

MD–Ligand–Receptor: A High-Performance Computing Tool for Characterizing Ligand–Receptor Binding Interactions in Molecular Dynamics Trajectories


 ,
,  and
and
Abstract
1. Introduction
2. Results
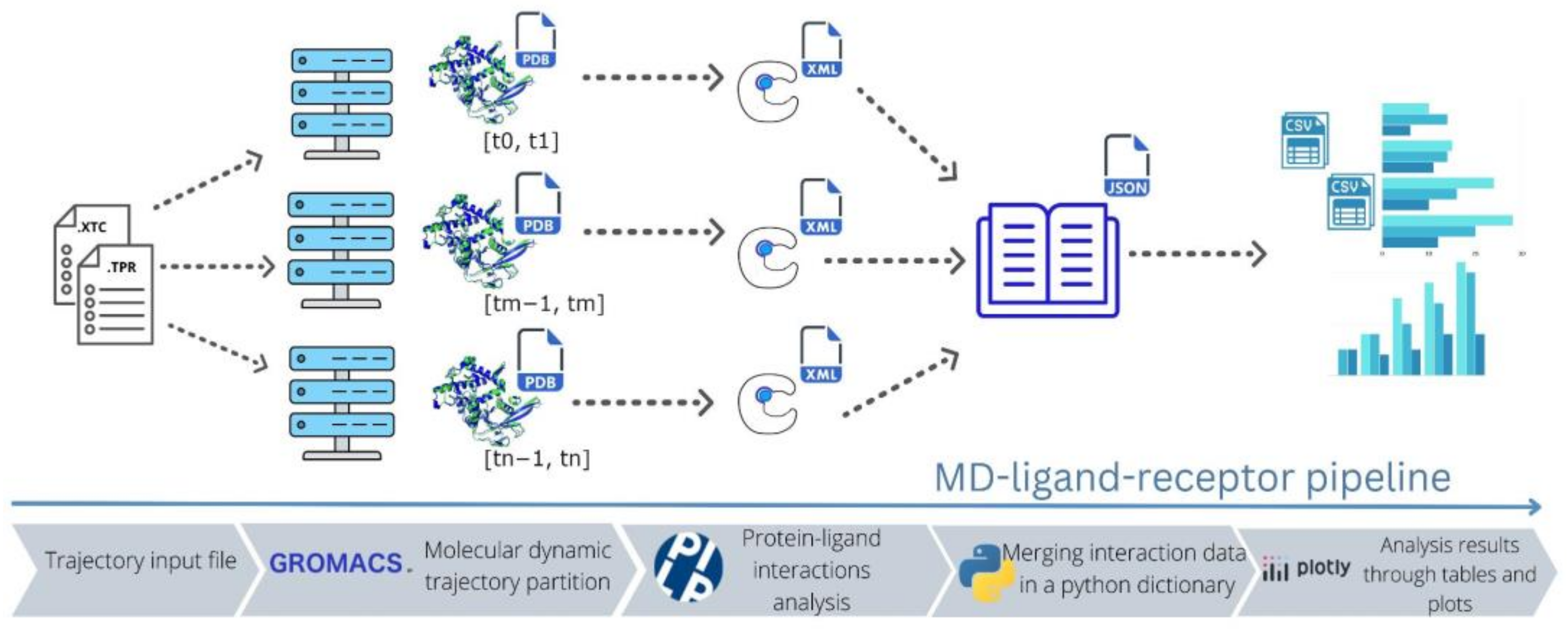
2.1. MD–Ligand–Receptor Workflow
2.2. MD–Ligand–Receptor Software Details
| Algorithm 1. MD-ligand-receptor; The pseudocode describes the steps implemented in the pipeline. | ||||||
| Require: (topology, trajectory); | Files describing the MD | |||||
| Require: (start, end); | Positive time interval T (ps) | |||||
| Require: time limit; | Set time limit for program execution | |||||
| 1: Procedure MD-ligand-receptor(topology, trajectory, start, end, time limit) | ||||||
| 2: #Initial stage | ||||||
| 3: comm = MPI.init() | # spawns MPI processes | |||||
| 4: rank = comm.Get_rank() | # gets MPI process id | |||||
| 5: t_start, t_end = comm.Scatter(start, end) | # process obtains t′ from time interval T | |||||
| 6: N_PDB ← 100 | # fixes PDB number per iteration | |||||
| 7: interaction_table ← dict() | ||||||
| 8: time ← 0 | ||||||
| 9: sub_t_start ← t_start | # initializes partial time interval sub_t for first iteration | |||||
| 10: sub_t_end ← sub_t_start + N_PDB | # sets end of sub_t interval | |||||
| 11: #Second stage | ||||||
| 12: while time < time_limit and sub_t_start ≤ t_end do | # iterates until all of t′ is covered | |||||
| 13: if sub_t_end > t_end then | ||||||
| 14: sub_t_end = t_end | ||||||
| 15: end if | ||||||
| 16: gmx_trjconv(sub_t_start, sub_t_end) | # splits the sub time interval in pdb files | |||||
| 17: #Third stage | ||||||
| 18: for pdb_file in directory do | ||||||
| 19: plip_analysis = PLIP(pdb_file) | # generates the xml file with interaction data | |||||
| 20: interaction_table = parse_xml(plip_analysis) | # extracts interactions from xml | |||||
| 21: end for | ||||||
| 22: remove(pdb_file, plip_analysis) | # deletes produced files to free disk space | |||||
| 23: sub_t_start += N_PDB | # updates sub_t_start for the next iteration | |||||
| 24: sub_t_end = sub_t_start + N_PDB | ||||||
| 25: time = timer() | ||||||
| 26: end while | ||||||
| 27: #Final stage | ||||||
| 28: interaction_table = comm.Gather(interaction_table) | # gathers the final result | |||||
| 29: if rank == 0 then | # the leader process merges all the interactions data | |||||
| 30: merge_tables(interaction_table) | ||||||
| 31: end if | ||||||
| 32: end procedure | ||||||
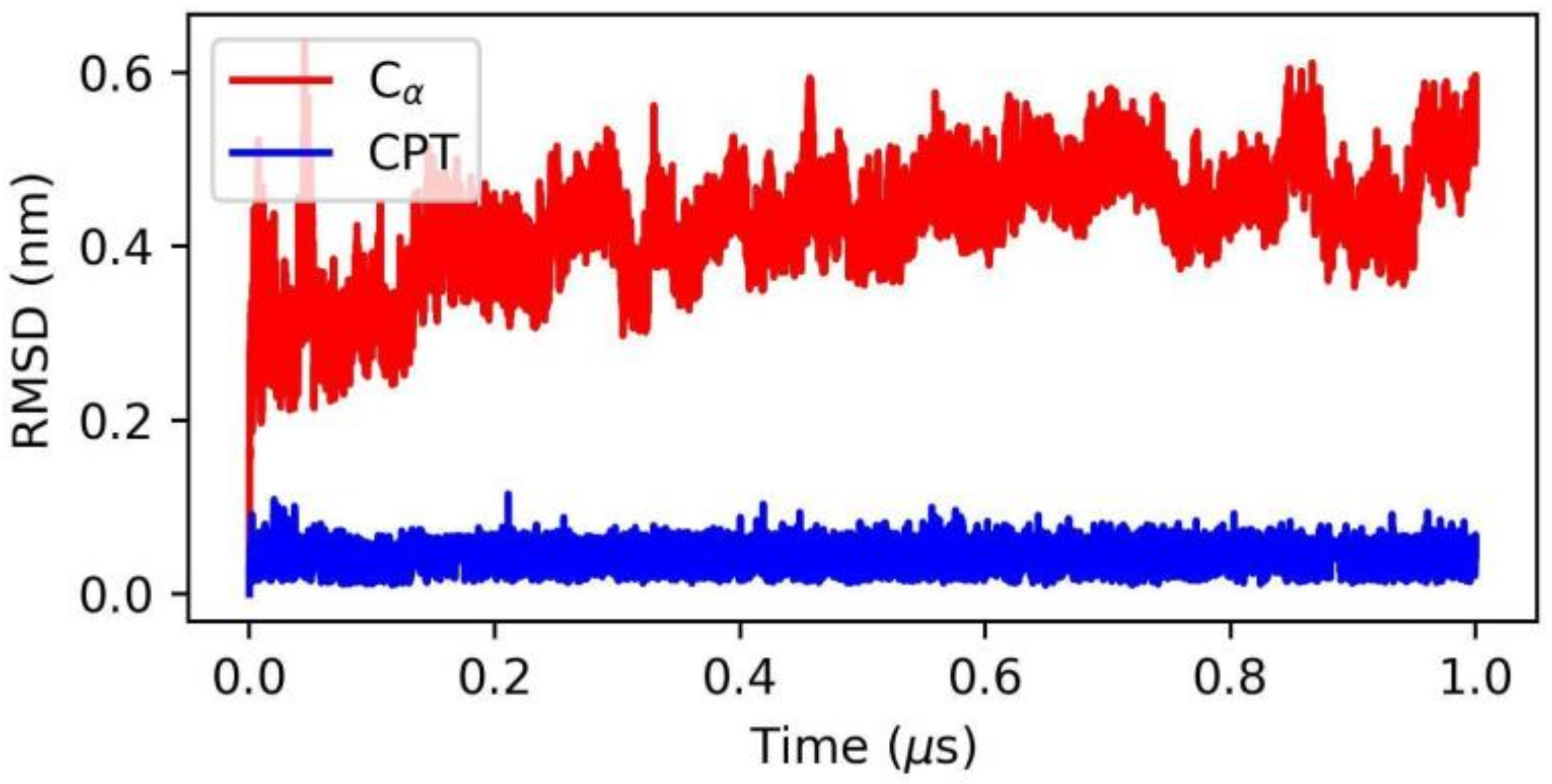
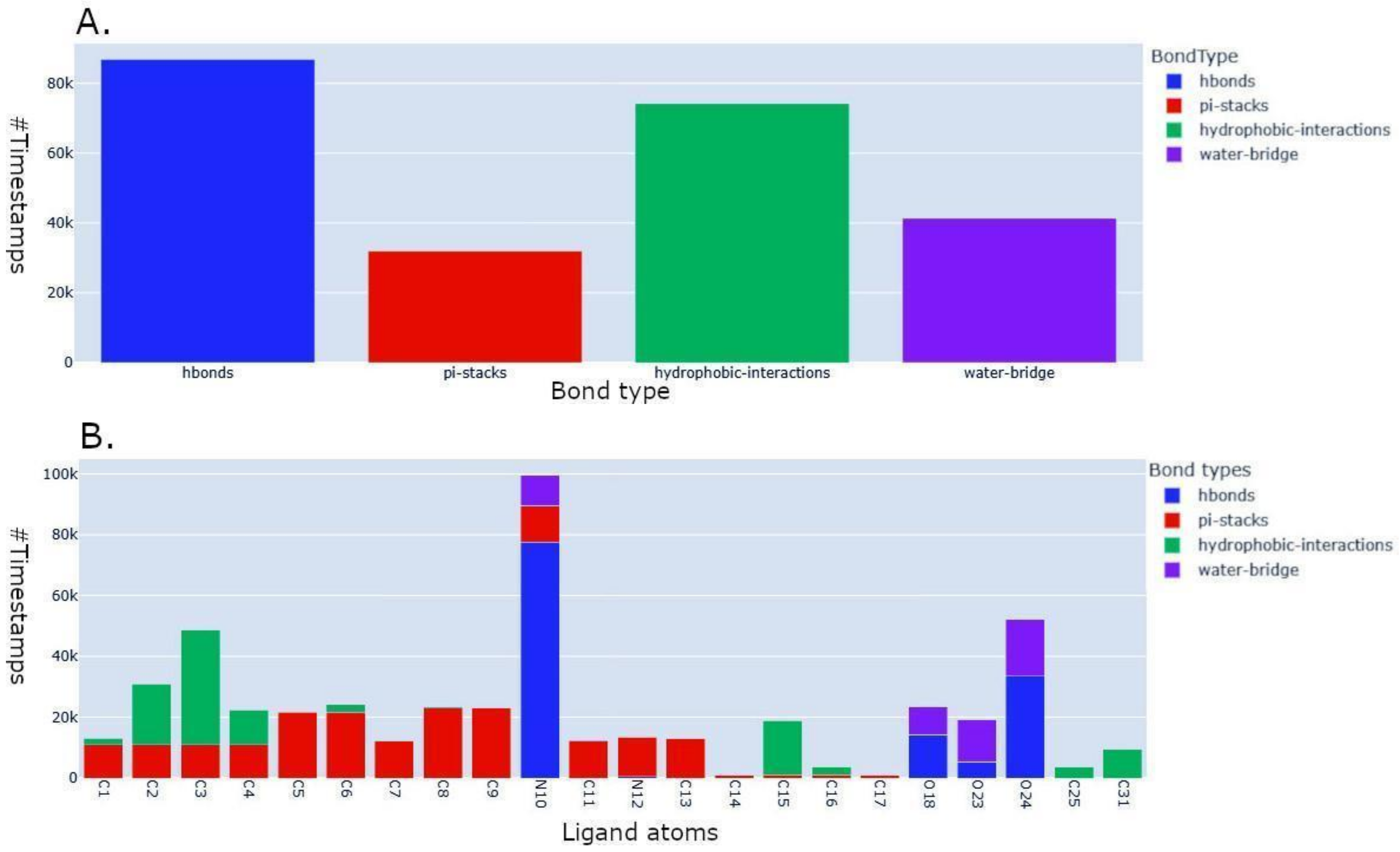
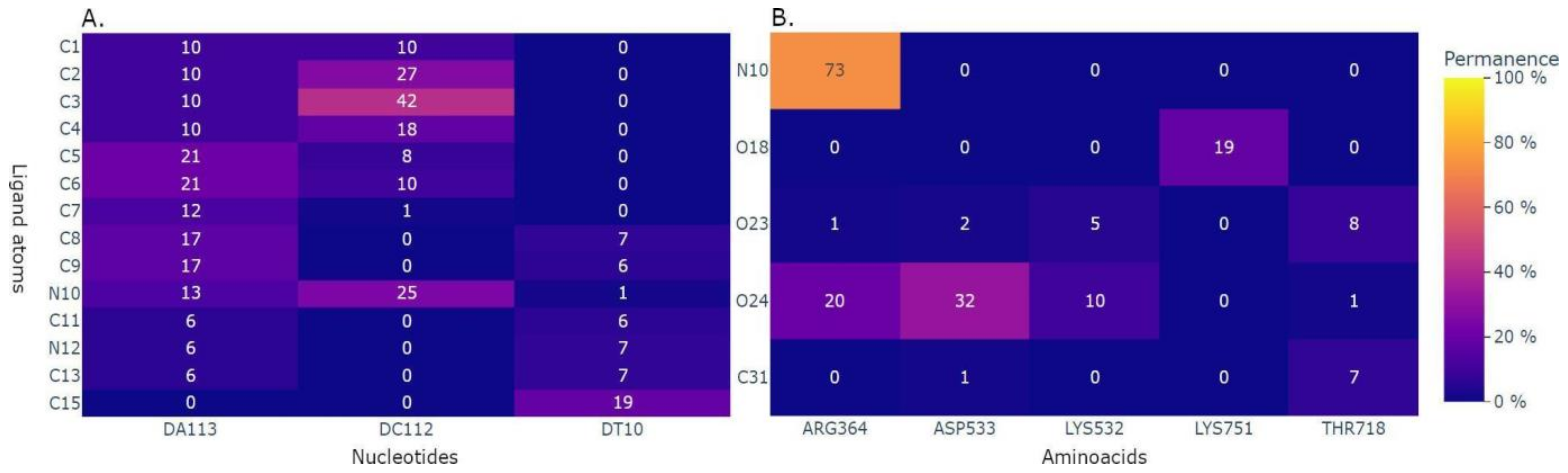
2.3. Application to the Study of Lrbi for Human Topoisomerase 1 and Camptothecin
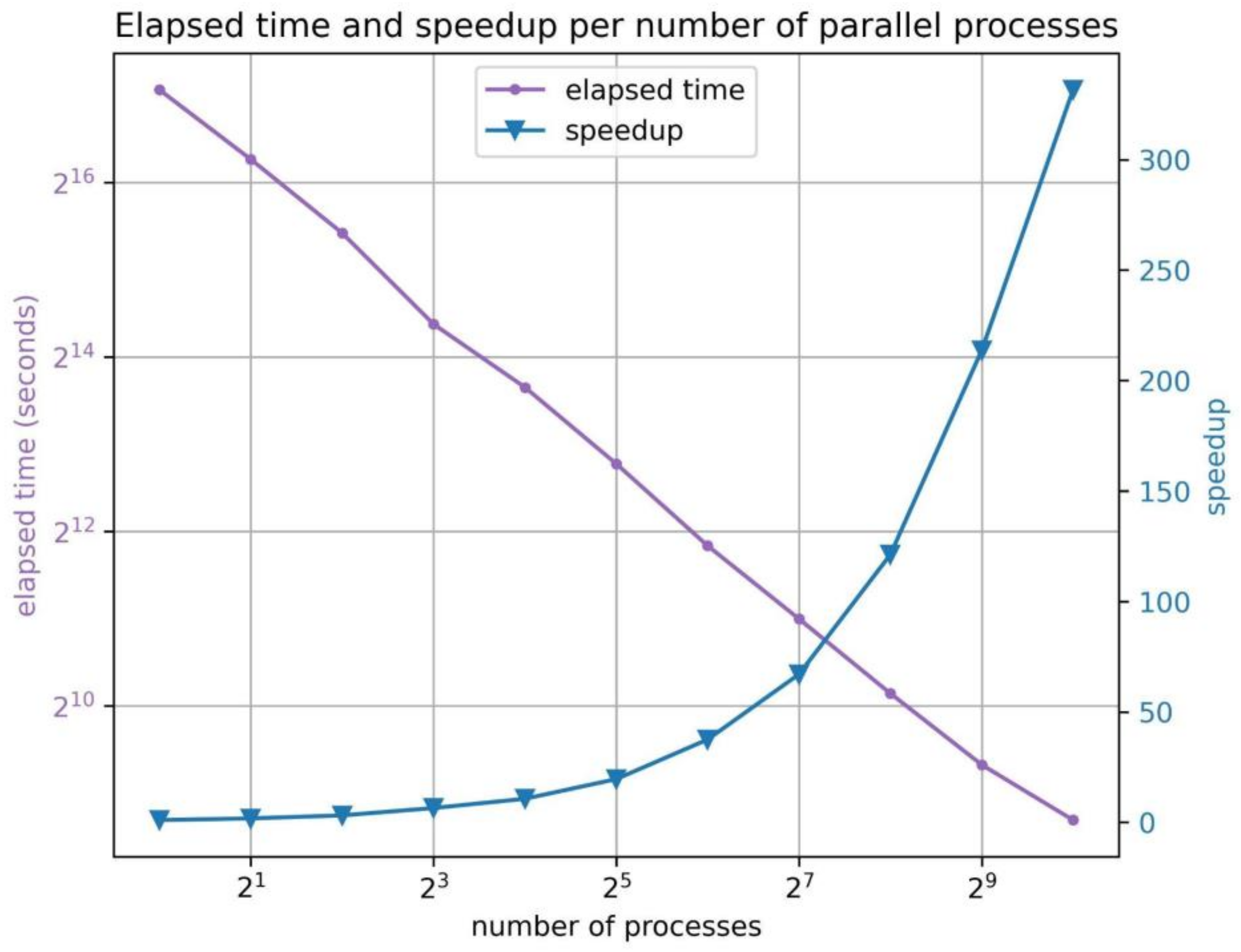
2.4. MD–Ligand–Receptor Benchmarks
2.5. MD–Ligand–Receptor Visualization Tools
3. Software Comparison
3.1. Useful Tools for Preprocessing
3.2. Comparing MDLR with Other Tools
4. Discussion
5. Materials and Methods
5.1. Software Included in the Pipeline
5.2. Programming Language and Parallel Libraries (MPI)
5.3. HPC Setup for Testing
5.4. Input Data Preparation and HPC Environment
5.5. Visualization Libraries
5.6. Molecular Docking and Dynamics Protocol Utilized in the Test Case
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, M.A. Protein-ligand interactions: Fundamentals. Methods Mol. Biol. 2013, 1008, 3–34. [Google Scholar] [PubMed]
- Das, N.R.; Sharma, T.; Toropov, A.A.; Toropova, A.P.; Tripathi, M.K.; Achary, P.G.R. Machine-learning technique, QSAR and molecular dynamics for hERG-drug interactions. J. Biomol. Struct. Dyn. 2023, 5, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Salimi, A.; Lim, J.H.; Jang, J.H.; Lee, J.Y. The use of machine learning modeling, virtual screening, molecular docking, and molecular dynamics simulations to identify potential VEGFR2 kinase inhibitors. Sci. Rep. 2022, 5, 18825. [Google Scholar] [CrossRef] [PubMed]
- Bruce, N.J.; Ganotra, G.K.; Kokh, D.B.; Sadiq, S.K.; Wade, R.C. New approaches for computing ligand-receptor binding kinetics. Curr. Opin. Struct. Biol. 2018, 49, 1–10. [Google Scholar] [CrossRef]
- Gentilucci, L.; Tolomelli, A.; De Marco, R.; Artali, R. Molecular docking of opiates and opioid peptides, a tool for the design of selective agonists and antagonists, and for the investigation of atypical ligand-receptor interactions. Curr. Med. Chem. 2012, 19, 1587–1601. [Google Scholar] [CrossRef] [PubMed]
- Duay, S.S.; Yap, R.C.Y.; Gaitano, A.L., 3rd; Santos, J.A.A.; Macalino, S.J.Y. Roles of Virtual Screening and Molecular Dynamics Simulations in Discovering and Understanding Antimalarial Drugs. Int. J. Mol. Sci. 2023, 26, 9289. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Dhakal, A.; McKay, C.; Tanner, J.J.; Cheng, J. Artificial intelligence in the prediction of protein-ligand interactions: Recent advances and future directions. Brief Bioinform. 2022, 17, bbab476. [Google Scholar] [CrossRef]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.; Kuang, Q.; Pu, X.; Ji, Y.; Zhang, Z.; Li, M. A comparative study of family specific protein-ligand complex affinity prediction based on random forest approach. J. Comput. Aided Mol. Des. 2015, 29, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Durantel, D.; Pasmant, E.; Lau, G.; Schinazi, R.F. COVID-19: Discovery, diagnostics and drug development Tarik. J. Hepatol 2020, 74, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Roshdy, E.; Mohamed, D.E.M.; Ali, T.F.S.; Gabr, G.A.; Jaragh-Alhadad, L.A.; Mekhemer, G.A.H.; Shawky, A.M.; Sidhom, P.A.; et al. In silico drug discovery of SIRT2 inhibitors from natural source as anticancer agents. Sci. Rep. 2023, 13, 2146. [Google Scholar] [CrossRef]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rodríguez, M.; Rosales-Hernández, M.C.; Mendieta-Wejebe, J.E.; Martínez-Archundia, M.; Basurto, J.C. Current Tools and Methods in Molecular Dynamics (MD) Simulations for Drug Design. Curr. Med. Chem. 2016, 23, 3909–3924. [Google Scholar] [CrossRef] [PubMed]
- Dubey, K.D.; Tiwari, R.K.; Ojha, R.P. Recent advances in protein−ligand interactions: Molecular dynamics simulations and binding free energy. Curr. Comput. Aided Drug Des. 2013, 9, 518–531. [Google Scholar] [CrossRef]
- Gabellone, S.; Piccinino, D.; Filippi, S.; Castrignanò, T.; Zippilli, C.; Del Buono, D.; Saladino, R. Lignin Nanoparticles Deliver Novel Thymine Biomimetic Photo-Adducts with Antimelanoma Activity. Int. J. Mol. Sci. 2022, 23, 915. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef]
- Castrignanò, T.; Chillemi, G.; Desideri, A. Structure and hydration of BamHI DNA recognition site: A molecular dynamics investigation. Biophys. J. 2000, 79, 1263–1272. [Google Scholar] [CrossRef]
- Chillemi, G.; Castrignanò, T.; Desideri, A. Structure and hydration of the DNA-human topoisomerase I covalent complex. Biophys. J. 2001, 81, 490–500. [Google Scholar] [CrossRef]
- Castrignanò, T.; Chillemi, G.; Varani, G.; Desideri, A. Molecular dynamics simulation of the RNA complex of a double-stranded RNA-binding domain reveals dynamic features of the intermolecular interface and its hydration. Biophys. J. 2002, 83, 3542–3552. [Google Scholar] [CrossRef] [PubMed]
- Rungruangmaitree, R.; Phoochaijaroen, S.; Chimprasit, A.; Saparpakorn, P.; Pootanakit, K.; Tanramluk, D. Structural analysis of the coronavirus main protease for the design of pan-variant inhibitors. Sci. Rep. 2023, 13, 7055. [Google Scholar] [CrossRef] [PubMed]
- Pirolli, D.; Righino, B.; Camponeschi, C.; Ria, F.; Di Sante, G.; De Rosa, M.C. Virtual screening and molecular dynamics simulations provide insight into repurposing drugs against SARS-CoV-2 variants Spike protein/ACE2 interface. Sci. Rep. 2023, 13, 1494. [Google Scholar] [CrossRef]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef]
- Kutzner, C.; Kniep, C.; Cherian, A.; Nordstrom, L.; Grubmülle, H.; de Groot, B.L.; Gapsys, V. GROMACS in the cloud: A global supercomputer to speed up alchemical drug design. J. Chem. Inf. Model. 2022, 62, 1691–1711. [Google Scholar] [CrossRef]
- Madeddu, F.; Di Martino, J.; Pieroni, M.; Del Buono, D.; Bottoni, P.; Botta, L.; Castrignanò, T.; Saladino, R. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of New Drugs against Human Topoisomerase I Receptor. Int. J. Mol. Sci. 2022, 23, 14652. [Google Scholar] [CrossRef] [PubMed]
- Grottesi, A.; Bešker, N.; Emerson, A.; Manelfi, C.; Beccari, A.R.; Frigerio, F.; Lindahl, E.; Cerchia, C.; Talarico, C. Computational studies of SARS-CoV-2 3CLpro: Insights from MD simulations. Int. J. Mol. Sci. 2020, 21, 5346. [Google Scholar] [CrossRef] [PubMed]
- Ghahremanian, S.; Rashidi, M.M.; Raeisi, K.; Toghraie, D. Molecular dynamics simulation approach for discovering potential inhibitors against SARS-CoV-2: A structural review. J. Mol. Liq. 2022, 354, 118901. [Google Scholar] [CrossRef]
- Castrignanò, T.; Gioiosa, S.; Flati, T.; Cestari, M.; Picardi, E.; Chiara, M.; Fratelli, M.; Amente, S.; Cirilli, M.; Tangaro, M.A.; et al. ELIXIR-IT HPC@ CINECA: High performance computing resources for the bioinformatics community. BMC Bioinform. 2020, 21, 352. [Google Scholar] [CrossRef]
- Petrini, A.; Mesiti, M.; Schubach, M.; Frasca, M.; Danis, D.; Re, M.; Grossi, G.; Cappelletti, L.; Castrignanò, T.; Robinson, P.N.; et al. parSMURF, a high-performance computing tool for the genome-wide detection of pathogenic variants. GigaScience 2020, 9, giaa052. [Google Scholar] [CrossRef]
- Chiara, M.; Gioiosa, S.; Chillemi, G.; D’Antonio, M.; Flati, T.; Picardi, E.; Zambelli, F.; Horner, D.S.; Pesole, G.; Castrignanò, T. CoVaCS: A consensus variant calling system. BMC Genom. 2018, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, M.; Picardi, E.; Castrignanò, T.; D’Erchia, A.M.; Pesole, G. Exploring the RNA editing potential of RNA-seq data by ExpEdit. RNA Bioinform. 2015, 1269, 327–338. [Google Scholar]
- Picardi, E.; D’Antonio, M.; Carrabino, D.; Castrignanò, T.; Pesole, G. ExpEdit: A webserver to explore human RNA editing in RNA-Seq experiments. Bioinformatics 2011, 27, 1311–1312. [Google Scholar] [CrossRef] [PubMed]
- Flati, T.; Gioiosa, S.; Spallanzani, N.; Tagliaferri, I.; Diroma, M.A.; Pesole, G.; Chillemi, G.; Picardi, E.; Castrignanò, T. HPC-REDItools: A novel HPC-aware tool for improved large scale RNA-editing analysis. BMC Bioinform. 2020, 21, 353. [Google Scholar] [CrossRef]
- Gioiosa, S.; Bolis, M.; Flati, T.; Massini, A.; Garattini, E.; Chillemi, G.; Fratelli, M.; Castrignanò, T. Massive NGS data analysis reveals hundreds of potential novel gene fusions in human cell lines. GigaScience 2018, 7, giy062. [Google Scholar] [CrossRef]
- Castrignanò, T.; Rizzi, R.; Talamo, I.G.; De Meo, P.D.; Anselmo, A.; Bonizzoni, P.; Pesole, G. ASPIC: A web resource for alternative splicing prediction and transcript isoforms characterization. Nucleic Acids Res. 2006, 34, W440–W443. [Google Scholar] [CrossRef]
- Van Zalm, P.; Viodé, A.; Smolen, K.; Fatou, B.; Hayati, A.N.; Schlaffner, C.N.; Levy, O.; Steen, J.; Steen, H. A Parallelization Strategy for the Time Efficient Analysis of Thousands of LC/MS Runs in High-Performance Computing Environment. J. Proteome Res. 2022, 21, 2810–2814. [Google Scholar] [CrossRef]
- Bartolini, B.; Chillemi, G.; Abbate, I.; Bruselles, A.; Rozera, G.; Castrignanò, T.; Paoletti, D.; Picardi, E.; Desideri, A.; Pesole, G.; et al. Assembly and characterization of pandemic influenza A H1N1 genome in nasopharyngeal swabs using high-throughput pyrosequencing. Microbiol.-Q. J. Microbiol. Sci. 2011, 34, 391. [Google Scholar]
- Abuín, J.M.; Lopes, N.; Ferreira, L.; Pena, T.F.; Schmidt, B. Big data in metagenomics: Apache spark vs. MPI. PLoS ONE 2020, 15, e0239741. [Google Scholar] [CrossRef]
- Di Matteo, F.; Frumenzio, G.; Chandramouli, B.; Grottesi, A.; Emerson, A.; Musiani, F. Computational Study of Helicase from SARS-CoV-2 in RNA-Free and Engaged Form. Int. J. Mol. Sci. 2022, 23, 14721. [Google Scholar] [CrossRef]
- Prandi, I.G.; Mavian, C.; Giombini, E.; Gruber, C.E.M.; Pietrucci, D.; Borocci, S.; Abid, N.; Beccari, A.R.; Talarico, C.; Chillemi, G. Structural Evolution of Delta (B. 1.617. 2) and Omicron (BA. 1) Spike Glycoproteins. Int. J. Mol. Sci. 2022, 23, 8680. [Google Scholar] [CrossRef] [PubMed]
- Castrignanò, T.; De Meo, P.D.; Carrabino, D.; Orsini, M.; Floris, M.; Tramontano, A. The MEPS server for identifying protein conformational epitopes. BMC Bioinform. 2007, 8, S6. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Dalcin, L.; Fang, Y.L.L. mpi4py: Status update after 12 years of development. Comput. Sci. Eng. 2021, 23, 47–54. [Google Scholar] [CrossRef]
- Dalcin, L.; Lisandro, D. Parallel distributed computing using Python. Adv. Water Resour. 2011, 34, 1124–1139. [Google Scholar] [CrossRef]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef]
- Botta, L.; Filippi, S.; Zippilli, C.; Cesarini, S.; Bizzarri, B.M.; Cirigliano, A.; Rinaldi, T.; Paiardini, A.; Fiorucci, D.; Saladino, R.; et al. Artemisinin Derivatives with Antimelanoma Activity Show Inhibitory Effect against Human DNA Topoisomerase 1. ACS Med. Chem. Lett. 2020, 11, 1035–1040. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Dequidt, A.; Devemy, J.; Padua, A.A. Thermalized Drude oscillators with the LAMMPS molecular dynamics simulator. J. Chem. Inf. Model. 2016, 56, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Karplus, M. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Vermaas, J.V.; Hardy, D.J.; Stone, J.E.; Tajkhorshid, E.; Kohlmeyer, A. TopoGromacs: Automated topology conversion from CHARMM to GROMACS within VMD. J. Chem. Inf. Model. 2016, 27, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Klein, C.; Swails, J.M.; Yin, J.; Gilson, M.K.; Mobley, D.L.; Zhong, E.D. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J. Comput. Aided Mol. Des. 2017, 31, 147–161. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- Münz, M.; Biggin, P.C. JGromacs: A Java package for analyzing protein simulations. J. Chem. Inf. Model. 2012, 23, 255–259. [Google Scholar] [CrossRef]
- Kokh, D.B.; Doser, B.; Richter, S.; Ormersbach, F.; Cheng, X.; Wade, R.C. A workflow for exploring ligand dissociation from a macromolecule: Efficient random acceleration molecular dynamics simulation and interaction fingerprint analysis of ligand trajectories. J. Chem. Phys. 2020, 153, 125102. [Google Scholar] [CrossRef]
- Schatz, K.; Franco-Moreno, J.J.; Schäfer, M.; Rose, A.S.; Ferrario, V.; Pleiss, J.; Krone, M. Visual Analysis of Large-Scale Protein-Ligand Interaction Data. Comput. Graph. Forum 2021, 40, 394–408. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Loschwitz, J.; Jäckering, A.; Keutmann, M.; Olagunju, M.; Olubiyi, O.O.; Strodel, B. Dataset of AMBER force field parameters of drugs, natural products and steroids for simulations using GROMACS. Data Brief 2021, 35, 106948. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Output File | File Description |
|---|---|
| bonds_complete.json | JSON dictionary containing all the recorded interactions |
| hbonds.csv | CSV file containing all the recorded hbonds interactions |
| hydrophobic-interactions.csv | CSV file containing all the recorded hydrophobic interactions |
| pi-stacks.csv | CSV file containing all the recorded pi stacks interactions |
| water-bridge.csv | CSV file containing all the recorded water bridge interactions |
| salt-bridges.csv | CSV file containing all the recorded salt bridges interactions |
| LIG_ATOMS.csv | CSV file containing all ligand’s atoms |
| RCPT_ATOMS.csv | CSV file containing all receptor’s atoms |
| # of Processes | 1 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Speedup | 1.0 | 1.7 | 3.1 | 6.4 | 10.6 | 19.5 | 37.5 | 67.0 | 121.2 | 213.7 | 331.6 |
| Elapsed time (s) | 136,948 | 78,788 | 43,857 | 21,266 | 12,874 | 7009 | 3654 | 2043 | 1130 | 641 | 413 |
| Resource | Initial Format | Final Format | Link | Reference |
|---|---|---|---|---|
| ACPYPE | AMBER | GROMACS | https://alanwilter.github.io/acpype/ accessed on 16 April 2023, Version 2022.7.21 | [53] |
| TopoGromacs | CHARMM | GROMACS | https://github.com/akohlmey/topotools/blob/master/topogromacs.tcl accessed on 16 April 2023, Version 1.8 | [54] |
| InterMol | LAMMPS DESMOND | GROMACS | https://github.com/shirtsgroup/InterMol accessed on 16 April 2023, Version 0.1.2 | [55] |
| MDTraj | NAMD TINKER DESMOND AMBER CHARMM LAMMPS | GROMACS | https://www.mdtraj.org/1.9.8.dev0/index.html accessed on 16 April 2023, Version 1.9.8.dev0 | [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pieroni, M.; Madeddu, F.; Di Martino, J.; Arcieri, M.; Parisi, V.; Bottoni, P.; Castrignanò, T. MD–Ligand–Receptor: A High-Performance Computing Tool for Characterizing Ligand–Receptor Binding Interactions in Molecular Dynamics Trajectories. Int. J. Mol. Sci. 2023, 24, 11671. https://doi.org/10.3390/ijms241411671
Pieroni M, Madeddu F, Di Martino J, Arcieri M, Parisi V, Bottoni P, Castrignanò T. MD–Ligand–Receptor: A High-Performance Computing Tool for Characterizing Ligand–Receptor Binding Interactions in Molecular Dynamics Trajectories. International Journal of Molecular Sciences. 2023; 24(14):11671. https://doi.org/10.3390/ijms241411671
Chicago/Turabian StylePieroni, Michele, Francesco Madeddu, Jessica Di Martino, Manuel Arcieri, Valerio Parisi, Paolo Bottoni, and Tiziana Castrignanò. 2023. "MD–Ligand–Receptor: A High-Performance Computing Tool for Characterizing Ligand–Receptor Binding Interactions in Molecular Dynamics Trajectories" International Journal of Molecular Sciences 24, no. 14: 11671. https://doi.org/10.3390/ijms241411671
APA StylePieroni, M., Madeddu, F., Di Martino, J., Arcieri, M., Parisi, V., Bottoni, P., & Castrignanò, T. (2023). MD–Ligand–Receptor: A High-Performance Computing Tool for Characterizing Ligand–Receptor Binding Interactions in Molecular Dynamics Trajectories. International Journal of Molecular Sciences, 24(14), 11671. https://doi.org/10.3390/ijms241411671