
Pim Kinases: Important Regulators of Cardiovascular Disease


Abstract
1. Introduction
2. Pim Kinases
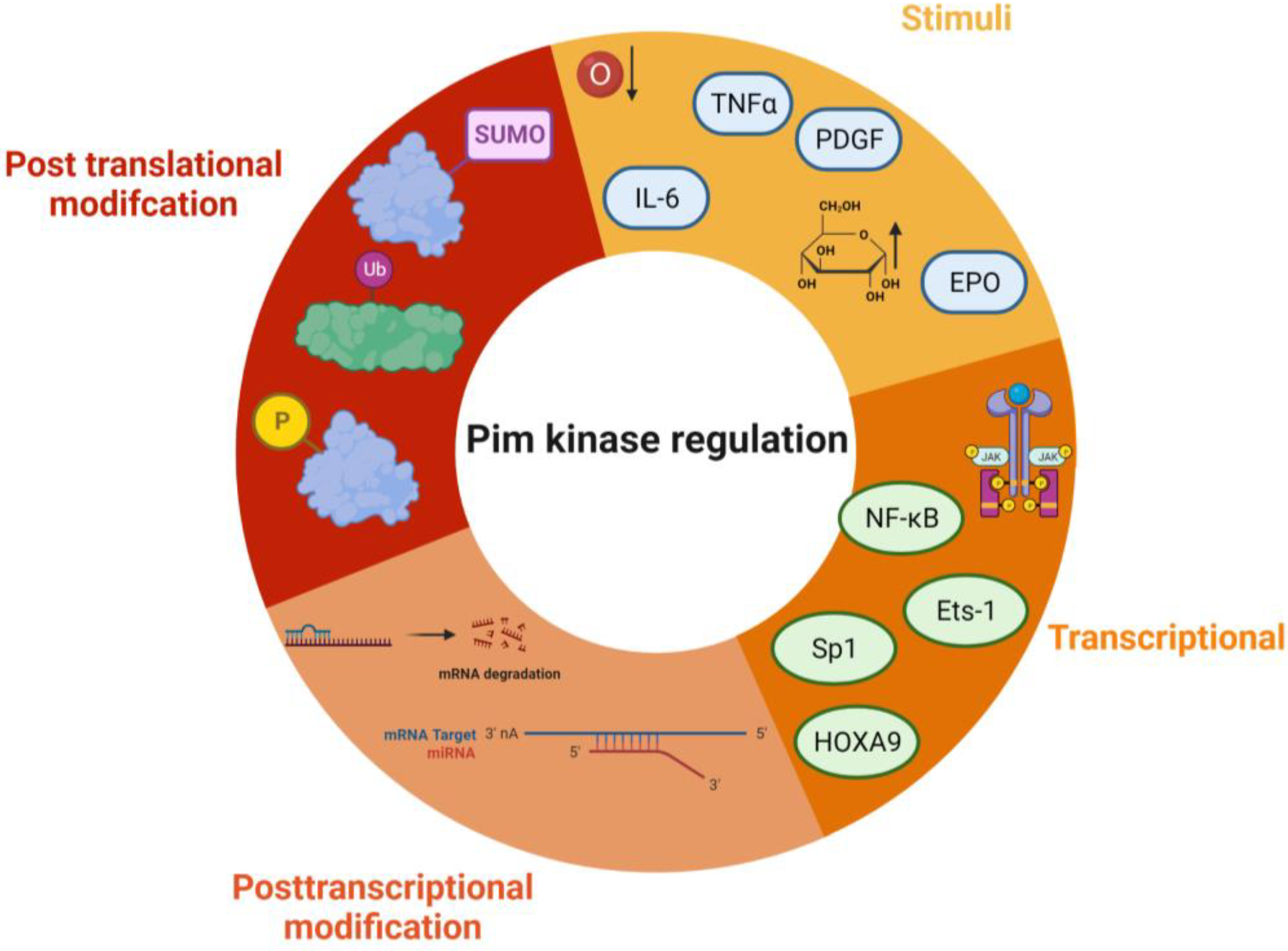
2.1. Structure, Regulation, and Localisation of Pim Kinases
2.2. Pim-1
2.3. Pim-2
2.4. Pim-3
3. Pim Kinase and Cardiovascular Disease
3.1. Hypertension
3.1.1. Vascular Smooth Muscle Cells
3.1.2. Endothelial Cells
3.2. Atherosclerosis
3.2.1. Cholesterol Metabolism
3.2.2. Endothelial Cells and Atherosclerosis
3.2.3. Monocytes and Macrophages
3.2.4. Vascular Smooth Muscle Cells and Atherosclerosis
3.3. Thrombosis
3.3.1. Endothelial Cells and Thrombosis
3.3.2. Platelets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Disease | Number of Patients | Cardiovascular Side Effects (% of Patients) | Clinical Trial Number/Reference |
|---|---|---|---|---|
| AZD1208 | Solid Tumour | 67 | Thrombocytopenia (5%) Platelet count decreased (25.7%) 3% atrial fibrillation | NCT01588548 [17] |
| AZD1208 | Acute myeloid leukaemia | 55 | Hypotension (31.3%) | NCT01489722 [17] |
| Pim447 | Multiple myeloma | 13 | Thrombocytopenia (76.9%) Electrocardiogram QT prolonged (15.4%) | NCT02160951 [18] |
| LGH447 (Pim447) | Multiple Myeloma | 77 | Thrombocytopenia (32.9%) Congestive cardiomyopathy (2.7%) Palpitations (1%) Atrial fibrillation (2.5%) Bradycardia (1%) Sinus bradycardia (2.5%) Tachycardia (53%) | NCT01456689 [19] |
| LGH447 (Pim447) | AML or High risk Myelodysplastic syndrome | 70 | Thrombocytopenia (32.4%) Platelet count decreased (9%) Disseminated intravascular coagulation (18%) Tachycardia (9%) Sinus bradycardia (9%) Electrocardiogram Qt Prolonged (13.5%) Hypertension (9%) Hypotension (18.8%) | NCT02078609 [134] |
| SGI-1776 | Relapsed/ Refractory Leukaemia | 14 | Prolonged QtC time Studies withdrawn with no results | NCT01239108 NCT00848601 [135] |
| TP3654 | Advanced solid tumours | 22 | No reported cardiovascular side effects | NCT03715504 [130] |
| TP3654 | Myelofibrosis | 8 | No reported cardiovascular side effects | NCT04176198 [136] |
| INCB053914 | Advanced haematological malignancies | 8 | Thrombocytopenia (15%) | NCT02587598 [137] |
3.4. Cardiac Dysfunction, Injury, and Recovery
Cardiomyocytes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef]
- Shahin, R.; Shaheen, O.; El-Dahiyat, F.; Habash, M.; Saffour, S. Research advances in kinase enzymes and inhibitors for cardiovascular disease treatment. Future Sci. OA 2017, 3, Fso204. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.; Gutierrez, J.A.; et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA 2016, 315, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Theo Cuypers, H.; Selten, G.; Quint, W.; Zijlstra, M.; Maandag, E.R.; Boelens, W.; van Wezenbeek, P.; Melief, C.; Berns, A. Murine leukemia virus-induced T-cell lymphomagenesis: Integration of proviruses in a distinct chromosomal region. Cell 1984, 37, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Aho, T.L.T.; Sandholm, J.; Peltola, K.J.; Mankonen, H.P.; Lilly, M.; Koskinen, P.J. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 2004, 571, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, A.; Campbell, D.G.; Toth, R.; McLauchlan, H.; Hastie, C.J.; Arthur, J.S. Pim kinases phosphorylate multiple sites on Bad and promote 14-3-3 binding and dissociation from Bcl-XL. BMC Cell Biol. 2006, 7, 1. [Google Scholar] [CrossRef]
- Yan, B.; Zemskova, M.; Holder, S.; Chin, V.; Kraft, A.; Koskinen, P.J.; Lilly, M. The PIM-2 Kinase Phosphorylates BAD on Serine 112 and Reverses BAD-induced Cell Death. J. Biol. Chem. 2003, 278, 45358–45367. [Google Scholar] [CrossRef]
- Li, Y.Y.; Popivanova, B.K.; Nagai, Y.; Ishikura, H.; Fujii, C.; Mukaida, N. Pim-3, a proto-oncogene with serine/threonine kinase activity, is aberrantly expressed in human pancreatic cancer and phosphorylates bad to block bad-mediated apoptosis in human pancreatic cancer cell lines. Cancer Res. 2006, 66, 6741–6747. [Google Scholar] [CrossRef]
- Morishita, D.; Katayama, R.; Sekimizu, K.; Tsuruo, T.; Fujita, N. Pim Kinases Promote Cell Cycle Progression by Phosphorylating and Down-regulating p27Kip1 at the Transcriptional and Posttranscriptional Levels. Cancer Res. 2008, 68, 5076–5085. [Google Scholar] [CrossRef]
- Santio, N.M.; Vahakoski, R.L.; Rainio, E.-M.; Sandholm, J.A.; Virtanen, S.S.; Prudhomme, M.; Anizon, F.; Moreau, P.; Koskinen, P.J. Pim-selective inhibitor DHPCC-9 reveals Pim kinases as potent stimulators of cancer cell migration and invasion. Mol. Cancer 2010, 9, 279. [Google Scholar] [CrossRef]
- Arrouchi, H.; Lakhlili, W.; Ibrahimi, A. A review on PIM kinases in tumors. Bioinformation 2019, 15, 40–45. [Google Scholar] [CrossRef]
- Bellon, M.; Nicot, C. Targeting Pim kinases in hematological cancers: Molecular and clinical review. Mol. Cancer 2023, 22, 18. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Guo, Y.; Wang, H.; Fu, R. An overview of pim kinase as a target in multiple myeloma. Cancer Med. 2023, 12, 11746–11759. [Google Scholar] [CrossRef]
- Keane, N.A.; Reidy, M.; Natoni, A.; Raab, M.S.; O’Dwyer, M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 2015, 5, e325. [Google Scholar] [CrossRef]
- Qian, K.C.; Wang, L.; Hickey, E.R.; Studts, J.; Barringer, K.; Peng, C.; Kronkaitis, A.; Li, J.; White, A.; Mische, S.; et al. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J. Biol. Chem. 2005, 280, 6130–6137. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Cinalli, R.M.; Master, S.R.; Chodosh, L.A.; Thompson, C.B. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003, 17, 1841–1854. [Google Scholar] [CrossRef]
- Cortes, J.; Tamura, K.; DeAngelo, D.J.; de Bono, J.; Lorente, D.; Minden, M.; Uy, G.L.; Kantarjian, H.; Chen, L.S.; Gandhi, V.; et al. Phase I studies of AZD1208, a proviral integration Moloney virus kinase inhibitor in solid and haematological cancers. Br. J. Cancer 2018, 118, 1425–1433. [Google Scholar] [CrossRef]
- Iida, S.; Sunami, K.; Minami, H.; Hatake, K.; Sekiguchi, R.; Natsume, K.; Ishikawa, N.; Rinne, M.; Taniwaki, M. A phase I, dose-escalation study of oral PIM447 in Japanese patients with relapsed and/or refractory multiple myeloma. Int. J. Hematol. 2021, 113, 797–806. [Google Scholar] [CrossRef]
- Raab, M.S.; Thomas, S.K.; Ocio, E.M.; Guenther, A.; Goh, Y.-T.; Talpaz, M.; Hohmann, N.; Zhao, S.; Xiang, F.; Simon, C.; et al. The first-in-human study of the pan-PIM kinase inhibitor PIM447 in patients with relapsed and/or refractory multiple myeloma. Leukemia 2019, 33, 2924–2933. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Petryszak, R.; Keays, M.; Tang, Y.A.; Fonseca, N.A.; Barrera, E.; Burdett, T.; Füllgrabe, A.; Fuentes, A.M.; Jupp, S.; Koskinen, S.; et al. Expression Atlas update—An integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016, 44, D746–D752. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PIM kinase inhibitors: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2019, 172, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mandiyan, V.; Suzuki, Y.; Zhang, C.; Rice, J.; Tsai, J.; Artis, D.R.; Ibrahim, P.; Bremer, R. Crystal Structures of Proto-oncogene Kinase Pim1: A Target of Aberrant Somatic Hypermutations in Diffuse Large Cell Lymphoma. J. Mol. Biol. 2005, 348, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Russo, S.; Amos, A.; Pagano, N.; Bregman, H.; Debreczeni, J.E.; Lee, W.H.; von Delft, F.; Meggers, E.; Knapp, S. Crystal structure of the PIM2 kinase in complex with an organoruthenium inhibitor. PLoS ONE 2009, 4, e7112. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Jacobs, M.D.; Black, J.; Futer, O.; Swenson, L.; Hare, B.; Fleming, M.; Saxena, K. Pim-1 Ligand-bound Structures Reveal the Mechanism of Serine/Threonine Kinase Inhibition by LY294002*. J. Biol. Chem. 2005, 280, 13728–13734. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef]
- Ishchenko, A.; Zhang, L.; Le Brazidec, J.-Y.; Fan, J.; Chong, J.H.; Hingway, A.; Raditsis, A.; Singh, L.; Elenbaas, B.; Hong, V.S.; et al. Structure-based design of low-nanomolar PIM kinase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 474–480. [Google Scholar] [CrossRef]
- Palaty, C.K.; Kalmar, G.; Tai, G.; Oh, S.; Amankawa, L.; Affolter, M.; Aebersold, R.; Pelech, S.L. Identification of the Autophosphorylation Sites of theXenopus laevis Pim-1 Proto-oncogene-encoded Protein Kinase*. J. Biol. Chem. 1997, 272, 10514–10521. [Google Scholar] [CrossRef]
- Bullock, A.N.; Debreczeni, J.; Amos, A.L.; Knapp, S.; Turk, B.E. Structure and Substrate Specificity of the Pim-1 Kinase*. J. Biol. Chem. 2005, 280, 41675–41682. [Google Scholar] [CrossRef]
- Zhou, J.; Shao, L.; Yu, J.; Huang, J.; Feng, Q. PDGF-BB promotes vascular smooth muscle cell migration by enhancing Pim-1 expression via inhibiting miR-214. Ann. Transl. Med. 2021, 9, 1728. [Google Scholar] [CrossRef]
- Willert, M.; Augstein, A.; Poitz, D.M.; Schmeisser, A.; Strasser, R.H.; Braun-Dullaeus, R.C. Transcriptional regulation of Pim-1 kinase in vascular smooth muscle cells and its role for proliferation. Basic Res. Cardiol. 2010, 105, 267–277. [Google Scholar] [CrossRef]
- Wang, K.; Deng, X.; Shen, Z.; Jia, Y.; Ding, R.; Li, R.; Liao, X.; Wang, S.; Ha, Y.; Kong, Y.; et al. High glucose promotes vascular smooth muscle cell proliferation by upregulating proto-oncogene serine/threonine-protein kinase Pim-1 expression. Oncotarget 2017, 8, 88320–88331. [Google Scholar] [CrossRef]
- Warfel, N.A.; Sainz, A.G.; Song, J.H.; Kraft, A.S. PIM Kinase Inhibitors Kill Hypoxic Tumor Cells by Reducing Nrf2 Signaling and Increasing Reactive Oxygen Species. Mol. Cancer Ther. 2016, 15, 1637–1647. [Google Scholar] [CrossRef]
- Zhang, Y.; Lei, W.; Yan, W.; Li, X.; Wang, X.; Zhao, Z.; Hui, J.; Shen, Z.; Yang, J. microRNA-206 is involved in survival of hypoxia preconditioned mesenchymal stem cells through targeting Pim-1 kinase. Stem Cell Res. Ther. 2016, 7, 61. [Google Scholar] [CrossRef]
- Block, K.M.; Hanke, N.T.; Maine, E.A.; Baker, A.F. IL-6 stimulates STAT3 and Pim-1 kinase in pancreatic cancer cell lines. Pancreas 2012, 41, 773–781. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Y.; Qian, H.; Zhang, P.; Huang, C. Pim protein kinase-3 is regulated by TNF-α and promotes endothelial cell sprouting. Mol. Cells 2011, 32, 235–241. [Google Scholar] [CrossRef]
- Liang, C.; Li, Y.-Y. Use of regulators and inhibitors of Pim-1, a serine/threonine kinase, for tumour therapy (Review). Mol. Med. Rep. 2014, 9, 2051–2060. [Google Scholar] [CrossRef]
- Yuka Nagata, K.T. Thrombopoietin induces activation of at least two distinct signaling pathways. FEBS Lett. 1995, 377, 497–501. [Google Scholar] [CrossRef]
- Wernig, G.; Gonneville, J.R.; Crowley, B.J.; Rodrigues, M.S.; Reddy, M.M.; Hudon, H.E.; Walz, C.; Reiter, A.; Podar, K.; Royer, Y.; et al. The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood 2008, 111, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Miura, O.; Miura, Y.; Nakamura, N.; Quelle, F.W.; Witthuhn, B.A.; Ihle, J.N.; Aoki, N. Induction of Tyrosine Phosphorylation of Vav and Expression of Pim-1 Correlates With Jak2-Mediated Growth Signaling From the Erythropoietin Receptor. Blood 1994, 84, 4135–4141. [Google Scholar] [CrossRef] [PubMed]
- Mikkers, H.; Nawijn, M.; Allen, J.; Brouwers, C.; Verhoeven, E.; Jonkers, J.; Berns, A. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol. Cell. Biol. 2004, 24, 6104–6115. [Google Scholar] [CrossRef] [PubMed]
- Saris, C.J.; Domen, J.; Berns, A. The pim-1 oncogene encodes two related protein-serine/threonine kinases by alternative initiation at AUG and CUG. EMBO J. 1991, 10, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Wingett, D.; Reeves, R.; Magnuson, N.S. Stability changes in pim-1 proto-oncogene mRNA after mitogen stimulation of normal lymphocytes. J. Immunol. 1991, 147, 3653–3659. [Google Scholar] [CrossRef]
- Qian, Z.; Zhang, L.; Chen, J.; Li, Y.; Kang, K.; Qu, J.; Wang, Z.; Zhai, Y.; Li, L.; Gou, D. MiR-328 targeting PIM-1 inhibits proliferation and migration of pulmonary arterial smooth muscle cells in PDGFBB signaling pathway. Oncotarget 2016, 7, 54998–55011. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, W.; Guo, C.; Huang, T. GATA1 regulates the microRNA-328-3p/PIM1 axis via circular RNA ITGB1 to promote renal ischemia/reperfusion injury in HK-2 cells. Int. J. Mol. Med. 2022, 50, 5156. [Google Scholar] [CrossRef]
- Jalali, S.; Ramanathan, G.K.; Parthasarathy, P.T.; Aljubran, S.; Galam, L.; Yunus, A.; Garcia, S.; Cox, R.R., Jr.; Lockey, R.F.; Kolliputi, N. Mir-206 regulates pulmonary artery smooth muscle cell proliferation and differentiation. PLoS ONE 2012, 7, e46808. [Google Scholar] [CrossRef]
- Chen, J.; Yin, H.; Jiang, Y.; Radhakrishnan, S.K.; Huang, Z.P.; Li, J.; Shi, Z.; Kilsdonk, E.P.; Gui, Y.; Wang, D.Z.; et al. Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 368–375. [Google Scholar] [CrossRef]
- Xia, X.; Liang, Y.; Zheng, W.; Lin, D.; Sun, S. miR-410-5p promotes the development of diabetic cardiomyopathy by suppressing PIM1-induced anti-apoptosis. Mol. Cell. Probes 2020, 52, 101558. [Google Scholar] [CrossRef]
- Zheng, D.; Yu, Y.; Li, M.; Wang, G.; Chen, R.; Fan, G.C.; Martin, C.; Xiong, S.; Peng, T. Inhibition of MicroRNA 195 Prevents Apoptosis and Multiple-Organ Injury in Mouse Models of Sepsis. J. Infect. Dis. 2016, 213, 1661–1670. [Google Scholar] [CrossRef]
- Zhang, Z.; Lv, M.; Wang, X.; Zhao, Z.; Jiang, D.; Wang, L. LncRNA LUADT1 sponges miR-195 to prevent cardiac endothelial cell apoptosis in sepsis. Mol. Med. 2020, 26, 112. [Google Scholar] [CrossRef]
- Kim, O.; Jiang, T.; Xie, Y.; Guo, Z.; Chen, H.; Qiu, Y. Synergism of cytoplasmic kinases in IL6-induced ligand-independent activation of androgen receptor in prostate cancer cells. Oncogene 2004, 23, 1838–1844. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Katayama, K.; Noguchi, K.; Sugimoto, Y. Protein kinase C alpha-mediated phosphorylation of PIM-1L promotes the survival and proliferation of acute myeloid leukemia cells. Biochem. Biophys. Res. Commun. 2018, 503, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.S.; Chatham, L.; Sleigh, R.; Meek, D.W. A functional SUMO-motif in the active site of PIM1 promotes its degradation via RNF4, and stimulates protein kinase activity. Sci. Rep. 2017, 7, 3598. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Chen, X.P.; Vuong, B.Q.; Fay, S.; Rothman, P.B. Protein Phosphatase 2A Regulates the Stability of Pim Protein Kinases*. J. Biol. Chem. 2003, 278, 4800–4805. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Chen, X.P.; Losman, J.A.; Cowan, S.; Donahue, E.; Fay, S.; Vuong, B.Q.; Nawijn, M.C.; Capece, D.; Cohan, V.L.; Rothman, P. Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 2175–2180. [Google Scholar] [CrossRef]
- Mizuno, K.; Shirogane, T.; Shinohara, A.; Iwamatsu, A.; Hibi, M.; Hirano, T. Regulation of Pim-1 by Hsp90. Biochem. Biophys. Res. Commun. 2001, 281, 663–669. [Google Scholar] [CrossRef]
- Shay, K.P.; Wang, Z.; Xing, P.X.; McKenzie, I.F.; Magnuson, N.S. Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin-proteasome pathway. Mol. Cancer Res. MCR 2005, 3, 170–181. [Google Scholar] [CrossRef]
- Adam, K.; Lambert, M.; Lestang, E.; Champenois, G.; Dusanter-Fourt, I.; Tamburini, J.; Bouscary, D.; Lacombe, C.; Zermati, Y.; Mayeux, P. Control of Pim2 kinase stability and expression in transformed human haematopoietic cells. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Toth, R.K.; Solomon, R.; Warfel, N.A. Stabilization of PIM Kinases in Hypoxia Is Mediated by the Deubiquitinase USP28. Cells 2022, 11, 1006. [Google Scholar] [CrossRef]
- Nagarajan, L.; Louie, E.; Tsujimoto, Y.; ar-Rushdi, A.; Huebner, K.; Croce, C.M. Localization of the human pim oncogene (PIM) to a region of chromosome 6 involved in translocations in acute leukemias. Proc. Natl. Acad. Sci. USA 1986, 83, 2556–2560. [Google Scholar] [CrossRef]
- Liang, H.; Hittelman, W.; Nagarajan, L. Ubiquitous Expression and Cell Cycle Regulation of the Protein Kinase PIM-1. Arch. Biochem. Biophys. 1996, 330, 259–265. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, K.; Dai, B.; Guo, Z.; Jiang, T.; Chen, H.; Qiu, Y. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene 2006, 25, 70–78. [Google Scholar] [CrossRef]
- Linn, D.E.; Yang, X.; Xie, Y.; Alfano, A.; Deshmukh, D.; Wang, X.; Shimelis, H.; Chen, H.; Li, W.; Xu, K.; et al. Differential regulation of androgen receptor by PIM-1 kinases via phosphorylation-dependent recruitment of distinct ubiquitin E3 ligases. J. Biol. Chem. 2012, 287, 22959–22968. [Google Scholar] [CrossRef]
- Hoefnagel, J.J.; Dijkman, R.; Basso, K.; Jansen, P.M.; Hallermann, C.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. Distinct types of primary cutaneous large B-cell lymphoma identified by gene expression profiling. Blood 2005, 105, 3671–3678. [Google Scholar] [CrossRef]
- Bachmann, M.; Kosan, C.; Xing, P.X.; Montenarh, M.; Hoffmann, I.; Möröy, T. The oncogenic serine/threonine kinase Pim-1 directly phosphorylates and activates the G2/M specific phosphatase Cdc25C. Int. J. Biochem. Cell Biol. 2006, 38, 430–443. [Google Scholar] [CrossRef]
- Mochizuki, T.; Kitanaka, C.; Noguchi, K.; Muramatsu, T.; Asai, A.; Kuchino, Y. Physical and Functional Interactions between Pim-1 Kinase and Cdc25A Phosphatase: IMPLICATIONS FOR THE Pim-1-MEDIATED ACTIVATION OF THE c-Myc SIGNALING PATHWAY*. J. Biol. Chem. 1999, 274, 18659–18666. [Google Scholar] [CrossRef]
- Bachmann, M.; Hennemann, H.; Xing, P.X.; Hoffmann, I.; Möröy, T. The oncogenic serine/threonine kinase Pim-1 phosphorylates and inhibits the activity of Cdc25C-associated kinase 1 (C-TAK1): A novel role for Pim-1 at the G2/M cell cycle checkpoint. J. Biol. Chem. 2004, 279, 48319–48328. [Google Scholar] [CrossRef]
- van Lohuizen, M.; Verbeek, S.; Krimpenfort, P.; Domen, J.; Saris, C.; Radaszkiewicz, T.; Berns, A. Predisposition to lymphomagenesis in pim-1 transgenic mice: Cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell 1989, 56, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Li, X.; Magnuson, N.S. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 2008, 27, 4809–4819. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Huglo, A.V.; Furic, L. PIM activity in tumours: A key node of therapy resistance. Adv. Biol. Regul. 2018, 67, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Santio, N.M.; Eerola, S.K.; Paatero, I.; Yli-Kauhaluoma, J.; Anizon, F.; Moreau, P.; Tuomela, J.; Härkönen, P.; Koskinen, P.J. Pim Kinases Promote Migration and Metastatic Growth of Prostate Cancer Xenografts. PLoS ONE 2015, 10, e0130340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiu, J.; Ren, C.; Yu, Z. Protein kinase PIM2: A simple PIM family kinase with complex functions in cancer metabolism and therapeutics. J. Cancer 2021, 12, 2570–2581. [Google Scholar] [CrossRef]
- Dakin, L.A.; Block, M.H.; Chen, H.; Code, E.; Dowling, J.E.; Feng, X.; Ferguson, A.D.; Green, I.; Hird, A.W.; Howard, T.; et al. Discovery of novel benzylidene-1,3-thiazolidine-2,4-diones as potent and selective inhibitors of the PIM-1, PIM-2, and PIM-3 protein kinases. Bioorg. Med. Chem. Lett. 2012, 22, 4599–4604. [Google Scholar] [CrossRef]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A.; et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014, 123, 905–913. [Google Scholar] [CrossRef]
- Mukaida, N.; Wang, Y.-Y.; Li, Y.-Y. Roles of Pim-3, a novel survival kinase, in tumorigenesis. Cancer Sci. 2011, 102, 1437–1442. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Li, Y.Y.; Zheng, H.; Omura, K.; Fujii, C.; Tsuneyama, K.; Mukaida, N. Proto-oncogene, Pim-3 with serine/threonine kinase activity, is aberrantly expressed in human colon cancer cells and can prevent Bad-mediated apoptosis. Cancer Sci. 2007, 98, 321–328. [Google Scholar] [CrossRef]
- Beharry, Z.; Mahajan, S.; Zemskova, M.; Lin, Y.-W.; Tholanikunnel, B.G.; Xia, Z.; Smith, C.D.; Kraft, A.S. The Pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 528–533. [Google Scholar] [CrossRef]
- Ha, S.; Iqbal, N.J.; Mita, P.; Ruoff, R.; Gerald, W.L.; Lepor, H.; Taneja, S.S.; Lee, P.; Melamed, J.; Garabedian, M.J.; et al. Phosphorylation of the androgen receptor by PIM1 in hormone refractory prostate cancer. Oncogene 2013, 32, 3992–4000. [Google Scholar] [CrossRef]
- Li, Y.Y.; Mukaida, N. Pathophysiological roles of Pim-3 kinase in pancreatic cancer development and progression. World J. Gastroenterol. 2014, 20, 9392–9404. [Google Scholar] [CrossRef]
- Brault, L.; Gasser, C.; Bracher, F.; Huber, K.; Knapp, S.; Schwaller, J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 2010, 95, 1004–1015. [Google Scholar] [CrossRef]
- Zhu, N.; Ramirez, L.M.; Lee, R.L.; Magnuson, N.S.; Bishop, G.A.; Gold, M.R. CD40 signaling in B cells regulates the expression of the Pim-1 kinase via the NF-kappa B pathway. J. Immunol. 2002, 168, 744–754. [Google Scholar] [CrossRef]
- Hu, Y.L.; Passegué, E.; Fong, S.; Largman, C.; Lawrence, H.J. Evidence that the Pim1 kinase gene is a direct target of HOXA9. Blood 2007, 109, 4732–4738. [Google Scholar] [CrossRef]
- Li, Y.Y.; Wu, Y.; Tsuneyama, K.; Baba, T.; Mukaida, N. Essential contribution of Ets-1 to constitutive Pim-3 expression in human pancreatic cancer cells. Cancer Sci. 2009, 100, 396–404. [Google Scholar] [CrossRef]
- Dorsam, S.T.; Ferrell, C.M.; Dorsam, G.P.; Derynck, M.K.; Vijapurkar, U.; Khodabakhsh, D.; Pau, B.; Bernstein, H.; Haqq, C.M.; Largman, C.; et al. The transcriptome of the leukemogenic homeoprotein HOXA9 in human hematopoietic cells. Blood 2004, 103, 1676–1684. [Google Scholar] [CrossRef]
- WHO. Cardiovascular Diseases. Available online: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 18 April 2023).
- de Vries, M.; Heijink, I.H.; Gras, R.; den Boef, L.E.; Reinders-Luinge, M.; Pouwels, S.D.; Hylkema, M.N.; van der Toorn, M.; Brouwer, U.; van Oosterhout, A.J.M.; et al. Pim1 kinase protects airway epithelial cells from cigarette smoke-induced damage and airway inflammation. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 307, L240–L251. [Google Scholar] [CrossRef]
- Chittenden, T.W.; Sherman, J.A.; Xiong, F.; Hall, A.E.; Lanahan, A.A.; Taylor, J.M.; Duan, H.; Pearlman, J.D.; Moore, J.H.; Schwartz, S.M.; et al. Transcriptional profiling in coronary artery disease: Indications for novel markers of coronary collateralization. Circulation 2006, 114, 1811–1820. [Google Scholar] [CrossRef]
- Renard, S.; Paulin, R.; Breuils-Bonnet, S.; Simard, S.; Pibarot, P.; Bonnet, S.; Provencher, S. Pim-1: A new biomarker in pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 74–81. [Google Scholar] [CrossRef]
- Archacki, S.R.; Angheloiu, G.; Tian, X.-L.; Tan, F.L.; DiPaola, N.; Shen, G.-Q.; Moravec, C.; Ellis, S.; Topol, E.J.; Wang, Q. Identification of new genes differentially expressed in coronary artery disease by expression profiling. Physiol. Genom. 2003, 15, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Lampron, M.C.; Vitry, G.; Nadeau, V.; Grobs, Y.; Paradis, R.; Samson, N.; Tremblay, È.; Boucherat, O.; Meloche, J.; Bonnet, S.; et al. PIM1 (Moloney Murine Leukemia Provirus Integration Site) Inhibition Decreases the Nonhomologous End-Joining DNA Damage Repair Signaling Pathway in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Aldred, M.A. DNA Damage and Repair in Pulmonary Arterial Hypertension. Genes 2020, 11, 1224. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Leong, P.K.; Ho, Y.F.; Hsu, L.C.; Lu, P.H.; Chen, C.S.; Guh, J.H. Pim-1 knockdown potentiates paclitaxel-induced apoptosis in human hormone-refractory prostate cancers through inhibition of NHEJ DNA repair. Cancer Lett. 2012, 319, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Jie, W.; Wang, X.; Zhang, Y.; Guo, J.; Kuang, D.; Zhu, P.; Wang, G.; Ao, Q. SDF-1α/CXCR4 axis is involved in glucose-potentiated proliferation and chemotaxis in rat vascular smooth muscle cells. Int. J. Exp. Pathol. 2010, 91, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Brault, L.; Gasser, C.; Bullock, A.N.; Dechow, T.; Woetzel, S.; Pogacic, V.; Villa, A.; Ehret, S.; Berridge, G.; et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J. Exp. Med. 2009, 206, 1957–1970. [Google Scholar] [CrossRef]
- Carlson, D.A.; Singer, M.R.; Sutherland, C.; Redondo, C.; Alexander, L.T.; Hughes, P.F.; Knapp, S.; Gurley, S.B.; Sparks, M.A.; MacDonald, J.A.; et al. Targeting Pim Kinases and DAPK3 to Control Hypertension. Cell Chem. Biol. 2018, 25, 1195–1207.e32. [Google Scholar] [CrossRef]
- Álvarez-Santos, M.D.; Álvarez-González, M.; Estrada-Soto, S.; Bazán-Perkins, B. Regulation of Myosin Light-Chain Phosphatase Activity to Generate Airway Smooth Muscle Hypercontractility. Front. Physiol. 2020, 11, 701. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Chen, M.; Yi, B.; Zhu, N.; Wei, X.; Zhang, G.-X.; Huang, S.; Sun, J. Pim1 kinase promotes angiogenesis through phosphorylation of endothelial nitric oxide synthase at Ser-633. Cardiovasc. Res. 2016, 109, 141–150. [Google Scholar] [CrossRef]
- Katsube, A.; Hayashi, H.; Kusuhara, H. Pim-1L Protects Cell Surface–Resident ABCA1 From Lysosomal Degradation in Hepatocytes and Thereby Regulates Plasma High-Density Lipoprotein Level. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2304–2314. [Google Scholar] [CrossRef]
- Hooper, A.J.; Hegele, R.A.; Burnett, J.R. Tangier disease: Update for 2020. Curr. Opin. Lipidol. 2020, 31, 80–84. [Google Scholar] [CrossRef]
- Nofer, J.R.; Herminghaus, G.; Brodde, M.; Morgenstern, E.; Rust, S.; Engel, T.; Seedorf, U.; Assmann, G.; Bluethmann, H.; Kehrel, B.E. Impaired platelet activation in familial high density lipoprotein deficiency (Tangier disease). J. Biol. Chem. 2004, 279, 34032–34037. [Google Scholar] [CrossRef]
- Thomas, M.; Lange-Grünweller, K.; Weirauch, U.; Gutsch, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 2012, 31, 918–928. [Google Scholar] [CrossRef]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef]
- Friedl, J.; Puhlmann, M.; Bartlett, D.L.; Libutti, S.K.; Turner, E.N.; Gnant, M.F.; Alexander, H.R. Induction of permeability across endothelial cell monolayers by tumor necrosis factor (TNF) occurs via a tissue factor-dependent mechanism: Relationship between the procoagulant and permeability effects of TNF. Blood 2002, 100, 1334–1339. [Google Scholar] [CrossRef]
- Gao, B.; Saba, T.M.; Tsan, M.F. Role of alpha(v)beta(3)-integrin in TNF-alpha-induced endothelial cell migration. Am. J. Physiol. Cell Physiol. 2002, 283, C1196–C1205. [Google Scholar] [CrossRef]
- Atluri, P.; Woo, Y.J. Pro-Angiogenic Cytokines as Cardiovascular Therapeutics. BioDrugs 2008, 22, 209–222. [Google Scholar] [CrossRef]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox. Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- O’Brien, E.R.; Garvin, M.R.; Dev, R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M. Angiogenesis in human coronary atherosclerotic plaques. Am. J. Pathol. 1994, 145, 883–894. [Google Scholar]
- Hellings, W.E.; Peeters, W.; Moll, F.L.; Piers, S.R.; van Setten, J.; Van der Spek, P.J.; de Vries, J.P.; Seldenrijk, K.A.; De Bruin, P.C.; Vink, A.; et al. Composition of carotid atherosclerotic plaque is associated with cardiovascular outcome: A prognostic study. Circulation 2010, 121, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Tang, J.; Wang, Y.; Yu, M.; Zhao, L.; Yang, H.; Zhang, P.; Ma, Y. PI3K-like kinases restrain Pim gene expression in endothelial cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Mühleder, S.; Fernández-Chacón, M.; Garcia-Gonzalez, I.; Benedito, R. Endothelial sprouting, proliferation, or senescence: Tipping the balance from physiology to pathology. Cell. Mol. Life Sci. 2021, 78, 1329–1354. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Khotina, V.A.; Omelchenko, A.V.; Kalmykov, V.A.; Orekhov, A.N. The Role of the VEGF Family in Atherosclerosis Development and Its Potential as Treatment Targets. Int. J. Mol. Sci. 2022, 23, 931. [Google Scholar] [CrossRef] [PubMed]
- Zippo, A.; De Robertis, A.; Bardelli, M.; Galvagni, F.; Oliviero, S. Identification of Flk-1 target genes in vasculogenesis: Pim-1 is required for endothelial and mural cell differentiation in vitro. Blood 2004, 103, 4536–4544. [Google Scholar] [CrossRef]
- Liao, M.; Hu, F.; Qiu, Z.; Li, J.; Huang, C.; Xu, Y.; Cheng, X. Pim-2 kinase inhibits inflammation by suppressing the mTORC1 pathway in atherosclerosis. Aging 2021, 13, 22412–22431. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Paulin, R.; Courboulin, A.; Meloche, J.; Mainguy, V.; Dumas de la Roque, E.; Saksouk, N.; Côté, J.; Provencher, S.; Sussman, M.A.; Bonnet, S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 2011, 123, 1205–1215. [Google Scholar] [CrossRef]
- Walpen, T.; Peier, M.; Haas, E.; Kalus, I.; Schwaller, J.; Battegay, E.; Humar, R. Loss of pim1 imposes a hyperadhesive phenotype on endothelial cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2012, 30, 1083–1096. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, H.; Min, X.; Wang, Y.; Tang, J.; Cheng, J.; Li, D.; Chen, X.; Cheng, F.; Wang, N.; et al. Pim-3 is expressed in endothelial cells and promotes vascular tube formation. J. Cell. Physiol. 2009, 220, 82–90. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Wang, Z.; Davitt, C.; McKenzie, I.F.; Xing, P.X.; Magnuson, N.S. Pim-1 associates with protein complexes necessary for mitosis. Chromosoma 2002, 111, 80–95. [Google Scholar] [CrossRef]
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet signaling. Handb. Exp. Pharmacol. 2012, 59–85. [Google Scholar] [CrossRef]
- Bye, A.P.; Unsworth, A.J.; Gibbins, J.M. Platelet signaling: A complex interplay between inhibitory and activatory networks. J. Thromb. Haemost. JTH 2016, 14, 918–930. [Google Scholar] [CrossRef]
- Unsworth, A.J.; Bye, A.P.; Sage, T.; Gaspar, R.S.; Eaton, N.; Drew, C.; Stainer, A.; Kriek, N.; Volberding, P.J.; Hutchinson, J.L.; et al. Antiplatelet properties of Pim kinase inhibition are mediated through disruption of thromboxane A2 receptor signaling. Haematologica 2021, 106, 1968–1978. [Google Scholar] [CrossRef]
- Vinholt, P.J.; Hvas, A.M.; Frederiksen, H.; Bathum, L.; Jørgensen, M.K.; Nybo, M. Platelet count is associated with cardiovascular disease, cancer and mortality: A population-based cohort study. Thromb. Res. 2016, 148, 136–142. [Google Scholar] [CrossRef]
- Laird, P.W.; van der Lugt, N.M.; Clarke, A.; Domen, J.; Linders, K.; McWhir, J.; Berns, A.; Hooper, M. In vivo analysis of Pim-1 deficiency. Nucleic Acids Res. 1993, 21, 4750–4755. [Google Scholar] [CrossRef]
- An, N.; Lin, Y.W.; Mahajan, S.; Kellner, J.N.; Wang, Y.; Li, Z.; Kraft, A.S.; Kang, Y. Pim1 serine/threonine kinase regulates the number and functions of murine hematopoietic stem cells. Stem. Cells 2013, 31, 1202–1212. [Google Scholar] [CrossRef]
- An, N.; Kraft, A.S.; Kang, Y. Abnormal hematopoietic phenotypes in Pim kinase triple knockout mice. J. Hematol. Oncol. 2013, 6, 12. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Dillon, P.M.; Anthony, S.P.; Janat-Amsbury, M.; Ashenbramer, N.; Warner, S.L.; Mouritsen, L.; Wade, M.L.; Whatcott, C.; Bearss, D.; et al. A phase I, first-in-human, open-label, dose-escalation, safety, pharmacokinetic, and pharmacodynamic study of oral TP-3654 administered daily for 28 days to patients with advanced solid tumors. J. Clin. Oncol. 2020, 38, 3586. [Google Scholar] [CrossRef]
- Ghanavat, M.; Ebrahimi, M.; Rafieemehr, H.; Maniati, M.; Behzad, M.M.; Shahrabi, S. Thrombocytopenia in solid tumors: Prognostic significance. Oncol. Rev. 2019, 13, 413. [Google Scholar] [CrossRef]
- Fritz, E.; Ludwig, H.; Scheithauer, W.; Sinzinger, H. Shortened Platelet Half-life in Multiple Myeloma. Blood 1986, 68, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Nath, D.; Yang, Y.; Le, B.T.; Rahman, M.F.-U.; Faughnan, P.; Wang, Z.; Stuver, M.; He, R.; Tan, W.; et al. Genetic ablation of Pim1 or pharmacologic inhibition with TP-3654 ameliorates myelofibrosis in murine models. Leukemia 2022, 36, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Novartis Pharmaceuticals, I. A Safety and Efficacy Study of LGH447 in Patients with Acute Myeloid Leukemia (AML) or High Risk Myelodysplastic Syndrome (MDS). 2014. Available online: https://classic.clinicaltrials.gov/show/NCT02078609 (accessed on 5 July 2023).
- Astex Pharmaceuticals, I. Study of SGI-1776, a PIM Kinase Inhibitor, in Subjects with Relapsed/Refractory Leukemias. Available online: https://ClinicalTrials.gov/show/NCT01239108 (accessed on 5 July 2023).
- El Chaer, F.; McCloskey, J.; Rein, L.A.M.; Brown, R.A.; Green, S.D.; Pu, J.J.; Shirane, S.; Shimoda, K.; Ichii, M.; Yuda, J.; et al. Preliminary Data from the Phase I/II Study of TP-3654, a Selective Oral PIM1 Kinase Inhibitor, in Patients with Myelofibrosis Previously Treated with or Ineligible for JAK Inhibitor Therapy. Blood 2022, 140, 594–595. [Google Scholar] [CrossRef]
- Byrne, M.; Donnellan, W.; Patel, M.R.; Zeidan, A.M.; Cherry, M.; Baer, M.R.; Fathi, A.T.; Kaplan, J.; Zhou, F.; Zheng, F.; et al. Preliminary Results from an Ongoing Phase 1/2 Study of INCB053914, a Pan-Proviral Integration Sites for Moloney Virus (PIM) Kinase Inhibitor, in Patients with Advanced Hematologic Malignancies. Blood 2017, 130, 2585. [Google Scholar] [CrossRef]
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An Overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar]
- Gerber, Y.; Weston, S.A.; Enriquez-Sarano, M.; Berardi, C.; Chamberlain, A.M.; Manemann, S.M.; Jiang, R.; Dunlay, S.M.; Roger, V.L. Mortality Associated With Heart Failure After Myocardial Infarction. Circ. Heart Fail. 2016, 9, e002460. [Google Scholar] [CrossRef]
- Ridolfi, R.L.; Hutchins, G.M. The relationship between coronary artery lesions and myocardial infarcts: Ulceration of atherosclerotic plaques precipitating coronary thrombosis. Am. Heart J. 1977, 93, 468–486. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef]
- Golforoush, P.; Yellon, D.M.; Davidson, S.M. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic Res. Cardiol. 2020, 115, 73. [Google Scholar] [CrossRef]
- Muraski, J.A.; Rota, M.; Misao, Y.; Fransioli, J.; Cottage, C.; Gude, N.; Esposito, G.; Delucchi, F.; Arcarese, M.; Alvarez, R.; et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat. Med. 2007, 13, 1467–1475. [Google Scholar] [CrossRef]
- Din, S.; Konstandin, M.H.; Johnson, B.; Emathinger, J.; Völkers, M.; Toko, H.; Collins, B.; Ormachea, L.; Samse, K.; Kubli, D.A.; et al. Metabolic dysfunction consistent with premature aging results from deletion of Pim kinases. Circ. Res. 2014, 115, 376–387. [Google Scholar] [CrossRef]
- Mohsin, S.; Khan, M.; Nguyen, J.; Alkatib, M.; Siddiqi, S.; Hariharan, N.; Wallach, K.; Monsanto, M.; Gude, N.; Dembitsky, W.; et al. Rejuvenation of human cardiac progenitor cells with Pim-1 kinase. Circ. Res. 2013, 113, 1169–1179. [Google Scholar] [CrossRef]
- Borillo, G.A.; Mason, M.; Quijada, P.; Völkers, M.; Cottage, C.; McGregor, M.; Din, S.; Fischer, K.; Gude, N.; Avitabile, D.; et al. Pim-1 Kinase Protects Mitochondrial Integrity in Cardiomyocytes. Circ. Res. 2010, 106, 1265–1274. [Google Scholar] [CrossRef]
- Ebeid, D.E.; Khalafalla, F.G.; Broughton, K.M.; Monsanto, M.M.; Esquer, C.Y.; Sacchi, V.; Hariharan, N.; Korski, K.I.; Moshref, M.; Emathinger, J.; et al. Pim1 maintains telomere length in mouse cardiomyocytes by inhibiting TGFβ signalling. Cardiovasc. Res. 2021, 117, 201–211. [Google Scholar] [CrossRef]
- Yeh, J.K.; Wang, C.Y. Telomeres and Telomerase in Cardiovascular Diseases. Genes 2016, 7, 58. [Google Scholar] [CrossRef]
- An, N.; Cen, B.; Cai, H.; Song, J.H.; Kraft, A.; Kang, Y. Pim1 kinase regulates c-Kit gene translation. Exp. Hematol. Oncol. 2016, 5, 31. [Google Scholar] [CrossRef]
- Ebeid, D.E.; Firouzi, F.; Esquer, C.Y.; Navarrete, J.M.; Wang, B.J.; Gude, N.A.; Sussman, M.A. PIM1 Promotes Survival of Cardiomyocytes by Upregulating c-Kit Protein Expression. Cells 2020, 9, 2001. [Google Scholar] [CrossRef]
- Smith, A.L.; Ellison, F.M.; McCoy, J.P., Jr.; Chen, J. c-Kit expression and stem cell factor-induced hematopoietic cell proliferation are up-regulated in aged B6D2F1 mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 448–456. [Google Scholar] [CrossRef]
- Zhu, N.; Yi, B.; Guo, Z.; Zhang, G.; Huang, S.; Qin, Y.; Zhao, X.; Sun, J. Pim-1 Kinase Phosphorylates Cardiac Troponin I and Regulates Cardiac Myofilament Function. Cell. Physiol. Biochem. 2018, 45, 2174–2186. [Google Scholar] [CrossRef] [PubMed]
- Muraski, J.A.; Fischer, K.M.; Wu, W.; Cottage, C.T.; Quijada, P.; Mason, M.; Din, S.; Gude, N.; Alvarez, R.; Rota, M.; et al. Pim-1 kinase antagonizes aspects of myocardial hypertrophy and compensation to pathological pressure overload. Proc. Natl. Acad. Sci. USA 2008, 105, 13889–13894. [Google Scholar] [CrossRef]





| MicroRNA | Target | Cell Type | Result | Ref |
|---|---|---|---|---|
| MiR-328 | Pim-1 | Pulmonary artery smooth muscle cells. Human umbilical vein endothelial cells | Overexpression of MiR-328 increases the level of Pim-1 and promotes proliferation. Inhibition of MiRNA-328 promotes angiogenesis in high glucose conditions by increasing Pim-1 expression. | [46,47] |
| MiR-214 | Pim-1 | Bone marrow-derived mesenchymal stem cells Vascular smooth muscle cells | PDGF increases the expression of this MiRNA, resulting in Pim-1 knockdown. | [32] |
| MiR-206 | Pim-1 | Mesenchymal stem cells in hypoxia Pulmonary artery smooth muscle cells. | Reduction in miR-206 causes increased proliferation and reduced apoptosis, which is protective in infarcted hearts. | [36,48] |
| Mir-1 | Pim-1 | Vascular smooth muscle cells | Myocardin increases the expression of Mir-1, which results in the inhibition of VSMC proliferation via the knockdown of Pim-1. | [49] |
| miR 410-5-p | Pim-1 | Diabetic cardiomyopathy | MiR-410-5p inhibition decreases high glucose-induced myocardial apoptosis, preventing cardiomyopathy through the upregulation of Pim-1. | [50] |
| MiR-195 | Pim-1 | Sepsis—endothelial cells | Inhibition of miR-195 results in increased Pim-1, which is protective in a model of sepsis. | [51,52] |
| Pim-1 | Pim-2 | Pim-3 | |
|---|---|---|---|
| Isoforms | 44 kDa 34 kDa [42,64] | 34 kDa 37 kDa 40 kDa [7,61,73] | 34 kDa [78] |
| Isoform differences | Preferential of targets as opposed to others for Pim-1L and Pim-1S [66] such as Pim-1S favouring the androgen receptor [81] and Pim-1L phosphorylating Etk [65] | Largest isoform less active [7,61] | N/A |
| Protein localisation | Pim-1L plasma membrane Pim-1S cytosolic [65] | Cytoplasmic [61] | Cytoplasm [82] |
| Chromosome [83] | 6 [63] | X [83] | 22 [83] |
| Sequence similarity [24,78] | Pim-1 Pim-2 66%, Pim-1 Pim-3 77% [82], Pim-2 Pim-3 44% (Figure 2) | ||
| Structural differences | C-terminal helix missing in Pim-2 Six proline residues in the last structure [24]. | ||
| Transcriptional regulation differences | NF-KappaB [84]. HOX9A upregulates Pim-1 [85] | Not dependent on NF-KappaB, but Sp1 and Ets-1 [86]. HOX9A downregulates Pim-3 [87]. | |
| Degradation | Ubiquitination [59,60,62] | Ubiquitin independent in normoxia [61]. Under hypoxia, ubiquitination takes place [62] | Believed to be Ubiquitinated similarly to Pim-1 [62] |
| Consensus peptide sequence (AKRRRRHPSGPPTA) binding affinity [31] | 40–60 nM | 640 nmol/L | 40–60 nM |
| Kd for consensus sequence [31] | 0.058 µM | 0.64 µM | 0.039 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nock, S.; Karim, E.; Unsworth, A.J. Pim Kinases: Important Regulators of Cardiovascular Disease. Int. J. Mol. Sci. 2023, 24, 11582. https://doi.org/10.3390/ijms241411582
Nock S, Karim E, Unsworth AJ. Pim Kinases: Important Regulators of Cardiovascular Disease. International Journal of Molecular Sciences. 2023; 24(14):11582. https://doi.org/10.3390/ijms241411582
Chicago/Turabian StyleNock, Sophie, Eima Karim, and Amanda J. Unsworth. 2023. "Pim Kinases: Important Regulators of Cardiovascular Disease" International Journal of Molecular Sciences 24, no. 14: 11582. https://doi.org/10.3390/ijms241411582
APA StyleNock, S., Karim, E., & Unsworth, A. J. (2023). Pim Kinases: Important Regulators of Cardiovascular Disease. International Journal of Molecular Sciences, 24(14), 11582. https://doi.org/10.3390/ijms241411582