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. 2016, 24, 104–117. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Tomas, C.; Brown, A.; Strassheim, V.; Elson, J.L.; Newton, J.; Manning, P. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 2017, 12, e0186802. [Google Scholar] [CrossRef]
- Tomas, C.; Elson, J.L.; Strassheim, V.; Newton, J.L.; Walker, M. The effect of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) severity on cellular bioenergetic function. PLoS ONE 2020, 15, e0231136. [Google Scholar] [CrossRef] [PubMed]
- Tomas, C.; Brown, A.E.; Newton, J.L.; Elson, J.L. Mitochondrial complex activity in permeabilised cells of chronic fatigue syndrome patients using two cell types. PeerJ 2019, 7, e6500. [Google Scholar] [CrossRef]
- Fernandez-Guerra, P.; Gonzalez-Ebsen, A.C.; Boonen, S.E.; Courraud, J.; Gregersen, N.; Mehlsen, J.; Palmfeldt, J.; Olsen, R.K.J.; Brinth, L.S. Bioenergetic and Proteomic Profiling of Immune Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Patients: An Exploratory Study. Biomolecules 2021, 11, 961. [Google Scholar] [CrossRef] [PubMed]
- Castro-Marrero, J.; Cordero, M.D.; Sáez-Francas, N.; Jimenez-Gutierrez, C.; Aguilar-Montilla, F.J.; Aliste, L.; Alegre-Martin, J. Could mitochondrial dysfunction be a differentiating marker between chronic fatigue syndrome and fibromyalgia? Antioxid. Redox Signal. 2013, 19, 1855–1860. [Google Scholar] [CrossRef]
- Lawson, N.; Hsieh, C.H.; March, D.; Wang, X. Elevated Energy Production in Chronic Fatigue Syndrome Patients. J. Nat. Sci. 2016, 2, e221. [Google Scholar]
- Smits, B.; van den Heuvel, L.; Knoop, H.; Kusters, B.; Janssen, A.; Borm, G.; Bleijenberg, G.; Rodenburg, R.; van Engelen, B. Mitochondrial enzymes discriminate between mitochondrial disorders and chronic fatigue syndrome. Mitochondrion 2011, 11, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, R.C.; Kurk, R.M.; Visser, F.C.; Sluiter, W.; Scholte, H.R. Patients with chronic fatigue syndrome performed worse than controls in a controlled repeated exercise study despite a normal oxidative phosphorylation capacity. J. Transl. Med. 2010, 8, 93. [Google Scholar] [CrossRef]
- Missailidis, D.; Annesley, S.J.; Allan, C.Y.; Sanislav, O.; Lidbury, B.A.; Lewis, D.P.; Fisher, P.R. An Isolated Complex V Inefficiency and Dysregulated Mitochondrial Function in Immortalized Lymphocytes from ME/CFS Patients. Int. J. Mol. Sci. 2020, 21, 1074. [Google Scholar] [CrossRef]
- Missailidis, D.; Sanislav, O.; Allan, C.Y.; Smith, P.K.; Annesley, S.J.; Fisher, P.R. Dysregulated Provision of Oxidisable Substrates to the Mitochondria in ME/CFS Lymphoblasts. Int. J. Mol. Sci. 2021, 22, 2046. [Google Scholar] [CrossRef] [PubMed]
- Sweetman, E.; Kleffmann, T.; Edgar, C.; de Lange, M.; Vallings, R.; Tate, W. A SWATH-MS analysis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome peripheral blood mononuclear cell proteomes reveals mitochondrial dysfunction. J. Transl. Med. 2020, 18, 365. [Google Scholar] [CrossRef]
- Mandarano, A.H.; Maya, J.; Giloteaux, L.; Peterson, D.L.; Maynard, M.; Gottschalk, C.G.; Hanson, M.R. Myalgic encephalomyelitis/chronic fatigue syndrome patients exhibit altered T Cell Metabolism and cytokine associations. J. Clin. Investig. 2020, 130, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Maya, J.; Leddy, S.M.; Gottschalk, C.G.; Peterson, D.L.; Hanson, M.R. Altered Fatty Acid Oxidation in Lymphocyte Populations of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2023, 24, 2010. [Google Scholar] [CrossRef] [PubMed]
- Curriu, M.; Carrillo, J.; Massanella, M.; Rigau, J.; Alegre, J.; Puig, J.; Garcia-Quintana, A.M.; Castro-Marrero, J.; Negredo, E.; Clotet, B.; et al. Screening NK-, B- and T-cell phenotype and function in patients suffering from Chronic Fatigue Syndrome. J. Transl. Med. 2013, 11, 68. [Google Scholar] [CrossRef]
- Brenu, E.W.; Huth, T.K.; Hardcastle, S.L.; Fuller, K.; Kaur, M.; Johnston, S.; Ramos, S.B.; Staines, D.R.; Marshall-Gradisnik, S.M. Role of adaptive and innate immune cells in chronic fatigue syndrome/myalgic encephalomyelitis. Int. Immunol. 2014, 26, 233–242. [Google Scholar] [CrossRef]
- Brenu, E.W.; van Driel, M.L.; Staines, D.R.; Ashton, K.J.; Ramos, S.B.; Keane, J.; Klimas, N.G.; Marshall-Gradisnik, S.M. Immunological abnormalities as potential biomarkers in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J. Transl. Med. 2011, 9, 81. [Google Scholar] [CrossRef]
- Karhan, E.; Gunter, C.L.; Ravanmehr, V.; Horne, M.; Kozhaya, L.; Renzullo, S.; Placek, L.; George, J.; Robinson, P.N.; Vernon, S.D.; et al. Perturbation of effector and regulatory T cell subsets in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). bioRxiv 2019. [Google Scholar] [CrossRef]
- Bantug, G.R.; Galluzzi, L.; Kroemer, G.; Hess, C. The spectrum of T Cell Metabolism in health and disease. Nat. Rev. Immunol. 2018, 18, 19–34. [Google Scholar] [CrossRef]
- Germain, A.; Giloteaux, L.; Moore, G.E.; Levine, S.M.; Chia, J.K.; Keller, B.A.; Stevens, J.; Franconi, C.J.; Mao, X.; Shungu, D.C.; et al. Plasma metabolomics reveals disrupted response and recovery following maximal exercise in myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight 2022, 7, e157621. [Google Scholar] [CrossRef]
- Germain, A.; Barupal, D.K.; Levine, S.M.; Hanson, M.R. Comprehensive Circulatory Metabolomics in ME/CFS Reveals Disrupted Metabolism of Acyl Lipids and Steroids. Metabolites 2020, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Germain, A.; Ruppert, D.; Levine, S.M.; Hanson, M.R. Metabolic profiling of a myalgic encephalomyelitis/chronic fatigue syndrome discovery cohort reveals disturbances in fatty acid and lipid metabolism. Mol. Biosyst. 2017, 13, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.W.; McGregor, N.R.; Lewis, D.P.; Butt, H.L.; Gooley, P.R. Metabolic profiling reveals anomalous energy metabolism and oxidative stress pathways in chronic fatigue syndrome patients. Metabolomics 2015, 11, 1626–1639. [Google Scholar] [CrossRef]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; Bright, A.T.; Alaynick, W.A.; Wang, L.; Baxter, A.; Nathan, N.; Anderson, W.; Gordon, E. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, E5472–E5480. [Google Scholar] [CrossRef]
- Yamano, E.; Sugimoto, M.; Hirayama, A.; Kume, S.; Yamato, M.; Jin, G.; Tajima, S.; Goda, N.; Iwai, K.; Fukuda, S.; et al. Index markers of chronic fatigue syndrome with dysfunction of TCA and urea cycles. Sci. Rep. 2016, 6, 34990. [Google Scholar] [CrossRef]
- Nguyen, T.; Staines, D.; Johnston, S.; Marshall-Gradisnik, S. Reduced glycolytic reserve in isolated natural killer cells from Myalgic encephalomyelitis/ chronic fatigue syndrome patients: A preliminary investigation. Asian Pac. J. Allergy Immunol. 2019, 37, 102–108. [Google Scholar] [CrossRef]
- Mandarano, A.H. Altered Immune Metabolism in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome; Cornell University: Ithaca, NY, USA, 2019. [Google Scholar]
- Strayer, D.; Scott, V.; Carter, W. Low NK Cell Activity in Chronic Fatigue Syndrome (CFS) and Relationship to Symptom Severity. J. Clin. Cell. Immunol. 2015, 6, 2. [Google Scholar] [CrossRef]
- Fletcher, M.A.; Maher, K.J.; Klimas, N.G. Natural killer cell function in chronic fatigue syndrome. Clin. Appl. Immunol. Rev. 2002, 2, 129–139. [Google Scholar] [CrossRef]
- Fletcher, M.A.; Zeng, X.R.; Maher, K.; Levis, S.; Hurwitz, B.; Antoni, M.; Broderick, G.; Klimas, N.G. Biomarkers in chronic fatigue syndrome: Evaluation of natural killer cell function and dipeptidyl peptidase IV/CD26. PLoS ONE 2010, 5, e10817. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.; Murray, C.; Buchwald, D.; Levine, H.; Cheney, P.; Peterson, D.; Komaroff, A.L.; Ritz, J. Phenotypic and functional deficiency of natural killer cells in patients with chronic fatigue syndrome. J. Immunol. 1987, 139, 3306–3313. [Google Scholar] [CrossRef] [PubMed]
- Niavarani, S.R.; Lawson, C.; Bakos, O.; Boudaud, M.; Batenchuk, C.; Rouleau, S.; Tai, L.-H. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer 2019, 19, 823. [Google Scholar] [CrossRef]
- Kobayashi, T.; Lam, P.Y.; Jiang, H.; Bednarska, K.; Gloury, R.; Murigneux, V.; Tay, J.; Jacquelot, N.; Li, R.; Tuong, Z.K.; et al. Increased lipid metabolism impairs NK cell function and mediates adaptation to the lymphoma environment. Blood 2020, 136, 3004–3017. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Powell, J.D. Feeding an army: The metabolism of T cells in activation, anergy, and exhaustion. Mol. Immunol. 2015, 68, 492–496. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef]
- ElTanbouly, M.A.; Noelle, R.J. Rethinking peripheral T cell tolerance: Checkpoints across a T cell’s journey. Nat. Rev. Immunol. 2021, 21, 257–267. [Google Scholar] [CrossRef]
- Gunasinghe, S.D.; Peres, N.G.; Goyette, J.; Gaus, K. Biomechanics of T Cell Dysfunctions in Chronic Diseases. Front. Immunol. 2021, 12, 600829. [Google Scholar] [CrossRef]
- Salaman, M.R.; Gould, K.G. Breakdown of T-cell ignorance: The tolerance failure responsible for mainstream autoimmune diseases? J. Transl. Autoimmun. 2020, 3, 100070. [Google Scholar] [CrossRef]
- Parish, I.A.; Heath, W.R. Too dangerous to ignore: Self-tolerance and the control of ignorant autoreactive T cells. Immunol. Cell Biol. 2008, 86, 146–152. [Google Scholar] [CrossRef]
- Abbas, A.A.; Akbar, A.N. Induction of T Cell Senescence by Cytokine Induced Bystander Activation. Front. Aging 2021, 2, 714239. [Google Scholar] [CrossRef]
- Fülöp, T.; Larbi, A.; Pawelec, G. Human T cell aging and the impact of persistent viral infections. Front. Immunol. 2013, 4, 271. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P. Diverse functional autoantibodies in patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef]
- Gabibov, A.G.; Belogurov, J.A.A.; Lomakin, Y.A.; Zakharova, M.Y.; Avakyan, M.E.; Dubrovskaya, V.V.; Smirnov, I.V.; Ivanov, A.S.; Molnar, A.A.; Gurtsevitch, V.E. Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J. 2011, 25, 4211–4221. [Google Scholar] [CrossRef]
- Konstantinov, K.; Von Mikecz, A.; Buchwald, D.; Jones, J.; Gerace, L.; Tan, E. Autoantibodies to nuclear envelope antigens in chronic fatigue syndrome. J. Clin. Investig. 1996, 98, 1888–1896. [Google Scholar] [CrossRef]
- Nishikai, M.; Tomomatsu, S.; Hankins, R.; Takagi, S.; Miyachi, K.; Kosaka, S.; Akiya, K. Autoantibodies to a 68/48 kDa protein in chronic fatigue syndrome and primary fibromyalgia: A possible marker for hypersomnia and cognitive disorders. Rheumatology 2001, 40, 806–810. [Google Scholar] [CrossRef]
- Loebel, M.; Grabowski, P.; Heidecke, H.; Bauer, S.; Hanitsch, L.G.; Wittke, K.; Meisel, C.; Reinke, P.; Volk, H.-D.; Fluge, Ø. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav. Immun. 2016, 52, 32–39. [Google Scholar] [CrossRef]
- Tanaka, S.; Kuratsune, H.; Hidaka, Y.; Hakariya, Y.; Tatsumi, K.-I.; Takano, T.; Kanakura, Y.; Amino, N. Autoantibodies against muscarinic cholinergic receptor in chronic fatigue syndrome. Int. J. Mol. Med. 2003, 12, 225–230. [Google Scholar] [CrossRef]
- Mikecz, A.V.; Konstantinov, K.; Buchwald, D.S.; Gerace, L.; Tan, E.M. High frequency of autoantibodies to insoluble cellular antigens in patients with chronic fatigue syndrome. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1997, 40, 295–305. [Google Scholar] [CrossRef]
- Freitag, H.; Szklarski, M.; Lorenz, S.; Sotzny, F.; Bauer, S.; Philippe, A.; Kedor, C.; Grabowski, P.; Lange, T.; Riemekasten, G.; et al. Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Clin. Med. 2021, 10, 3675. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef]
- Chiodetti, L.; Choi, S.; Barber, D.L.; Schwartz, R.H. Adaptive Tolerance and Clonal Anergy Are Distinct Biochemical States. J. Immunol. 2006, 176, 2279–2291. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Lechner, O.; Lauber, J.; Franzke, A.; Sarukhan, A.; von Boehmer, H.; Buer, J. Fingerprints of anergic T cells. Curr. Biol. 2001, 11, 587–595. [Google Scholar] [CrossRef]
- Choi, S.; Schwartz, R.H. Molecular mechanisms for adaptive tolerance and other T cell anergy models. Semin. Immunol. 2007, 19, 140–152. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Lins, D.C.; Mescher, M.F. Signal 3 determines tolerance versus full activation of naive CD8 T cells: Dissociating proliferation and development of effector function. J. Exp. Med. 2003, 197, 1141–1151. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Alegre, M.-L.; Thompson, C.B. CTLA-4 is not required for induction of CD8+ T cell anergy in vivo. J. Immunol. 2001, 167, 4936–4941. [Google Scholar] [CrossRef]
- Vacchio, M.S.; Hodes, R.J. CD28 costimulation is required for in vivo induction of peripheral tolerance in CD8 T cells. J. Exp. Med. 2003, 197, 19–26. [Google Scholar] [CrossRef]
- Sim, J.H.; Kim, J.-H.; Park, A.K.; Lee, J.; Kim, K.-M.; Shin, H.M.; Kim, M.; Choi, K.; Choi, E.Y.; Kang, I.; et al. IL-7Rαlow CD8+ T Cells from Healthy Individuals Are Anergic with Defective Glycolysis. J. Immunol. 2020, 205, 2968–2978. [Google Scholar] [CrossRef]
- Miggelbrink, A.M.; Jackson, J.D.; Lorrey, S.J.; Srinivasan, E.S.; Waibl-Polania, J.; Wilkinson, D.S.; Fecci, P.E. CD4 T-Cell Exhaustion: Does It Exist and What Are Its Roles in Cancer? Clin. Cancer Res. 2021, 27, 5742–5752. [Google Scholar] [CrossRef]
- Lu, Y.-J.; Barreira-Silva, P.; Boyce, S.; Powers, J.; Cavallo, K.; Behar, S.M. CD4 T cell help prevents CD8 T cell exhaustion and promotes control of Mycobacterium tuberculosis infection. Cell Rep. 2021, 36, 109696. [Google Scholar] [CrossRef]
- Beltra, J.-C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e828. [Google Scholar] [CrossRef]
- Callender, L.A.; Carroll, E.C.; Bober, E.A.; Akbar, A.N.; Solito, E.; Henson, S.M. Mitochondrial mass governs the extent of human T cell senescence. Aging Cell 2020, 19, e13067. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Zheng, Y.; Delgoffe, G.M.; Meyer, C.F.; Chan, W.; Powell, J.D. Anergic T cells are metabolically anergic. J. Immunol. 2009, 183, 6095–6101. [Google Scholar] [CrossRef]
- Duré, M.; Macian, F. IL-2 signaling prevents T cell anergy by inhibiting the expression of anergy-inducing genes. Mol. Immunol. 2009, 46, 999–1006. [Google Scholar] [CrossRef]
- Sears, J.D.; Waldron, K.J.; Wei, J.; Chang, C.-H. Targeting metabolism to reverse T-cell exhaustion in chronic viral infections. Immunology 2021, 162, 135–144. [Google Scholar] [CrossRef]
- Thomann, A.S.; Schneider, T.; Cyran, L.; Eckert, I.N.; Kerstan, A.; Lutz, M.B. Conversion of Anergic T Cells into Foxp3- IL-10+ Regulatory T Cells by a Second Antigen Stimulus In Vivo. Front. Immunol. 2021, 12, 704578. [Google Scholar] [CrossRef]
- Brooks, D.G.; Trifilo, M.J.; Edelmann, K.H.; Teyton, L.; McGavern, D.B.; Oldstone, M.B.A. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 2006, 12, 1301–1309. [Google Scholar] [CrossRef]
- Ejrnaes, M.; Filippi, C.M.; Martinic, M.M.; Ling, E.M.; Togher, L.M.; Crotty, S.; von Herrath, M.G. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006, 203, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Hornig, M.; Montoya, J.G.; Klimas, N.G.; Levine, S.; Felsenstein, D.; Bateman, L.; Peterson, D.L.; Gottschalk, C.G.; Schultz, A.F.; Che, X.; et al. Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci. Adv. 2015, 1, e1400121. [Google Scholar] [CrossRef]
- Montoya, J.G.; Holmes, T.H.; Anderson, J.N.; Maecker, H.T.; Rosenberg-Hasson, Y.; Valencia, I.J.; Chu, L.; Younger, J.W.; Tato, C.M.; Davis, M.M. Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc. Natl. Acad. Sci. USA 2017, 114, E7150–E7158. [Google Scholar] [CrossRef]
- Patarca, R. Cytokines and chronic fatigue syndrome. Ann. N. Y. Acad. Sci. 2001, 933, 185–200. [Google Scholar] [CrossRef]
- Fletcher, M.A.; Zeng, X.R.; Barnes, Z.; Levis, S.; Klimas, N.G. Plasma cytokines in women with chronic fatigue syndrome. J. Transl. Med. 2009, 7, 96. [Google Scholar] [CrossRef]
- Brenu, E.W.; van Driel, M.L.; Staines, D.R.; Ashton, K.J.; Hardcastle, S.L.; Keane, J.; Tajouri, L.; Peterson, D.; Ramos, S.B.; Marshall-Gradisnik, S.M. Longitudinal investigation of natural killer cells and cytokines in chronic fatigue syndrome/myalgic encephalomyelitis. J. Transl. Med. 2012, 10, 88. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef]
- Bettonville, M.; d’Aria, S.; Weatherly, K.; Porporato, P.E.; Zhang, J.; Bousbata, S.; Sonveaux, P.; Braun, M.Y. Long-term antigen exposure irreversibly modifies metabolic requirements for T cell function. eLife 2018, 7, e30938. [Google Scholar] [CrossRef]
- Gottschalk, G.; Peterson, D.; Knox, K.; Maynard, M.; Whelan, R.J.; Roy, A. Elevated ATG13 in serum of patients with ME/CFS stimulates oxidative stress response in microglial cells via activation of receptor for advanced glycation end products (RAGE). Mol. Cell. Neurosci. 2022, 120, 103731. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Mora, E.; Carrillo, J.; Urrea, V.; Rigau, J.; Alegre, J.; Cabrera, C.; Oltra, E.; Castro-Marrero, J.; Blanco, J. Impact of Long-Term Cryopreservation on Blood Immune Cell Markers in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Implications for Biomarker Discovery. Front. Immunol. 2020, 11, 2974. [Google Scholar] [CrossRef]
- Jeffrey, M.G.; Nathanson, L.; Aenlle, K.; Barnes, Z.M.; Baig, M.; Broderick, G.; Klimas, N.G.; Fletcher, M.A.; Craddock, T.J.A. Treatment Avenues in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Split-gender Pharmacogenomic Study of Gene-expression Modules. Clin. Ther. 2019, 41, 815–835.e816. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.R.; Petty, R.; Burke, B.; Gough, J.; Fear, D.; Sinclair, L.I.; Mattey, D.L.; Richards, S.C.M.; Montgomery, J.; Baldwin, D.A.; et al. Gene Expression Subtypes in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J. Infect. Dis. 2008, 197, 1171–1184. [Google Scholar] [CrossRef]
- Kerr, J. Early Growth Response Gene Upregulation in Epstein–Barr Virus (EBV)-Associated Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Biomolecules 2020, 10, 1484. [Google Scholar] [CrossRef]
- Blauensteiner, J.; Bertinat, R.; León, L.E.; Riederer, M.; Sepúlveda, N.; Westermeier, F. Altered endothelial dysfunction-related miRs in plasma from ME/CFS patients. Sci. Rep. 2021, 11, 10604. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef]
- Bengsch, B.; Seigel, B.; Ruhl, M.; Timm, J.; Kuntz, M.; Blum, H.E.; Pircher, H.; Thimme, R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on Exhausted HCV-Specific CD8+ T Cells Is Linked to Antigen Recognition and T Cell Differentiation. PLoS Pathog. 2010, 6, e1000947. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Wherry, E.J. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007, 15, 143–146. [Google Scholar] [CrossRef]
- Janelle, V.; Delisle, J.S. T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers 2021, 13, 598. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; Wang, Y.; DePeaux, K.; et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef]
- Jammes, Y.; Steinberg, J.G.; Delliaux, S. Chronic fatigue syndrome: Acute infection and history of physical activity affect resting levels and response to exercise of plasma oxidant/antioxidant status and heat shock proteins. J. Intern. Med. 2012, 272, 74–84. [Google Scholar] [CrossRef]
- Richards, R.S.; Roberts, T.K.; McGregor, N.R.; Dunstan, R.H.; Butt, H.L. Blood parameters indicative of oxidative stress are associated with symptom expression in chronic fatigue syndrome. Redox Rep. 2000, 5, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Fujita, M. Fluctuation of serum vitamin E (α-tocopherol) concentrations during exacerbation and remission phases in patients with chronic fatigue syndrome. Heart Vessel. 2010, 25, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, A.; Unutmaz, D. Revisiting Immune Exhaustion During HIV Infection. Curr. HIV/AIDS Rep. 2011, 8, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, S.L.; Brenu, E.W.; Johnston, S.; Nguyen, T.; Huth, T.; Wong, N.; Ramos, S.; Staines, D.; Marshall-Gradisnik, S. Characterisation of cell functions and receptors in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (CFS/ME). BMC Immunol. 2015, 16, 35. [Google Scholar] [CrossRef]
- Helliwell, A.M.; Sweetman, E.C.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Changes in DNA methylation profiles of myalgic encephalomyelitis/chronic fatigue syndrome patients reflect systemic dysfunctions. Clin. Epigenet. 2020, 12, 167. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Gelson, W.T.; Das, A.; Fletcher, J.M.; Davies, S.E.; Curran, M.D.; Vowler, S.L.; Maini, M.K.; Akbar, A.N.; Alexander, G.J. CD4+ T-lymphocyte telomere length is related to fibrosis stage, clinical outcome and treatment response in chronic hepatitis C virus infection. J. Hepatol. 2010, 53, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Klenerman, P.; Hill, A. T cells and viral persistence: Lessons from diverse infections. Nat. Immunol. 2005, 6, 873–879. [Google Scholar] [CrossRef]
- Lopes, A.R.; Kellam, P.; Das, A.; Dunn, C.; Kwan, A.; Turner, J.; Peppa, D.; Gilson, R.J.; Gehring, A.; Bertoletti, A. Bim-mediated deletion of antigen-specific CD8+ T cells in patients unable to control HBV infection. J. Clin. Investig. 2008, 118, 1835–1845. [Google Scholar] [CrossRef]
- Judge, S.J.; Murphy, W.J.; Canter, R.J. Characterizing the Dysfunctional NK Cell: Assessing the Clinical Relevance of Exhaustion, Anergy, and Senescence. Front. Cell. Infect. Microbiol. 2020, 10, 49. [Google Scholar] [CrossRef]
- Zhao, Y.; Shao, Q.; Peng, G. Exhaustion and senescence: Two crucial dysfunctional states of T cells in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 27–35. [Google Scholar] [CrossRef]
- Moreira, A.; Gross, S.; Kirchberger, M.C.; Erdmann, M.; Schuler, G.; Heinzerling, L. Senescence markers: Predictive for response to checkpoint inhibitors. Int. J. Cancer 2019, 144, 1147–1150. [Google Scholar] [CrossRef]
- Bandres, E.; Merino, J.; Vazquez, B.; Inoges, S.; Moreno, C.; Subira, M.; Sanchez-Ibarrola, A. The increase of IFN-γ production through aging correlates with the expanded CD8+highCD28− CD57+ subpopulation. Clin. Immunol. 2000, 96, 230–235. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Dewald, H.K.; Vasilopoulos, T.; Gittens-Williams, L.; Fitzgerald-Bocarsly, P.; Herbig, U. Senescence-associated β-galactosidase reveals the abundance of senescent CD8+ T cells in aging humans. Aging Cell 2021, 20, e13344. [Google Scholar] [CrossRef]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- van Leeuwen, E.M.M.; Remmerswaal, E.B.M.; Vossen, M.T.M.; Rowshani, A.T.; Wertheim-van Dillen, P.M.E.; van Lier, R.A.W.; ten Berge, I.J.M. Emergence of a CD4+CD28− Granzyme B+, Cytomegalovirus-Specific T Cell Subset after Recovery of Primary Cytomegalovirus Infection. J. Immunol. 2004, 173, 1834–1841. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Hodge, S. Steroid resistant CD8+ CD28null NKT-like pro-inflammatory cytotoxic cells in chronic obstructive pulmonary disease. Front. Immunol. 2016, 7, 617. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef] [PubMed]
- Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8+CD28− Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A.L.d.; Silva, L.C.; Fernandes, J.R.; Benard, G. Preventing or reversing immunosenescence: Can exercise be an immunotherapy? Immunotherapy 2013, 5, 879–893. [Google Scholar] [CrossRef]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Henel, G.; Sawai, H.; Singh, K.; Lee, E.B.; Pryshchep, S.; Weyand, C.M. Costimulatory Pathways in Rheumatoid Synovitis and T-Cell Senescence. Ann. N. Y. Acad. Sci. 2005, 1062, 182–194. [Google Scholar] [CrossRef]
- González-Osuna, L.; Sierra-Cristancho, A.; Rojas, C.; Cafferata, E.A.; Melgar-Rodríguez, S.; Cárdenas, A.M.; Vernal, R. Premature Senescence of T-cells Favors Bone Loss During Osteolytic Diseases. A New Concern in the Osteoimmunology Arena. Aging Dis. 2021, 12, 1150–1161. [Google Scholar] [CrossRef]
- Alonso-Arias, R.; Moro-García, M.A.; López-Vázquez, A.; Rodrigo, L.; Baltar, J.; García, F.M.S.; Jaurrieta, J.J.S.; López-Larrea, C. NKG2D expression in CD4+ T lymphocytes as a marker of senescence in the aged immune system. Age 2011, 33, 591–605. [Google Scholar] [CrossRef]
- Moro-García, M.; Alonso-Arias, R.; Lopez-Larrea, C. When Aging Reaches CD4+ T-Cells: Phenotypic and Functional Changes. Front. Immunol. 2013, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Borsa, M.; Simon, A.K. Hallmarks and detection techniques of cellular senescence and cellular ageing in immune cells. Aging Cell 2021, 20, e13316. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Ding, S.; Liu, S.; Li, Y.; Sun, L. Human umbilical cord-derived mesenchymal stem cell therapy ameliorates lupus through increasing CD4+ T cell senescence via MiR-199a-5p/Sirt1/p53 axis. Theranostics 2021, 11, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D.; Azevedo, R.I.; Henson, S.M.; Libri, V.; Riddell, N.E.; Macaulay, R.; Kipling, D.; Soares, M.V.; Battistini, L.; Akbar, A.N. Reversible senescence in human CD4+ CD45RA+ CD27− memory T cells. J. Immunol. 2011, 187, 2093–2100. [Google Scholar] [CrossRef]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef]
- van der Windt, G.J.W.; Everts, B.; Chang, C.-H.; Jonathan, D.C.; Tori, C.F.; Amiel, E.; Edward, J.P.; Erika, L.P. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef]
- Liu, X.; Hartman, C.L.; Li, L.; Albert, C.J.; Si, F.; Gao, A.; Huang, L.; Zhao, Y.; Lin, W.; Hsueh, E.C.; et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci. Transl. Med. 2021, 13, eaaz6314. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Henson, S.M. CD8+ T-cell senescence: No role for mTOR. Biochem. Soc. Trans. 2015, 43, 734–739. [Google Scholar] [CrossRef]
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28− and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Cliff, J.M.; King, E.C.; Lee, J.-S.; Sepúlveda, N.; Wolf, A.-S.; Kingdon, C.; Bowman, E.; Dockrell, H.M.; Nacul, L.; Lacerda, E.; et al. Cellular Immune Function in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Front. Immunol. 2019, 10, 796. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, P.; Urra, J.M. Decreased Expression of the CD57 Molecule in T Lymphocytes of Patients with Chronic Fatigue Syndrome. Mol. Neurobiol. 2019, 56, 6581–6585. [Google Scholar] [CrossRef]
- Rajeevan, M.S.; Murray, J.; Oakley, L.; Lin, J.-M.S.; Unger, E.R. Association of chronic fatigue syndrome with premature telomere attrition. J. Transl. Med. 2018, 16, 44. [Google Scholar] [CrossRef]
- Trivedi, M.S.; Oltra, E.; Sarria, L.; Rose, N.; Beljanski, V.; Fletcher, M.A.; Klimas, N.G.; Nathanson, L. Identification of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome-associated DNA methylation patterns. PLoS ONE 2018, 13, e0201066. [Google Scholar] [CrossRef]
- Maes, M.; Mihaylova, I.; Kubera, M.; Bosmans, E. Not in the mind but in the cell: Increased production of cyclo-oxygenase-2 and inducible NO synthase in chronic fatigue syndrome. Neuroendocrinol. Lett. 2007, 28, 463–469. [Google Scholar]
- Morris, G.; Maes, M. Increased nuclear factor-κB and loss of p53 are key mechanisms in Myalgic Encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med. Hypotheses 2012, 79, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Vidard, L.; Dureuil, C.; Baudhuin, J.; Vescovi, L.; Durand, L.; Sierra, V.; Parmantier, E. CD137 (4-1BB) Engagement Fine-Tunes Synergistic IL-15– and IL-21–Driven NK Cell Proliferation. J. Immunol. 2019, 203, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Ardolino, M.; Azimi, C.S.; Iannello, A.; Trevino, T.N.; Horan, L.; Zhang, L.; Deng, W.; Ring, A.M.; Fischer, S.; Garcia, K.C.; et al. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J. Clin. Investig. 2014, 124, 4781–4794. [Google Scholar] [CrossRef]
- Tseng, H.-C.; Bui, V.; Man, Y.-G.; Cacalano, N.; Jewett, A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell–Cell Contact and Secreted Factors. Front. Immunol. 2014, 5, 269. [Google Scholar] [CrossRef]
- Tseng, H.C.; Cacalano, N.; Jewett, A. Split anergized Natural Killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget 2015, 6, 8947–8959. [Google Scholar] [CrossRef] [PubMed]
- Christine, A.B.; Khuong, B.N.; Gary, C.P.; Leslie, P.C.A.; Salazar-Mather, T.P. NATURAL KILLER CELLS IN ANTIVIRAL DEFENSE: Function and Regulation by Innate Cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef]
- Orr, M.T.; Lanier, L.L. Natural Killer Cell Education and Tolerance. Cell 2010, 142, 847–856. [Google Scholar] [CrossRef]
- Wiesmayr, S.; Webber, S.A.; Macedo, C.; Popescu, I.; Smith, L.; Luce, J.; Metes, D. Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur. J. Immunol. 2012, 42, 541–550. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Seo, H.; Jeon, I.; Kim, B.-S.; Park, M.; Bae, E.-A.; Song, B.; Koh, C.-H.; Shin, K.-S.; Kim, I.-K.; Choi, K.; et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat. Commun. 2017, 8, 15776. [Google Scholar] [CrossRef]
- MacFarlane, A.W., IV; Jillab, M.; Plimack, E.R.; Hudes, G.R.; Uzzo, R.G.; Litwin, S.; Dulaimi, E.; Al-Saleem, T.; Campbell, K.S. PD-1 Expression on Peripheral Blood Cells Increases with Stage in Renal Cell Carcinoma Patients and Is Rapidly Reduced after Surgical Tumor Resection. Cancer Immunol. Res. 2014, 2, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Esen, F.; Deniz, G.; Aktas, E.C. PD-1, CTLA-4, LAG-3, and TIGIT: The roles of immune checkpoint receptors on the regulation of human NK cell phenotype and functions. Immunol. Lett. 2021, 240, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, N.A.P.; DeGolier, K.; Haberthur, K.; Chinn, H.; Moyes, K.W.; Bouchlaka, M.N.; Walker, K.L.; Capitini, C.M.; Crane, C.A. An Uncoupling of Canonical Phenotypic Markers and Functional Potency of Ex Vivo-Expanded Natural Killer Cells. Front. Immunol. 2018, 9, 150. [Google Scholar] [CrossRef]
- Benson, D.M.J.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti–PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.-C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, A.J.; McElmurry, R.; Chu, S.; Hinderlie, P.; Bendzick, L.; Geller, M.A.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 2018, 3, e96219. [Google Scholar] [CrossRef]
- Gill, S.; Vasey, A.E.; De Souza, A.; Baker, J.; Smith, A.T.; Kohrt, H.E.; Florek, M.; Gibbs, K.D.J.; Tate, K.; Ritchie, D.S.; et al. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood 2012, 119, 5758–5768. [Google Scholar] [CrossRef]
- Hazeldine, J.; Lord, J.M. The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Res. Rev. 2013, 12, 1069–1078. [Google Scholar] [CrossRef]
- Gounder, S.S.; Abdullah, B.J.J.; Radzuanb, N.E.I.B.M.; Zain, F.D.B.M.; Sait, N.B.M.; Chua, C.; Subramani, B. Effect of Aging on NK Cell Population and Their Proliferation at Ex Vivo Culture Condition. Anal. Cell. Pathol. 2018, 2018, 7871814. [Google Scholar] [CrossRef]
- Beli, E.; Duriancik, D.M.; Clinthorne, J.F.; Lee, T.; Kim, S.; Gardner, E.M. Natural killer cell development and maturation in aged mice. Mech. Ageing Dev. 2014, 135, 33–40. [Google Scholar] [CrossRef]
- Nair, S.; Fang, M.; Sigal, L.J. The natural killer cell dysfunction of aged mice is due to the bone marrow stroma and is not restored by IL-15/IL-15Rα treatment. Aging Cell 2015, 14, 180–190. [Google Scholar] [CrossRef]
- Streltsova, M.A.; Erokhina, S.A.; Kanevskiy, L.M.; Lee, D.A.; Telford, W.G.; Sapozhnikov, A.M.; Kovalenko, E.I. Analysis of NK cell clones obtained using interleukin-2 and gene-modified K562 cells revealed the ability of “senescent” NK cells to lose CD57 expression and start expressing NKG2A. PLoS ONE 2018, 13, e0208469. [Google Scholar] [CrossRef] [PubMed]
- Rivas, J.L.; Palencia, T.; Fernández, G.; García, M. Association of T and NK Cell Phenotype with the Diagnosis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Front. Immunol. 2018, 9, 1028. [Google Scholar] [CrossRef]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Human Diseases Associated with this State | Short Description |
|---|---|---|
| (Loss of) self-ignorance |
| Antigen-inexperienced autoreactive T cells that persist in the periphery due to low antigen expression or physical isolation. If ignorance is overcome, this can induce autoimmunity. |
| (Loss of) self-tolerance |
| T cells that express high-affinity receptors to self-antigens, and will induce cell death once activated. If the body cannot regulate these cells, the breakdown of this state can cause autoimmunity. |
| Anergy (and the loss of this mechanism) |
| T cells that are unable to mount an immune response, driven by sub-optimal co-stimulation (CD28) and/or an increased inhibitory receptor abundance (CTLA4) during a primed cell state. While an anergic T cell can be found in chronic viral infections and cancer, a breakdown of this tolerance mechanism can induce autoimmunity. |
| Exhaustion |
| An unresponsive T cell state characterized by a progressive loss of effector functions, and high levels of inhibitory factors during prolonged antigen exposure. |
| Senescence |
| An irreversible T cell state that occurs during aging, chronic infection, and cancer, differing from exhaustion by having low levels of inhibitory receptors. Proliferation is lost, apoptosis is inhibited, and the cell releases a mixture of molecules that can contribute to inflammatory pathways. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maya, J. Surveying the Metabolic and Dysfunctional Profiles of T Cells and NK Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2023, 24, 11937. https://doi.org/10.3390/ijms241511937
Maya J. Surveying the Metabolic and Dysfunctional Profiles of T Cells and NK Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. International Journal of Molecular Sciences. 2023; 24(15):11937. https://doi.org/10.3390/ijms241511937
Chicago/Turabian StyleMaya, Jessica. 2023. "Surveying the Metabolic and Dysfunctional Profiles of T Cells and NK Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome" International Journal of Molecular Sciences 24, no. 15: 11937. https://doi.org/10.3390/ijms241511937
APA StyleMaya, J. (2023). Surveying the Metabolic and Dysfunctional Profiles of T Cells and NK Cells in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. International Journal of Molecular Sciences, 24(15), 11937. https://doi.org/10.3390/ijms241511937