
Roles of Lipopolysaccharide Glycosyltransferases in Maintenance of Helicobacter pylori Morphology, Cell Wall Permeability, and Antimicrobial Susceptibilities

 ,
,
Abstract
1. Introduction
2. Results
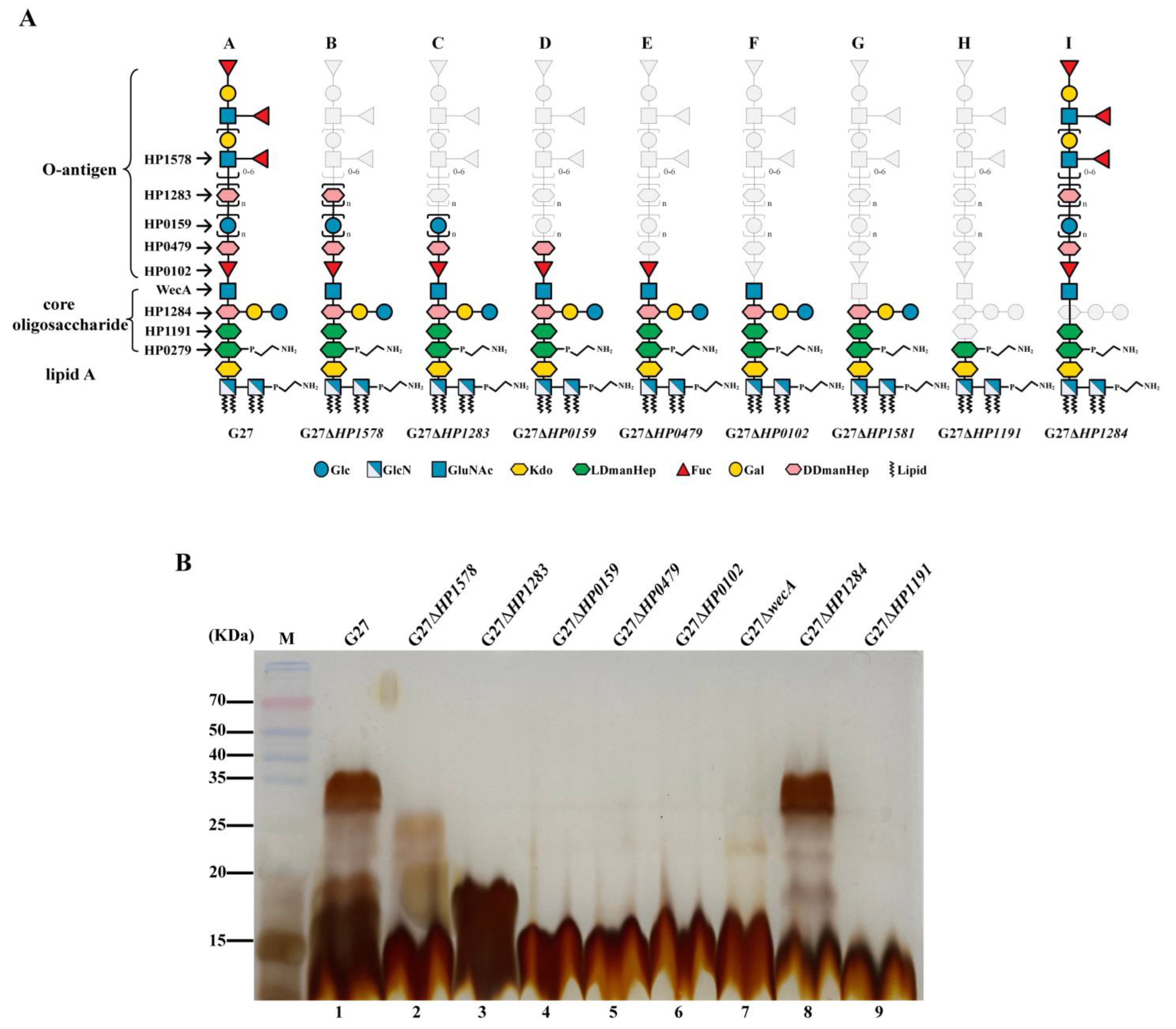
2.1. Deletion of LPS Glycosyltransferase Genes except for HP0279 Leads to Various Degree of H. pylori LPS Truncation, but Does Not Interfere with Bacterial Fitness
2.2. Deletion of LPS Glycosyltransferase Genes Affects the Spiral and Rod-like Morphology
2.3. Deletion of LPS Glycosyltransferase Genes Affects H. pylori Sensitivity to Polymyxin B
2.4. Deletion of LPS Glycosyltransferase Genes Does Not Affect H. pylori Outer Membrane Permeability, but Increases Bacteria Sensitivity to Rifampicin
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, Growth Conditions, and Oligonucleotide Primers
4.2. Construction of wecA Gene Mutant in H. pylori 26695, J99, and P12
4.3. H. pylori LPS Microextraction for Silver Staining
4.4. Morphology Analysis of H. pylori Strains by Gram-Staining
4.5. Growth Analysis of H. pylori Strains
4.6. Outer Membrane Permeability Assay
4.7. Antimicrobial Susceptibility Testing
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ren, S.; Cai, P.; Liu, Y.; Wang, T.; Zhang, Y.; Li, Q.; Gu, Y.; Wei, L.; Yan, C.; Jin, G. Prevalence of Helicobacter pylori infection in China: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2021, 37, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Neddermann, M.; Maubach, G.; Naumann, M. Pathogenesis of Helicobacter pylori infection. Helicobacter 2016, 21 (Suppl. S1), 19–25. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Chey, W.D.; Leontiadis, G.I.; Howden, C.W.; Moss, S.F. ACG Clinical Guideline: Treatment of Helicobacter pylori Infection. Am. J. Gastroenterol. 2017, 112, 212–239, Erratum in Am. J. Gastroenterol. 2018, 113, 1102. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Megraud, F.; Rokkas, T.; Gisbert, J.P.; Liou, J.-M.; Schulz, C.; Gasbarrini, A.; Hunt, R.H.; Leja, M.; O’Morain, C.; et al. Management of Helicobacter pylori infection: The Maastricht VI/Florence consensus report. Gut 2022, 71, 1724–1762. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kao, J.Y.; Kanwal, F.; Gilger, M.; LoVecchio, F.; Moss, S.F.; Crowe, S.; Elfant, A.; Haas, T.; Hapke, R.J.; et al. Houston Consensus Conference on Testing for Helicobacter pylori Infection in the United States. Clin. Gastroenterol. Hepatol. 2018, 16, 992–1002.e6. [Google Scholar] [CrossRef]
- Silva, L.M.; Correia, V.G.; Moreira, A.S.; Domingues, M.R.M.; Ferreira, R.M.; Figueiredo, C.; Azevedo, N.F.; Marcos-Pinto, R.; Carneiro, F.; Magalhães, A.; et al. Helicobacter pylori lipopolysaccharide structural domains and their recognition by immune proteins revealed with carbohydrate microarrays. Carbohydr. Polym. 2021, 253, 117350. [Google Scholar] [CrossRef]
- Li, H.; Liao, T.; Debowski, A.W.; Tang, H.; Nilsson, H.O.; Stubbs, K.A.; Marshall, B.J.; Benghezal, M. Lipopolysaccharide Structure and Biosynthesis in Helicobacter pylori. Helicobacter 2016, 21, 445–461. [Google Scholar] [CrossRef]
- Simpson, B.W.; Trent, M.S. Pushing the envelope: LPS modifications and their consequences. Nat. Rev. Genet. 2019, 17, 403–416. [Google Scholar] [CrossRef]
- Cullen, T.W.; Giles, D.K.; Wolf, L.N.; Ecobichon, C.; Boneca, I.G.; Trent, M.S. Helicobacter pylori versus the Host: Remodeling of the Bacterial Outer Membrane Is Required for Survival in the Gastric Mucosa. PLoS Pathog. 2011, 7, e1002454. [Google Scholar] [CrossRef]
- Sijmons, D.; Guy, A.J.; Walduck, A.K.; Ramsland, P.A. Helicobacter pylori and the Role of Lipopolysaccharide Variation in Innate Immune Evasion. Front. Immunol. 2022, 13, 868225. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, T.; Liao, T.; Debowski, A.W.; Nilsson, H.-O.; Fulurija, A.; Haslam, S.M.; Mulloy, B.; Dell, A.; Stubbs, K.A.; et al. The redefinition of Helicobacter pylori lipopolysaccharide O-antigen and core-oligosaccharide domains. PLoS Pathog. 2017, 13, e1006280. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, K.; Logan, S.M.; Conlan, J.W.; Chandan, V.; Aubry, A.; Smirnova, N.; Ulrichsen, H.; Chan, K.H.; Griffith, D.W.; Harrison, B.A.; et al. Identification of a D-glycero-D-manno-heptosyltransferase gene from Helicobacter pylori. J. Bacteriol. 2005, 187, 5156–5165. [Google Scholar] [CrossRef] [PubMed]
- Chandan, V.; Logan, S.M.; Harrison, B.A.; Vinogradov, E.; Aubry, A.; Stupak, J.; Li, J.; Altman, E. Characterization of a waaF mutant of Helicobacter pylori strain 26695 provides evidence that an extended lipopolysaccharide structure has a limited role in the invasion of gastric cancer cells. Biochem. Cell Biol. 2007, 85, 582–590. [Google Scholar] [CrossRef]
- Altman, E.; Chandan, V.; Li, J.; Vinogradov, E. Lipopolysaccharide structures of Helicobacter pylori wild-type strain 26695 and 26695 HP0826: Kan mutant devoid of the O-chain polysaccharide component. Carbohydr. Res. 2011, 346, 2437–2444. [Google Scholar] [CrossRef]
- Altman, E.; Chandan, V.; Li, J.; Vinogradov, E. Lipopolysaccharide structure of Helicobacter pylori serogroup O:3. Carbohydr. Res. 2013, 378, 139–143. [Google Scholar] [CrossRef]
- Li, H.; Marceau, M.; Yang, T.; Liao, T.; Tang, X.; Hu, R.; Xie, Y.; Tang, H.; Tay, A.; Shi, Y.; et al. East-Asian Helicobacter pylori strains synthesize heptan-deficient lipopolysaccharide. PLoS Genet. 2019, 15, e1008497. [Google Scholar] [CrossRef]
- Moran, A.P.; Shiberu, B.; Ferris, J.A.; Knirel, Y.A.; Senchenkova, S.N.; Perepelov, A.V.; Jansson, P.E.; Goldberg, J.B. Role of Helicobacter pylori rfaJ genes (HP0159 and HP1416) in lipopolysaccharide synthesis. FEMS Microbiol. Lett. 2004, 241, 57–65. [Google Scholar] [CrossRef]
- Mobley, H.L.T.; Mendz, G.L.; Hazell, S.L. (Eds.) Helicobacter Pylori: Physiology and Genetics; ASM Press: Washington, DC, USA, 2001. [Google Scholar]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef]
- Pernitzsch, S.R.; Alzheimer, M.; Bremer, B.U.; Robbe-Saule, M.; De Reuse, H.; Sharma, C.M. Small RNA mediated gradual control of lipopolysaccharide biosynthesis affects antibiotic resistance in Helicobacter pylori. Nat. Commun. 2021, 12, 4433. [Google Scholar] [CrossRef]
- Matsuura, M. Structural Modifications of Bacterial Lipopolysaccharide that Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front. Immunol. 2013, 4, 109. [Google Scholar] [CrossRef] [PubMed]
- Holtrup, S.; Greger, M.; Mayer, B.; Specht, M.; Waidner, B. Insights Into the Helical Shape Complex of Helicobacter pylori. Front. Microbiol. 2022, 13, 929194. [Google Scholar] [CrossRef] [PubMed]
- Waidner, B.; Specht, M.; Dempwolff, F.; Haeberer, K.; Schaetzle, S.; Speth, V.; Kist, M.; Graumann, P.L. A Novel System of Cytoskeletal Elements in the Human Pathogen Helicobacter pylori. PLoS Pathog. 2009, 5, e1000669. [Google Scholar] [CrossRef]
- Specht, M.; Schätzle, S.; Graumann, P.L.; Waidner, B. Helicobacter pyloriPossesses Four Coiled-Coil-Rich Proteins That Form Extended Filamentous Structures and Control Cell Shape and Motility. J. Bacteriol. 2011, 193, 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Cabeen, M.T.; Murolo, M.A.; Briegel, A.; Bui, N.K.; Vollmer, W.; Ausmees, N.; Jensen, G.J.; Jacobs-Wagner, C. Mutations in the Lipopolysaccharide Biosynthesis Pathway Interfere with Crescentin-Mediated Cell Curvature in Caulobacter crescentus. J. Bacteriol. 2010, 192, 3368–3378. [Google Scholar] [CrossRef] [PubMed]
- Sycuro, L.K.; Pincus, Z.; Gutierrez, K.D.; Biboy, J.; Stern, C.A.; Vollmer, W.; Salama, N.R. Peptidoglycan crosslinking relaxation promotes Helicobacter pylori’s helical shape and stomach colonization. Cell 2010, 141, 822–833. [Google Scholar] [CrossRef]
- Salama, N.R. Cell morphology as a virulence determinant: Lessons from Helicobacter pylori. Curr. Opin. Microbiol. 2020, 54, 11–17. [Google Scholar] [CrossRef]
- Altman, E.; Chandan, V.; Larocque, S.; Aubry, A.; Logan, S.M.; Vinogradov, E.; Li, J. Effect of the HP0159 ORF mutation on the lipopolysaccharide structure and colonizing ability of Helicobacter pylori. FEMS Immunol. Med Microbiol. 2008, 53, 204–213. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Xu, J.; Huang, C.; Chen, J. Rcs Phosphorelay Responses to Truncated Lipopolysaccharide-Induced Cell Envelope Stress in Yersinia enterocolitica. Molecules 2020, 25, 5718. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Yethon, J.A.; Vinogradov, E.; Perry, M.B.; Whitfield, C. Mutation of the Lipopolysaccharide Core Glycosyltransferase Encoded by waaG Destabilizes the Outer Membrane of Escherichia coli by Interfering with Core Phosphorylation. J. Bacteriol. 2000, 182, 5620–5623. [Google Scholar] [CrossRef]
- Fernández, L.; Álvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Kocíncová, D.; Lam, J.S.; Martínez, J.L.; Hancock, R.E.W. Characterization of the Polymyxin B Resistome of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 110–119. [Google Scholar] [CrossRef]
- Lugtenberg, B.; Van, A.l.p.h.e.n.L. Molecular architecture and functioning of the outer membrane of Escherichia coli and other gram-negative bacteria. Biochim. Biophys. Acta 1983, 737, 51–115. [Google Scholar] [CrossRef] [PubMed]
- Trent, M.S.; Stead, C.M.; Tran, A.X.; Hankins, J.V. Diversity of endotoxin and its impact on pathogenesis. J. Endotoxin Res. 2006, 12, 205–223. [Google Scholar] [CrossRef]
- Chaudhary, N.; Aggarwal, B.; Saini, V.; Yavvari, P.S.; Sharma, P.; Srivastava, A.; Bajaj, A. Polyaspartate-derived synthetic antimicrobial polymer enhances the activity of rifampicin against multidrug-resistant Pseudomonas aeruginosa infections. Biomater. Sci. 2022, 10, 5158–5171. [Google Scholar] [CrossRef] [PubMed]
- Cetuk, H.; Anishkin, A.; Scott, A.J.; Rempe, S.B.; Ernst, R.K.; Sukharev, S. Partitioning of Seven Different Classes of Antibiotics into LPS Monolayers Supports Three Different Permeation Mechanisms through the Outer Bacterial Membrane. Langmuir ACS J. Surf. Colloids 2021, 37, 1372–1385. [Google Scholar] [CrossRef]
- Debowski, A.W.; Gauntlett, J.C.; Li, H.; Liao, T.; Sehnal, M.; Nilsson, H.-O.; Marshall, B.J.; Benghezal, M. Xer-cise inHelicobacter pylori: One-step Transformation for the Construction of Markerless Gene Deletions. Helicobacter 2012, 17, 435–443. [Google Scholar] [CrossRef]
- Luo, G.; Zhang, J.; Wang, H.; Sun, Y.; Cheng, B.; Xu, Z.; Zhang, Y.; Li, H.; Lu, W.; Nemeth, E.; et al. Human defensin-inspired discovery of peptidomimetic antibiotics. Proc. Natl. Acad. Sci. USA 2022, 119, e2117283119. [Google Scholar] [CrossRef]
- EUCAST Breakpoint Tables for Interpretation of MICs and Zone Diameters, Version 10.0. 2020. Available online: https://www.eucast.org/ (accessed on 1 March 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain or Plasmid Name | Description | LPS Biosynthesis | Reference or Source |
|---|---|---|---|
| H. pylori Strains | |||
| G27 | Wild-type strain, streptomycin resistant | Full-length LPS | [17] |
| G27ΔHP1578 | Clean deletion of HPG27_1515 in G27 | Deletion of Lewis antigen | [17] |
| G27ΔHP1283 | Clean deletion of HPG27_1235 in G27 | Deletion of Lewis antigen and heptan | [17] |
| G27ΔHP0159 | Clean deletion of HPG27_146 in G27 | Deletion of Lewis antigen, DD-heptan, and glucan | [17] |
| G27ΔHP0479 | Clean deletion of HPG27_437 in G27 | Deletion of Lewis antigen, DD-heptan, glucan, and LD-heptose of Trio | [17] |
| G27ΔHP0102 | Clean deletion of HPG27_94 in G27 | Deletion of Lewis antigen, DD-heptan, glucan, Trio LD-heptose and fucose | [17] |
| G27ΔwecA | Clean deletion of HPG27_1518 in G27 | Deletion of the entire O-antigen | [17] |
| G27ΔHP1284 | Clean deletion of HPG27_1236 in G27 | Deletion of side-chain and heptose III in the core oligosaccharide | [17] |
| G27ΔHP1191 | Clean deletion of HPG27_1136 in G27 | Deletion of the entire O-antigen, and side-chain, heptose III and heptose II in the core oligosaccharide | [17] |
| 26695 | Wild-type strain, streptomycin resistant | Full-length LPS | Stored in our lab |
| 26695ΔwecA | Clean deletion of HP1581 in 26695 | Deletion of the entire O-antigen | This work |
| J99 | Wild-type strain | Full-length LPS | Stored in our lab |
| J99ΔwecA | Deletion of wecA in J99 by replacing it with difH-rpsl-cat-difH | Deletion of the entire O-antigen | This work |
| P12 | Wild-type strain | Full-length LPS | Stored in our lab |
| P12ΔwecA | Deletion of wecA in P12 by replacing it with difH-rpsl-cat-difH | Deletion of the entire O-antigen | This work |
| Plasmids | |||
| pWecA-AB-difH-RC | pGEM®-T Easy vector containing sequences flanking HPG27_1518, with difH-rpsL-cat-difH inserted between the flanking sequences | [17] |
| Strains | Polymyxin B (μg/mL) | Rifampicin (μg/mL) | Clarithromycin (μg/mL) | Levofloxacin (μg/mL) | Amoxicillin (μg/mL) | Tetracycline (μg/mL) | Metronidazole (μg/mL) |
|---|---|---|---|---|---|---|---|
| G27 | 18.67 ± 7.05 | 6.67 ± 1.33 | 0.03 ± 0.01 | 0.23 ± 0.02 | 0.04 ± 0.01 | 0.27 ± 0.05 | 3.30 ± 0.33 |
| G27ΔHP1578 | 13.33 ± 2.67 | 1.83 ± 0.16 | 0.03 ± 0.01 | 0.18 ± 0.04 | 0.04 ± 0.01 | 0.27 ± 0.05 | 3.30 ± 0.33 |
| G27ΔHP1283 | 13.33 ± 2.67 | 1.42 ± 0.36 | 0.03 ± 0.01 | 0.21 ± 0.02 | 0.03 ± 0.01 | 0.27 ± 0.05 | 3.30 ± 0.33 |
| G27ΔHP0159 | 2.00 ± 0.00 | 2.50 ± 0.76 | 0.02 ± 0.01 | 0.19 ± 0.04 | 0.04 ± 0.01 | 0.23 ± 0.02 | 3.00 ± 0.57 |
| G27ΔHP0479 | 1.50 ± 0.50 | 0.83 ± 0.08 | 0.02 ± 0.01 | 0.17 ± 0.02 | 0.03 ± 0.00 | 0.27 ± 0.05 | 3.00 ± 0.57 |
| G27ΔHP0102 | 6.67 ± 1.33 | 0.66 ± 0.08 | 0.03 ± 0.01 | 0.17 ± 0.02 | 0.04 ± 0.01 | 0.27 ± 0.05 | 3.33 ± 0.33 |
| G27ΔwecA | 0.96 ± 0.52 | 0.42 ± 0.04 | 0.02 ± 0.01 | 0.21 ± 0.02 | 0.03 ± 0.00 | 0.27 ± 0.05 | 3.33 ± 0.66 |
| G27ΔHP1284 | 3.25 ± 2.37 | 0.58 ± 0.08 | 0.02 ± 0.01 | 0.19 ± 0.04 | 0.04 ± 0.01 | 0.27 ± 0.05 | 3.17 ± 0.83 |
| G27ΔHP1191 | 2.33 ± 0.88 | 0.58 ± 0.08 | 0.03 ± 0.01 | 0.17 ± 0.02 | 0.04 ± 0.01 | 0.27 ± 0.05 | 2.50 ± 0.76 |
| 26695 | 72.00 ± 30.29 | 5.33 ± 1.33 | 0.08 ± 0.02 | 0.33 ± 0.08 | 0.08 ± 0.02 | 0.21 ± 0.04 | 85.3 ± 21.3 |
| 26695ΔwecA | 2.25 ± 0.94 | 1.08 ± 0.51 | 0.08 ± 0.02 | 0.31 ± 0.09 | 0.08 ± 0.02 | 0.21 ± 0.04 | 85.3 ± 21.3 |
| J99 | 53.33 ± 10.67 | 5.33 ± 1.33 | 0.02 ± 0.01 | 0.09 ± 0.03 | 0.03 ± 0.01 | 0.10 ± 0.04 | 0.67 ± 0.17 |
| J99ΔwecA | 0.83 ± 0.17 | 2.33 ± 0.88 | 0.02 ± 0.01 | 0.09 ± 0.02 | 0.03 ± 0.01 | 0.09 ± 0.02 | 0.63 ± 0.19 |
| P12 | 7.67 ± 4.18 | 6.00 ± 1.15 | 0.03 ± 0.01 | 0.23 ± 0.02 | 0.02 ± 0.00 | 0.21 ± 0.02 | 0.83 ± 0.17 |
| P12ΔwecA | 0.31 ± 0.09 | 2.16 ± 0.93 | 0.02 ± 0.01 | 0.21 ± 0.04 | 0.02 ± 0.00 | 0.23 ± 0.02 | 0.83 ± 0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, X.; Yang, T.; Shen, Y.; Song, X.; Benghezal, M.; Marshall, B.J.; Tang, H.; Li, H. Roles of Lipopolysaccharide Glycosyltransferases in Maintenance of Helicobacter pylori Morphology, Cell Wall Permeability, and Antimicrobial Susceptibilities. Int. J. Mol. Sci. 2023, 24, 11381. https://doi.org/10.3390/ijms241411381
Tang X, Yang T, Shen Y, Song X, Benghezal M, Marshall BJ, Tang H, Li H. Roles of Lipopolysaccharide Glycosyltransferases in Maintenance of Helicobacter pylori Morphology, Cell Wall Permeability, and Antimicrobial Susceptibilities. International Journal of Molecular Sciences. 2023; 24(14):11381. https://doi.org/10.3390/ijms241411381
Chicago/Turabian StyleTang, Xiaoqiong, Tiankuo Yang, Yalin Shen, Xiaona Song, Mohammed Benghezal, Barry J. Marshall, Hong Tang, and Hong Li. 2023. "Roles of Lipopolysaccharide Glycosyltransferases in Maintenance of Helicobacter pylori Morphology, Cell Wall Permeability, and Antimicrobial Susceptibilities" International Journal of Molecular Sciences 24, no. 14: 11381. https://doi.org/10.3390/ijms241411381
APA StyleTang, X., Yang, T., Shen, Y., Song, X., Benghezal, M., Marshall, B. J., Tang, H., & Li, H. (2023). Roles of Lipopolysaccharide Glycosyltransferases in Maintenance of Helicobacter pylori Morphology, Cell Wall Permeability, and Antimicrobial Susceptibilities. International Journal of Molecular Sciences, 24(14), 11381. https://doi.org/10.3390/ijms241411381