
Multi-Drug Resistance in Bacterial Genomes—A Comprehensive Bioinformatic Analysis

 , , and
, , and 
Abstract
1. Introduction
2. Results
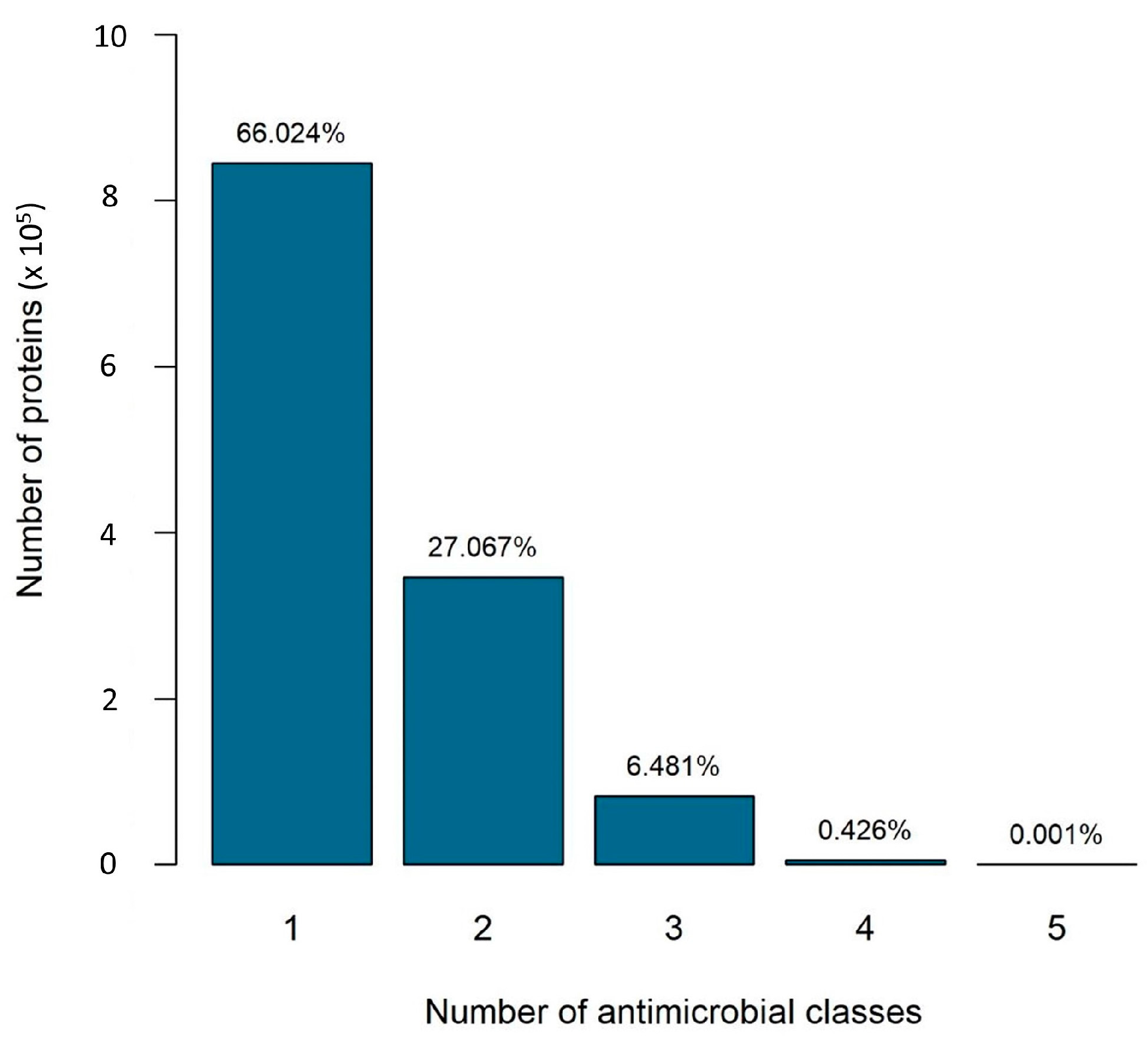
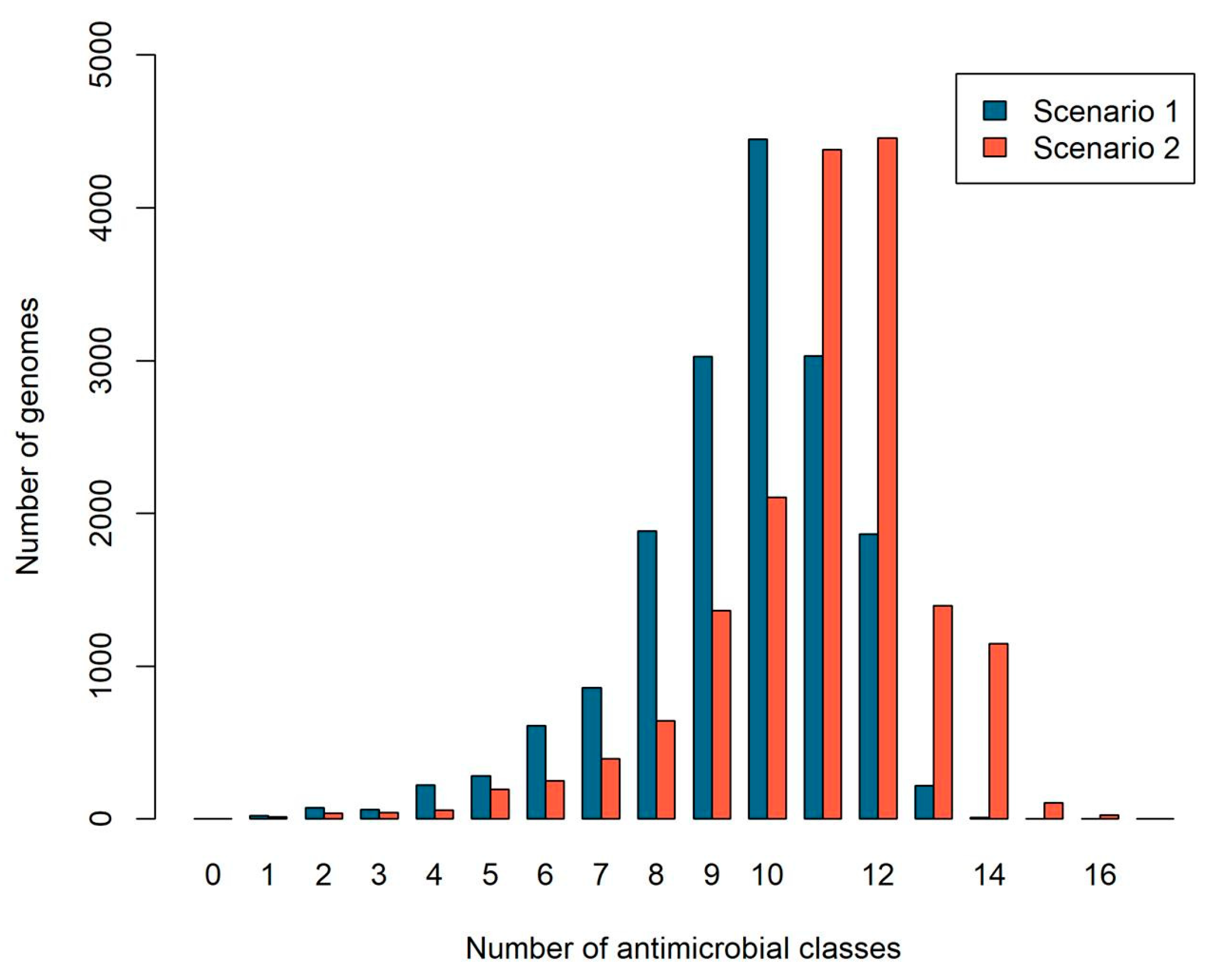
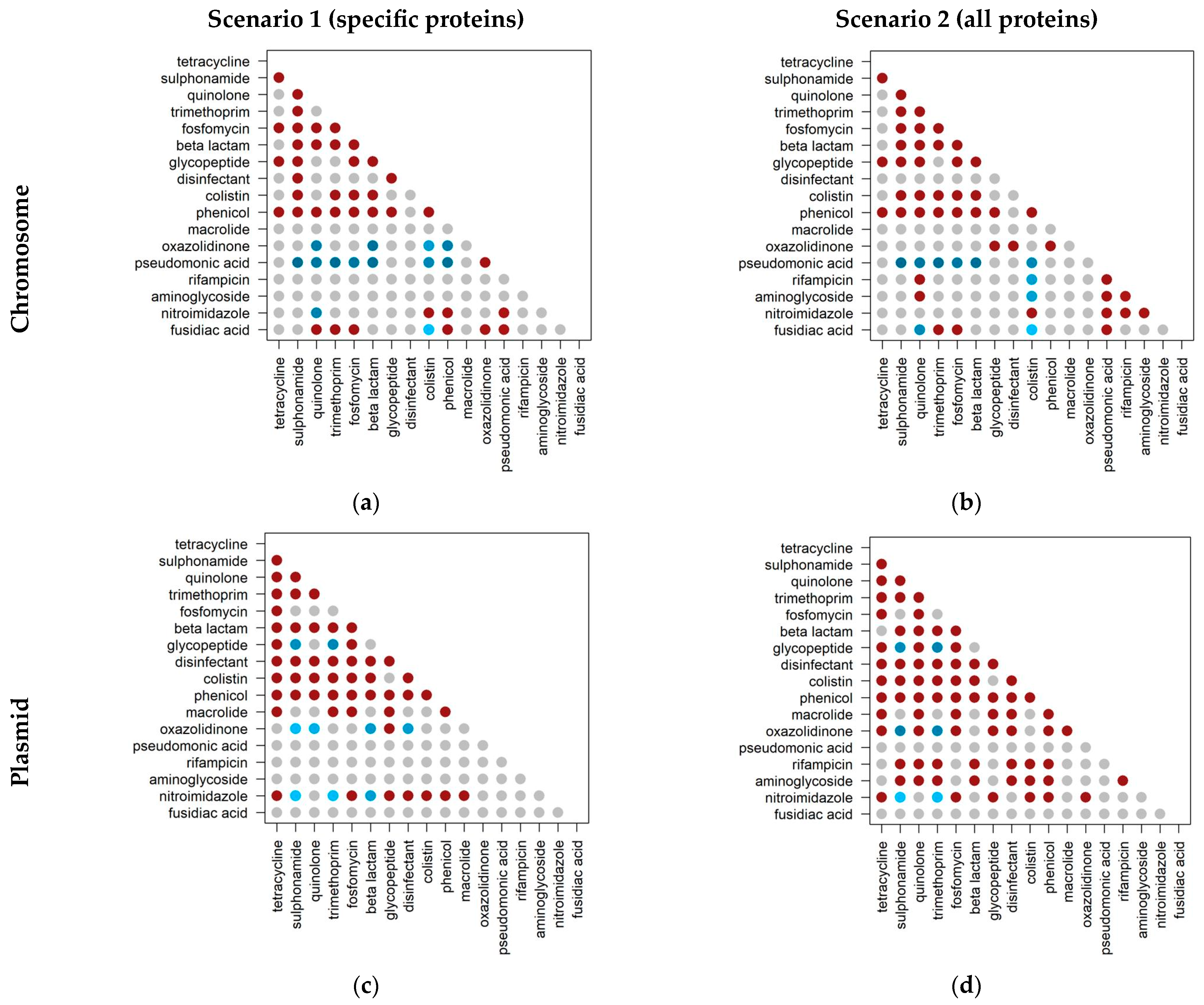
2.1. Specific versus Nonspecific Proteins
- Scenario 1 considers specific proteins: those that confer resistance to a single antibiotic resistance class;
- Scenario 2 considers all proteins: those that confer resistance to one or more classes.
2.2. The Number of Antibiotic Resistance Classes in Bacterial Genomes
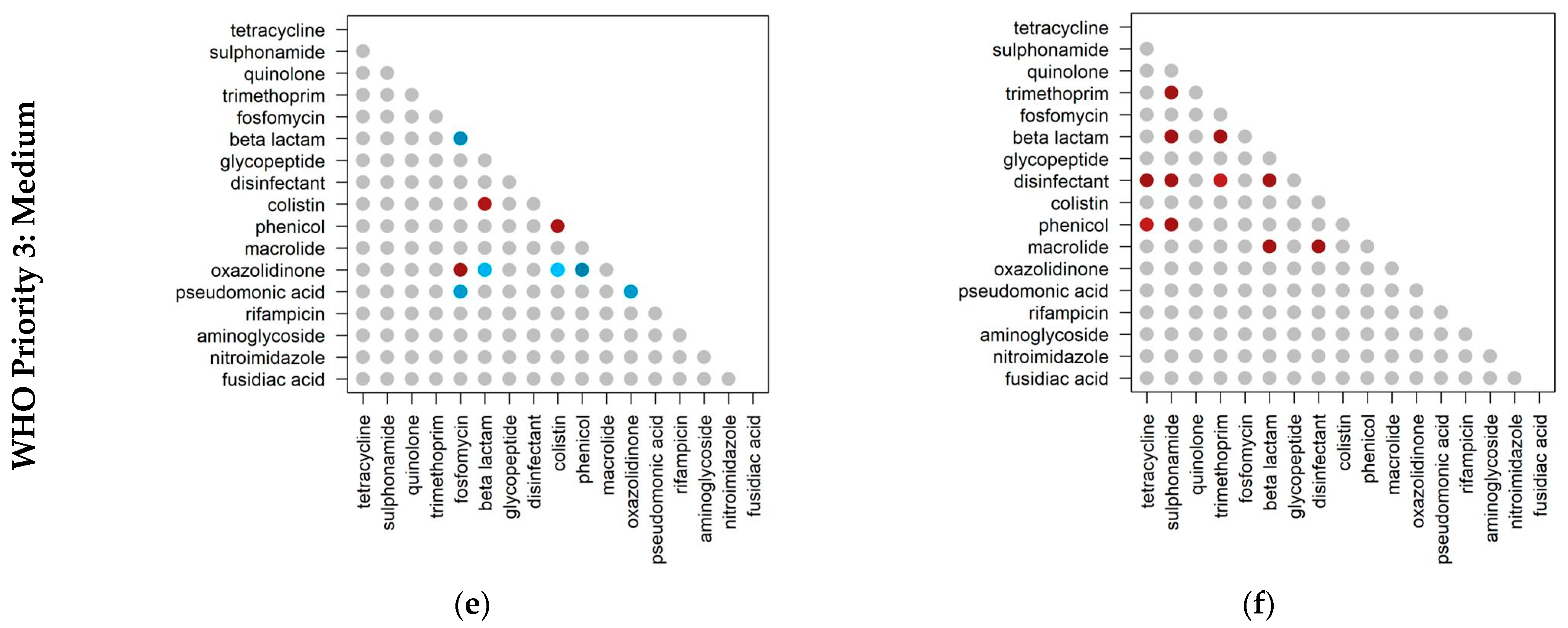
2.3. Co-Occurrence of Antibiotic Resistance Classes
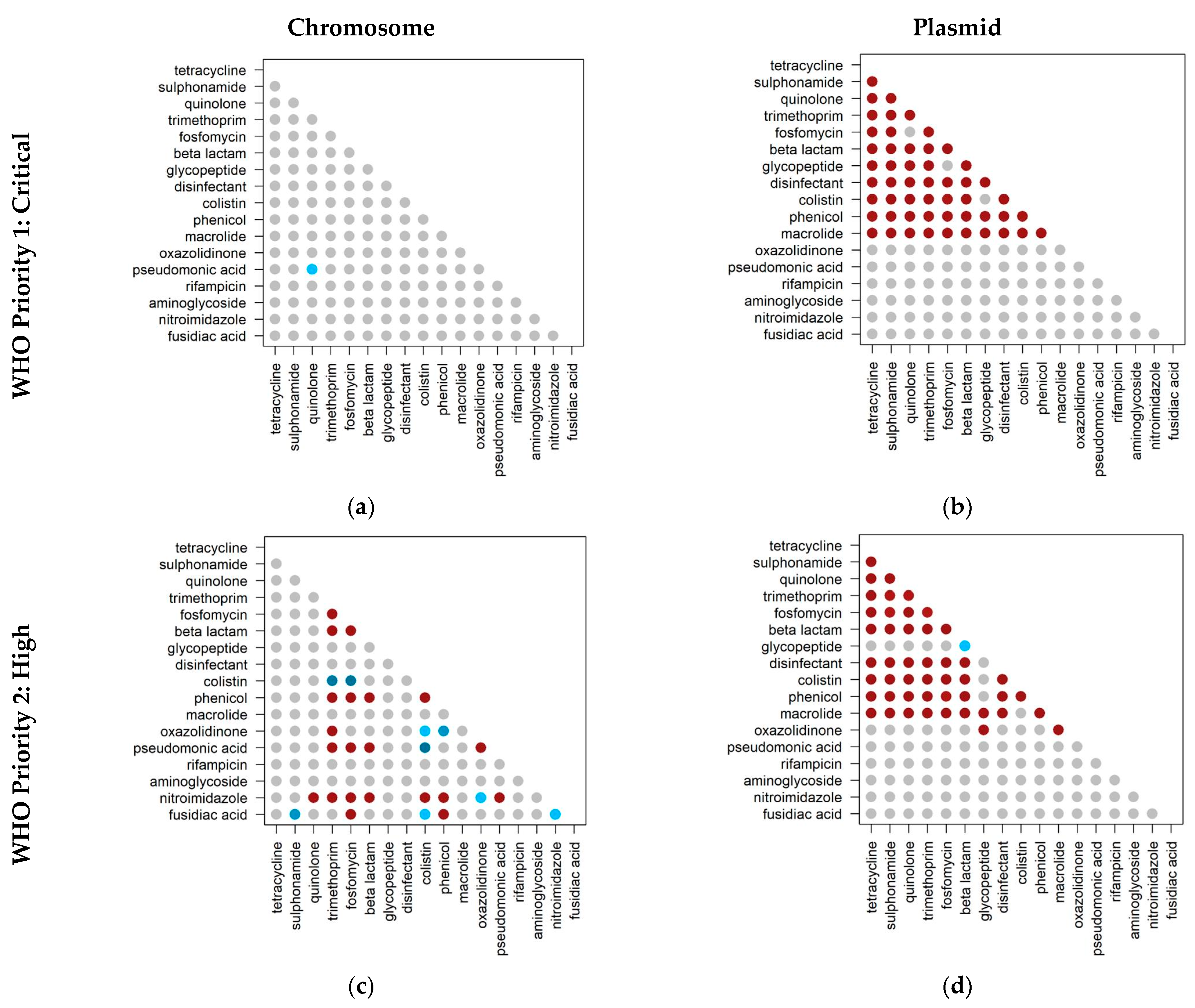
2.4. Co-Occurrence of Antibiotic Resistance Classes in Antibiotic-Resistant Priority Pathogens
3. Discussion
4. Materials and Methods
4.1. Dataset
- Download the FASTA format files of the protein sequence of all complete bacterial genomes present in the National Center for Biotechnology Information (NCBI) Reference Sequence (RefSeq) database—16,622 genomes (from https://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/, accessed on 19 October 2020).
- Format the protein sequence FASTA files belonging to the ResFinder database in the bash shell, using the BLAST manual as guidance. ResFinder is a comprehensive database divided into 17 classes of acquired genes and chromosomal mutations associated with antimicrobial resistance. The database was downloaded from https://bitbucket.org/genomicepidemiology/resfinder_db.git, and accessed on 30 October 2020 [44].
- Running BLASTP using a bash script, and non-default parameters. qcov was set to 0.6 for coverage, the e-value was set to 1 × 10−5 and the output format was set to ‘6’, which gives a tabular format to the BLAST results. The BLAST+ executable package (version ncbi-blast-2.9.0+) was downloaded from the NCBI website (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+, accessed on 11 November 2019) [45].
- A post-BLAST filter was applied to filter out hits with at least 30% identity. Each alignment that passed all filters was then divided into three categories based on its genomic location: (i) exclusively on the plasmid; (ii) exclusively on the chromosome; or (iii) both on the plasmid and on the chromosome.
4.2. Co-Occurrence Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adedeji, W.A. The Treasure Called Antibiotics. Ann. Ib. Postgrad. Med. 2016, 14, 56. [Google Scholar] [PubMed]
- WHO. New Report Calls for Urgent-Action to Avert Antimicrobial Resistance Crisis; WHO: Geneva, Switzerland, 2019.
- Zhu, G.; Wang, X.; Yang, T.; Su, J.; Qin, Y.; Wang, S.; Gillings, M.; Wang, C.; Ju, F.; Lan, B.; et al. Air Pollution Could Drive Global Dissemination of Antibiotic Resistance Genes. ISME J. 2021, 15, 270–281. [Google Scholar] [CrossRef]
- Sib, E.; Voigt, A.M.; Wilbring, G.; Schreiber, C.; Faerber, H.A.; Skutlarek, D.; Parcina, M.; Mahn, R.; Wolf, D.; Brossart, P.; et al. Antibiotic Resistant Bacteria and Resistance Genes in Biofilms in Clinical Wastewater Networks. Int. J. Hyg. Environ. Health 2019, 222, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Hatosy, S.M.; Martiny, A.C. The Ocean as a Global Reservoir of Antibiotic Resistance Genes. Appl. Env. Microbiol 2015, 81, 7593–7599. [Google Scholar] [CrossRef]
- Tacão, M.; Correia, A.; Henriques, I.S. Low Prevalence of Carbapenem-Resistant Bacteria in River Water: Resistance Is Mostly Related to Intrinsic Mechanisms. Microb. Drug Resist. 2015, 21, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, T.; Botelho, A. Metagenomics and Other Omics Approaches to Bacterial Communities and Antimicrobial Resistance Assessment in Aquacultures. Antibiotics 2021, 10, 787. [Google Scholar] [CrossRef]
- Silva, D.G.; Domingues, C.P.F.; Figueiredo, J.F.; Dionisio, F.; Botelho, A.; Nogueira, T. Estuarine Aquacultures at the Crossroads of Animal Production and Antibacterial Resistance: A Metagenomic Approach to the Resistome. Biology 2022, 11, 1681. [Google Scholar] [CrossRef]
- Wang, F.; Fu, Y.-H.; Sheng, H.-J.; Topp, E.; Jiang, X.; Zhu, Y.-G.; Tiedje, J.M. Antibiotic Resistance in the Soil Ecosystem: A One Health Perspective. Curr. Opin. Environ. Sci. Health 2021, 20, 100230. [Google Scholar] [CrossRef]
- Li, M.; Li, Z.; Zhong, Q.; Liu, J.; Han, G.; Li, Y.; Li, C. Antibiotic Resistance of Fecal Carriage of Escherichia Coli from Pig Farms in China: A Meta-Analysis. Env. Sci. Pollut. Res. 2022, 29, 22989–23000. [Google Scholar] [CrossRef]
- Periasamy, D.; Sundaram, A. A Novel Approach for Pathogen Reduction in Wastewater Treatment. J. Env. Health Sci. Eng. 2013, 11, 12. [Google Scholar] [CrossRef]
- Woolhouse, M.; Ward, M.; van Bunnik, B.; Farrar, J. Antimicrobial Resistance in Humans, Livestock and the Wider Environment. Phil. Trans. R. Soc. B 2015, 370, 20140083. [Google Scholar] [CrossRef]
- Debroas, D.; Siguret, C. Viruses as Key Reservoirs of Antibiotic Resistance Genes in the Environment. ISME J. 2019, 13, 2856–2867. [Google Scholar] [CrossRef] [PubMed]
- Shousha, A.; Awaiwanont, N.; Sofka, D.; Smulders, F.J.M.; Paulsen, P.; Szostak, M.P.; Humphrey, T.; Hilbert, F. Bacteriophages Isolated from Chicken Meat and the Horizontal Transfer of Antimicrobial Resistance Genes. Appl. Env. Microbiol. 2015, 81, 4600–4606. [Google Scholar] [CrossRef] [PubMed]
- Baker-Austin, C.; Wright, M.S.; Stepanauskas, R.; McArthur, J.V. Co-Selection of Antibiotic and Metal Resistance. Trends Microbiol. 2006, 14, 176–182. [Google Scholar] [CrossRef]
- Escudeiro, P.; Pothier, J.; Dionisio, F.; Nogueira, T. Antibiotic Resistance Gene Diversity and Virulence Gene Diversity Are Correlated in Human Gut and Environmental Microbiomes. mSphere 2019, 4, e00135-19. [Google Scholar] [CrossRef]
- Mazhar, S.H.; Li, X.; Rashid, A.; Su, J.; Xu, J.; Brejnrod, A.D.; Su, J.-Q.; Wu, Y.; Zhu, Y.-G.; Zhou, S.G.; et al. Co-Selection of Antibiotic Resistance Genes, and Mobile Genetic Elements in the Presence of Heavy Metals in Poultry Farm Environments. Sci. Total Environ. 2021, 755, 142702. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Shen, Q.; Liu, F.; Ma, J.; Xu, G.; Wang, Y.; Wu, M. Antibiotic Resistance Gene Abundances Associated with Antibiotics and Heavy Metals in Animal Manures and Agricultural Soils Adjacent to Feedlots in Shanghai; China. J. Hazard. Mater. 2012, 235–236, 178–185. [Google Scholar] [CrossRef]
- Song, J.; Rensing, C.; Holm, P.E.; Virta, M.; Brandt, K.K. Comparison of Metals and Tetracycline as Selective Agents for Development of Tetracycline Resistant Bacterial Communities in Agricultural Soil. Environ. Sci. Technol. 2017, 51, 3040–3047. [Google Scholar] [CrossRef]
- Icgen, B.; Yilmaz, F. Co-Occurrence of Antibiotic and Heavy Metal Resistance in Kızılırmak River Isolates. Bull Env. Contam Toxicol. 2014, 93, 735–743. [Google Scholar] [CrossRef]
- Amsaveni, R.; Sureshkumar, M.; Mary, J.R.; Indra, U.; Vivekanandhan, G. Screening of Multi-Drug and Metal Resistant Aeromonas Species from Diverse Sources. Am. J. Infect. Dis. 2015, 11, 41–47. [Google Scholar] [CrossRef]
- Domingues, C.P.F.; Rebelo, J.S.; Pothier, J.; Monteiro, F.; Nogueira, T.; Dionisio, F. The Perfect Condition for the Rising of Superbugs: Person-to-Person Contact and Antibiotic Use Are the Key Factors Responsible for the Positive Correlation between Antibiotic Resistance Gene Diversity and Virulence Gene Diversity in Human Metagenomes. Antibiotics 2021, 10, 605. [Google Scholar] [CrossRef]
- Dionisio, F.; Domingues, C.P.F.; Rebelo, J.S.; Monteiro, F.; Nogueira, T. The Impact of Non-Pathogenic Bacteria on the Spread of Virulence and Resistance Genes. IJMS 2023, 24, 1967. [Google Scholar] [CrossRef] [PubMed]
- Darmancier, H.; Domingues, C.P.F.; Rebelo, J.S.; Amaro, A.; Dionísio, F.; Pothier, J.; Serra, O.; Nogueira, T. Are Virulence and Antibiotic Resistance Genes Linked? A Comprehensive Analysis of Bacterial Chromosomes and Plasmids. Antibiotics 2022, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Kiyaga, S.; Kyany’a, C.; Muraya, A.W.; Smith, H.J.; Mills, E.G.; Kibet, C.; Mboowa, G.; Musila, L. Genetic Diversity, Distribution, and Genomic Characterization of Antibiotic Resistance and Virulence of Clinical Pseudomonas Aeruginosa Strains in Kenya. Front. Microbiol. 2022, 13, 835403. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Jiang, Y.; Zhou, J.; Yu, Y. A Global Perspective on the Convergence of Hypervirulence and Carbapenem Resistance in Klebsiella Pneumoniae. J. Glob. Antimicrob. Resist. 2021, 25, 26–34. [Google Scholar] [CrossRef]
- Coburn, P.S.; Baghdayan, A.S.; Dolan, G.; Shankar, N. Horizontal Transfer of Virulence Genes Encoded on the Enterococcus Faecalis Pathogenicity Island: Transfer of PAI-Encoded Virulence Genes in E. Faecalis. Mol. Microbiol. 2007, 63, 530–544. [Google Scholar] [CrossRef]
- Kumarasamy, K.K.; Toleman, M.A.; Walsh, T.R.; Bagaria, J.; Butt, F.; Balakrishnan, R.; Chaudhary, U.; Doumith, M.; Giske, C.G.; Irfan, S.; et al. Emergence of a New Antibiotic Resistance Mechanism in India, Pakistan, and the UK: A Molecular, Biological, and Epidemiological Study. Lancet Infect Dis. 2010, 10, 597–602. [Google Scholar] [CrossRef]
- Aarestrup, F.M.; Seyfarth, A.M.; Emborg, H.-D.; Pedersen, K.; Hendriksen, R.S.; Bager, F. Effect of Abolishment of the Use of Antimicrobial Agents for Growth Promotion on Occurrence of Antimicrobial Resistance in Fecal Enterococci from Food Animals in Denmark. Antimicrob. Agents Chemother. 2001, 45, 2054–2059. [Google Scholar] [CrossRef]
- Li, X.; Brejnrod, A.; Trivedi, U.; Russel, J.; Stokholm, J.; Thorsen, J.; Shah, S.A.; Vestergaard, G.A.; Rasmussen, M.A.; Nesme, J.; et al. Co-Localization of Antibiotic Resistance Genes Is Widespread in the Infant Gut Microbiome and Associates with an Immature Gut Microbial Composition. bioRxiv 2023. [Google Scholar] [CrossRef]
- Martiny, H.-M.; Munk, P.; Brinch, C.; Aarestrup, F.M.; Calle, M.L.; Petersen, T.N. Utilizing Co-Abundances of Antimicrobial Resistance Genes to Identify Potential Co-Selection in the Resistome. bioRxiv 2022. [Google Scholar] [CrossRef]
- Colinon, C.; Miriagou, V.; Carattoli, A.; Luzzaro, F.; Rossolini, G.M. Characterization of the IncA/C Plasmid PCC416 Encoding VIM-4 and CMY-4 β-Lactamases. J. Antimicrob. Chemother. 2007, 60, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids Carrying Antimicrobial Resistance Genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef]
- Carattoli, A.; Seiffert, S.N.; Schwendener, S.; Perreten, V.; Endimiani, A. Differentiation of IncL and IncM Plasmids Associated with the Spread of Clinically Relevant Antimicrobial Resistance. PLoS ONE 2015, 10, e0123063. [Google Scholar] [CrossRef]
- San Millan, A. Evolution of Plasmid-Mediated Antibiotic Resistance in the Clinical Context. Trends Microbiol. 2018, 26, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A. Plasmids and the Spread of Resistance. Int. J. Med. Microbiol. 2013, 303, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Daghrir, R.; Drogui, P. Tetracycline Antibiotics in the Environment: A Review. Env. Chem. Lett. 2013, 11, 209–227. [Google Scholar] [CrossRef]
- Wright, G. Bacterial Resistance to Antibiotics: Enzymatic Degradation and Modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef]
- Domingues, I.L.; Gama, J.A.; Carvalho, L.M.; Dionisio, F. Social Behaviour Involving Drug Resistance: The Role of Initial Density, Initial Frequency and Population Structure in Shaping the Effect of Antibiotic Resistance as a Public Good. Heredity 2017, 119, 295–301. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial Persistence as a Phenotypic Switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef]
- Levin, B.R.; Concepción-Acevedo, J.; Udekwu, K.I. Persistence: A Copacetic and Parsimonious Hypothesis for the Existence of Non-Inherited Resistance to Antibiotics. Curr. Opin. Microbiol. 2014, 21, 18–21. [Google Scholar] [CrossRef]
- Rebelo, J.S.; Domingues, C.P.; Monteiro, F.; Nogueira, T.; Dionisio, F. Bacterial Persistence Is Essential for Susceptible Cell Survival in Indirect Resistance, Mainly for Lower Cell Densities. PLoS ONE 2021, 16, e0246500. [Google Scholar] [CrossRef] [PubMed]
- Dionisio, F.; Baquero, F.; Fuertes, M. Psychological and Cultural Factors Influencing Antibiotic Prescription. Trends Microbiol. 2023, 31, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A Better Web Interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resistance Classes | Scenario 1 (Specific Proteins) Number of Genomes | Scenario 2 (All Proteins) Number of Genomes |
|---|---|---|
| Aminoglycoside | 0 | 887 |
| β-lactam | 13,200 | 13,200 |
| Colistin | 5993 | 5993 |
| Disinfectant | 16,363 | 16,492 |
| Fosfomycin | 11,306 | 11,306 |
| Fusidic acid | 1065 | 1065 |
| Glycopeptide | 16,101 | 16,101 |
| Macrolide | 16,610 | 16,613 |
| Nitroimidazole | 2143 | 2143 |
| Oxazolidinone | 3691 | 16,445 |
| Phenicol | 12,682 | 15,657 |
| Pseudomonic acid | 7946 | 7946 |
| Quinolone | 5223 | 11,640 |
| Rifampicin | 0 | 887 |
| Sulphonamide | 14,940 | 14,940 |
| Tetracycline | 16,193 | 16,338 |
| Trimethoprim | 14,416 | 14,416 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingues, C.P.F.; Rebelo, J.S.; Dionisio, F.; Nogueira, T. Multi-Drug Resistance in Bacterial Genomes—A Comprehensive Bioinformatic Analysis. Int. J. Mol. Sci. 2023, 24, 11438. https://doi.org/10.3390/ijms241411438
Domingues CPF, Rebelo JS, Dionisio F, Nogueira T. Multi-Drug Resistance in Bacterial Genomes—A Comprehensive Bioinformatic Analysis. International Journal of Molecular Sciences. 2023; 24(14):11438. https://doi.org/10.3390/ijms241411438
Chicago/Turabian StyleDomingues, Célia P. F., João S. Rebelo, Francisco Dionisio, and Teresa Nogueira. 2023. "Multi-Drug Resistance in Bacterial Genomes—A Comprehensive Bioinformatic Analysis" International Journal of Molecular Sciences 24, no. 14: 11438. https://doi.org/10.3390/ijms241411438
APA StyleDomingues, C. P. F., Rebelo, J. S., Dionisio, F., & Nogueira, T. (2023). Multi-Drug Resistance in Bacterial Genomes—A Comprehensive Bioinformatic Analysis. International Journal of Molecular Sciences, 24(14), 11438. https://doi.org/10.3390/ijms241411438