
Evolution of Vaccines Formulation to Tackle the Challenge of Anti-Microbial Resistant Pathogens

 ,
,
Abstract
1. Introduction
2. State-of-the-Art and Innovative Vaccines Formulation Strategies to Prevent AMR Pathogen-Induced Diseases
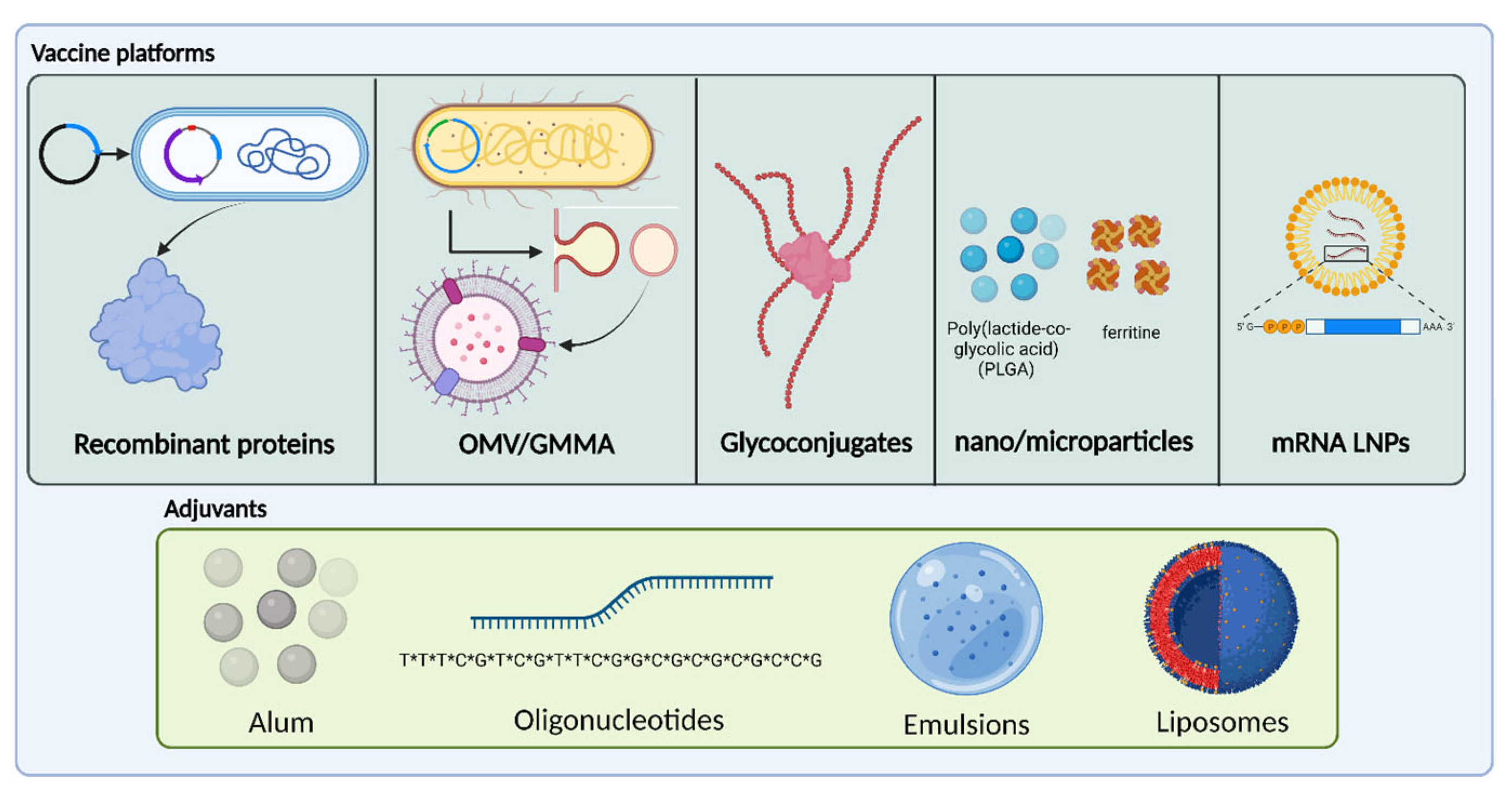
2.1. Formulation of Vaccines against AMR Pathogens
2.2. Recombinant Protein-Based Antigens for Bacterial Targets
2.2.1. Generalities of Protein-Based Anti-Bacterial Vaccines
2.2.2. Applications to AMR Pathogens
2.3. Membrane Vesicle-Based Antigens
2.3.1. Generalities on Membrane Vesicles
2.3.2. Application to AMR Pathogens
2.4. Glycoconjugate-Based Antigens
2.4.1. Generalities on Glycoconjugates
2.4.2. Glycoconjugate Vaccines Application to AMR Pathogens
2.5. RNA-Based Vaccines
2.6. Future Perspective of Nano and Micro Vaccines Drug Delivery Systems (DDSs) and Their Application against AMR Pathogens
2.6.1. Self-Assembling Protein Nanoparticles (SAPNs) for Vaccine Delivery
2.6.2. PLGA Based Nano and Microparticles DDS for Vaccines Delivery
3. Adjuvants for Improved Vaccines against Antimicrobial Resistance Pathogens
3.1. Aluminium Salts
3.1.1. Generalities on Aluminium Salts
3.1.2. Use of Aluminium-Based Adjuvants in Vaccines against AMR Pathogens
3.2. Oligonucleotide Adjuvants
3.2.1. Generalities on Oligonucleotides Adjuvants
3.2.2. Use of Oligonucleotides Adjuvants in Vaccines against AMR Pathogens
3.3. Emulsion Adjuvants
3.3.1. Generalities on Emulsion Adjuvants
3.3.2. Use of Emulsion Adjuvants in Vaccines against AMR Pathogen
3.4. Liposome-Based Adjuvants
3.4.1. Generalities on Liposome-Based Adjuvant
3.4.2. Use of Liposome-Based Adjuvant in Vaccines against AMR Pathogens
4. Alternative Vaccines Administration Routes in AMR Vaccines
5. Can Antimicrobial Resistance Lead to A New Era for Vaccine Analytics?
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J.W. Ageing populations: The challenges ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef]
- Carr, C.J. Chemotherapy: Yesterday, To-day, and To-morrow. The Linacre Lecture Delivered at Cambridge on May 6, 1946.; Alexander Fleming. Q. Rev. Biol. 1947, 22, 235. [Google Scholar] [CrossRef]
- Alekshun, M.N.; Levy, S.B. Molecular Mechanisms of Antibacterial Multidrug Resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Genet. 2007, 5, 175–186. [Google Scholar] [CrossRef]
- Blake, K.L.; O’Neill, A.J. Transposon library screening for identification of genetic loci participating in intrinsic susceptibility and acquired resistance to antistaphylococcal agents. J. Antimicrob. Chemother. 2012, 68, 12–16. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 464–473. [Google Scholar] [CrossRef]
- Castañeda-Barba, S.; Top, E.M.; Stalder, T. Plasmids, a molecular cornerstone of antimicrobial resistance in the One Health era. Nat. Rev. Microbiol. 2023. [CrossRef]
- Jansen, K.U.; Anderson, A.S. The role of vaccines in fighting antimicrobial resistance (AMR). Hum. Vaccines Immunother. 2018, 14, 2142–2149. [Google Scholar] [CrossRef]
- Europäisches Zentrum für die Prävention und die Kontrolle von Krankheiten. The Bacterial Challenge, Time to React: A Call to Narrow the Gap between Multidrug-Resistant Bacteria in the EU and the Development of New Antibacterial Agents; ECDC: Stockholm, Sweden, 2009.
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Tackling Drug-Resistant Infections Globally: Final Report and Recommendations the Review on Antimicrobial Resistance Chaired by Jim O’neill. 2016. Available online: https://wellcomecollection.org/works/thvwsuba (accessed on 6 April 2023).
- Micoli, F.; Bagnoli, F.; Rappuoli, R.; Serruto, D. The role of vaccines in combatting antimicrobial resistance. Nat. Rev. Genet. 2021, 19, 287–302. [Google Scholar] [CrossRef]
- Lipsitch, M.; Siber, G.R. How Can Vaccines Contribute to Solving the Antimicrobial Resistance Problem? mBio 2016, 7. [Google Scholar] [CrossRef]
- Chang, H.-H.; Cohen, T.; Grad, Y.H.; Hanage, W.P.; O’Brien, T.F.; Lipsitch, M. Origin and Proliferation of Multiple-Drug Resistance in Bacterial Pathogens. Microbiol. Mol. Biol. Rev. 2015, 79, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Bacterial Vaccines in Clinical and Preclinical Development 2021. 2022. Available online: http://apps.who.int/bookorders (accessed on 25 September 2022).
- Malarski, M.; Hasso-Agopsowicz, M.; Soble, A.; Mok, W.; Mathewson, S.; Vekemans, J. Vaccine impact on antimicrobial resistance to inform Gavi, the Vaccine Alliance’s 2018 Vaccine Investment Strategy: Report from an expert survey. F1000Research 2019, 8, 1685. [Google Scholar] [CrossRef] [PubMed]
- Odneal, K.E. Introduction to vaccine development. In Practical Aspects of Vaccine Development; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1–8. [Google Scholar] [CrossRef]
- Smith, L.C.; Nelson, P. Formulation design considerations and good practice for live attenuated vaccine development. In Practical Aspects of Vaccine Development; Elsevier: Amsterdam, The Netherlands, 2021; pp. 27–78. [Google Scholar] [CrossRef]
- Brito, L.A.; Malyala, P.; O’hagan, D.T. Vaccine adjuvant formulations: A pharmaceutical perspective. Semin. Immunol. 2013, 25, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Urgent Call for Better Use of Existing Vaccines and Development of New Vaccines to Tackle AMR. Available online: https://www.who.int/news/item/12-07-2022-urgent-call-for-better-use-of-existing-vaccines-and-development-of-new-vaccines-to-tackle-amr (accessed on 30 May 2023).
- Defendi, H.G.T.; Madeira, L.D.S.; Borschiver, S. Analysis of the COVID-19 Vaccine Development Process: An Exploratory Study of Accelerating Factors and Innovative Environments. J. Pharm. Innov. 2021, 17, 555–571. [Google Scholar] [CrossRef]
- Bill, R.M. Recombinant protein subunit vaccine synthesis in microbes: A role for yeast? J. Pharm. Pharmacol. 2014, 67, 319–328. [Google Scholar] [CrossRef]
- Plotkin, S.A. Vaccines: Past, present and future. Nat. Med. 2005, 11, S5–S11. [Google Scholar] [CrossRef]
- Ikonomou, L.; Schneider, Y.-J.; Agathos, S.N. Insect cell culture for industrial production of recombinant proteins. Appl. Microbiol. Biotechnol. 2003, 62, 1–20. [Google Scholar] [CrossRef] [PubMed]
- García-Fruitós, E. (Ed.) Insoluble Proteins; Springer: New York, NY, USA, 2015; Volume 1258. [Google Scholar] [CrossRef]
- Dertzbaugh, M.T. Genetically Engineered Vaccines: An Overview. Plasmid 1998, 39, 100–113. [Google Scholar] [CrossRef]
- Rodrigues, M.X.; Yang, Y.; de Souza, M.E.B., Jr.; do Carmo, S.J.; Bicalho, R.C. Development and evaluation of a new recombinant protein vaccine (YidR) against Klebsiella pneumoniae infection. Vaccine 2020, 38, 4640–4648. [Google Scholar] [CrossRef]
- Rosano, G.L.; Morales, E.S.; Ceccarelli, E.A. New tools for recombinant protein production in Escherichia coli: A 5-year update. Protein Sci. 2019, 28, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Sengupta, M.; Prakash, J.; Tripathy, B.C. Production of Recombinant Pharmaceutical Proteins. In Basic and Applied Aspects of Biotechnology; Springer: Singapore, 2017; pp. 77–101. [Google Scholar] [CrossRef]
- Clegg, J.; Soldaini, E.; McLoughlin, R.M.; Rittenhouse, S.; Bagnoli, F.; Phogat, S. Staphylococcus aureus Vaccine Research and Development: The Past, Present and Future, Including Novel Therapeutic Strategies. Front. Immunol. 2021, 12, 705360. [Google Scholar] [CrossRef] [PubMed]
- Moriel, D.G.; Bertoldi, I.; Spagnuolo, A.; Marchi, S.; Rosini, R.; Nesta, B.; Pastorello, I.; Corea, V.A.M.; Torricelli, G.; Cartocci, E.; et al. Identification of protective and broadly conserved vaccine antigens from the genome of extraintestinal pathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 2010, 107, 9072–9077. [Google Scholar] [CrossRef]
- Nascimento, I.P.; Leite, L.C.C. Recombinant vaccines and the development of new vaccine strategies. Braz. J. Med. Biol. Res. 2012, 45, 1102–1111. [Google Scholar] [CrossRef]
- Tedeschi, G.; Mangiagalli, M.; Chmielewska, S.; Lotti, M.; Natalello, A.; Brocca, S. Aggregation properties of a disordered protein are tunable by pH and depend on its net charge per residue. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 2543–2550. [Google Scholar] [CrossRef]
- Kim, H.L.; Mcauley, A.; Mcguire, J. Protein Effects on Surfactant Adsorption Suggest the Dominant Mode of Surfactant-Mediated Stabilization of Protein. J. Pharm. Sci. 2014, 103, 1337–1345. [Google Scholar] [CrossRef]
- Al-Hussein, A.; Gieseler, H. Investigation of Histidine Stabilizing Effects on LDH During Freeze-Drying. J. Pharm. Sci. 2013, 102, 813–826. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Zeng, H.; Li, J.-X.; Wang, B.; Meng, F.-Y.; Yang, F.; Gu, J.; Liang, H.-Y.; Hu, Y.-M.; Liu, P.; et al. Evaluation of a recombinant five-antigen Staphylococcus aureus vaccine: The randomized, single-centre phase 1a/1b clinical trials. Vaccine 2022, 40, 3216–3227. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.L.; Soema, P.C.; Slütter, B.; Ossendorp, F.; Jiskoot, W. PLGA particulate delivery systems for subunit vaccines: Linking particle properties to immunogenicity. Hum. Vaccines Immunother. 2016, 12, 1056–1069. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, L.; Stork, M.; van der Ley, P. Outer membrane vesicles as platform vaccine technology. Biotechnol. J. 2015, 10, 1689–1706. [Google Scholar] [CrossRef] [PubMed]
- Bitto, N.J.; Kaparakis-Liaskos, M. The Therapeutic Benefit of Bacterial Membrane Vesicles. Int. J. Mol. Sci. 2017, 18, 1287. [Google Scholar] [CrossRef]
- Dowling, J.K.; Mansell, A. Toll-like receptors: The swiss army knife of immunity and vaccine development. Clin. Transl. Immunol. 2016, 5, e85. [Google Scholar] [CrossRef]
- Kulp, A.; Kuehn, M.J. Biological Functions and Biogenesis of Secreted Bacterial Outer Membrane Vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef]
- Piccioli, D.; Bartolini, E.; Micoli, F. GMMA as a ‘plug and play’ technology to tackle infectious disease to improve global health: Context and perspectives for the future. Expert Rev. Vaccines 2021, 21, 163–172. [Google Scholar] [CrossRef]
- Scorza, F.B.; Colucci, A.M.; Maggiore, L.; Sanzone, S.; Rossi, O.; Ferlenghi, I.; Pesce, I.; Caboni, M.; Norais, N.; Di Cioccio, V.; et al. High Yield Production Process for Shigella Outer Membrane Particles. PLoS ONE 2012, 7, e35616. [Google Scholar] [CrossRef]
- Acevedo, R.; Fernã¡Ndez, S.; Zayas, C.; Acosta, A.; Sarmiento, M.E.; Ferro, V.A.; Rosenqvist, E.; Campa, C.; Cardoso, D.; Garcia, L.; et al. Bacterial Outer Membrane Vesicles and Vaccine Applications. Front. Immunol. 2014, 5, 121. [Google Scholar] [CrossRef]
- Frenck, R.W.; Conti, V.; Ferruzzi, P.; Ndiaye, A.G.; Parker, S.; McNeal, M.M.; Dickey, M.; Granada, J.P.; Cilio, G.L.; De Ryck, I.; et al. Efficacy, safety, and immunogenicity of the Shigella sonnei 1790GAHB GMMA candidate vaccine: Results from a phase 2b randomized, placebo-controlled challenge study in adults. EClinicalMedicine 2021, 39, 101076. [Google Scholar] [CrossRef]
- De Benedetto, G.; Alfini, R.; Cescutti, P.; Caboni, M.; Lanzilao, L.; Necchi, F.; Saul, A.; MacLennan, C.; Rondini, S.; Micoli, F. Characterization of O-antigen delivered by Generalized Modules for Membrane Antigens (GMMA) vaccine candidates against nontyphoidal Salmonella. Vaccine 2017, 35, 419–426. [Google Scholar] [CrossRef]
- Rossi, O.; Caboni, M.; Negrea, A.; Necchi, F.; Alfini, R.; Micoli, F.; Saul, A.; MacLennan, C.A.; Rondini, S.; Gerke, C. Toll-Like Receptor Activation by Generalized Modules for Membrane Antigens from Lipid A Mutants of Salmonella enterica Serovars Typhimurium and Enteritidis. Clin. Vaccine Immunol. 2016, 23, 304–314. [Google Scholar] [CrossRef]
- Huang, W.; Meng, L.; Chen, Y.; Dong, Z.; Peng, Q. Bacterial outer membrane vesicles as potential biological nanomaterials for antibacterial therapy. Acta Biomater. 2021, 140, 102–115. [Google Scholar] [CrossRef]
- Rossi, O.; Pesce, I.; Giannelli, C.; Aprea, S.; Caboni, M.; Citiulo, F.; Valentini, S.; Ferlenghi, I.; MacLennan, C.A.; D’Oro, U.; et al. Modulation of Endotoxicity of Shigella Generalized Modules for Membrane Antigens (GMMA) by Genetic Lipid A Modifications. J. Biol. Chem. 2014, 289, 24922–24935. [Google Scholar] [CrossRef] [PubMed]
- Mancini, F.; Micoli, F.; Necchi, F.; Pizza, M.; Scorza, F.B.; Rossi, O. GMMA-Based Vaccines: The Known and The Unknown. Front. Immunol. 2021, 12, 715393. [Google Scholar] [CrossRef]
- Prior, J.T.; Davitt, C.; Kurtz, J.; Gellings, P.; McLachlan, J.B.; Morici, L.A. Bacterial-Derived Outer Membrane Vesicles are Potent Adjuvants that Drive Humoral and Cellular Immune Responses. Pharmaceutics 2021, 13, 131. [Google Scholar] [CrossRef]
- Raso, M.M.; Gasperini, G.; Alfini, R.; Schiavo, F.; Aruta, M.G.; Carducci, M.; Forgione, M.C.; Martini, S.; Cescutti, P.; Necchi, F.; et al. GMMA and Glycoconjugate Approaches Compared in Mice for the Development of a Vaccine against Shigella flexneri Serotype 6. Vaccines 2020, 8, 160. [Google Scholar] [CrossRef]
- Micoli, F.; Rondini, S.; Alfini, R.; Lanzilao, L.; Necchi, F.; Negrea, A.; Rossi, O.; Brandt, C.; Clare, S.; Mastroeni, P.; et al. Comparative immunogenicity and efficacy of equivalent outer membrane vesicle and glycoconjugate vaccines against nontyphoidal Salmonella. Proc. Natl. Acad. Sci. USA 2018, 115, 10428–10433. [Google Scholar] [CrossRef]
- Rossi, O.; Aruta, M.G.; Acquaviva, A.; Mancini, F.; Micoli, F.; Necchi, F. Characterization of Competitive ELISA and Formulated Alhydrogel Competitive ELISA (FAcE) for Direct Quantification of Active Ingredients in GMMA-Based Vaccines. Methods Protoc. 2020, 3, 62. [Google Scholar] [CrossRef]
- Palmieri, E.; Arato, V.; Oldrini, D.; Ricchetti, B.; Aruta, M.G.; Pansegrau, W.; Marchi, S.; Giusti, F.; Ferlenghi, I.; Rossi, O.; et al. Stability of Outer Membrane Vesicles-Based Vaccines, Identifying the Most Appropriate Methods to Detect Changes in Vaccine Potency. Vaccines 2021, 9, 229. [Google Scholar] [CrossRef]
- Micoli, F.; Alfini, R.; Di Benedetto, R.; Necchi, F.; Schiavo, F.; Mancini, F.; Carducci, M.; Palmieri, E.; Balocchi, C.; Gasperini, G.; et al. GMMA Is a Versatile Platform to Design Effective Multivalent Combination Vaccines. Vaccines 2020, 8, 540. [Google Scholar] [CrossRef]
- Wai, S.N.; Lindmark, B.; Söderblom, T.; Takade, A.; Westermark, M.; Oscarsson, J.; Jass, J.; Richter-Dahlfors, A.; Mizunoe, Y.; Uhlin, B.E. Vesicle-Mediated Export and Assembly of Pore-Forming Oligomers of the Enterobacterial ClyA Cytotoxin. Cell 2003, 115, 25–35. [Google Scholar] [CrossRef]
- Muralinath, M.; Kuehn, M.J.; Roland, K.L.; Curtiss, R. Immunization with Salmonella enterica Serovar Typhimurium-Derived Outer Membrane Vesicles Delivering the Pneumococcal Protein PspA Confers Protection against Challenge with Streptococcus pneumoniae. Infect. Immun. 2011, 79, 887–894. [Google Scholar] [CrossRef]
- Biemans, R.; Micoli, F.; Romano, M.R. Glycoconjugate vaccines, production and characterization. In Recent Trends in Carbohydrate Chemistry; Elsevier: Amsterdam, The Netherlands, 2020; pp. 285–313. [Google Scholar] [CrossRef]
- Micoli, F.; Del Bino, L.; Alfini, R.; Carboni, F.; Romano, M.R.; Adamo, R. Glycoconjugate vaccines: Current approaches towards faster vaccine design. Expert Rev. Vaccines 2019, 18, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Defourny, K.A.Y.; Smid, E.J.; Abee, T. Gram-Positive Bacterial Extracellular Vesicles and Their Impact on Health and Disease. Front. Microbiol. 2018, 9, 1502. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Thompson, C.D.; Weidenmaier, C.; Lee, J.C. Release of Staphylococcus aureus extracellular vesicles and their ap-plication as a vaccine platform. Nat. Commun. 2018, 9, 1379. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Choi, D.Y.; Kim, D.K.; Kim, J.W.; Park, J.O.; Kim, S.; Kim, S.H.; Desiderio, D.M.; Kim, Y.K.; Kim, K.P.; et al. Gram-positive bacteria produce membrane vesicles: Proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef] [PubMed]
- García, Y.R.; Sohn, W.-Y.; Seib, K.L.; Taha, M.-K.; Vázquez, J.A.; de Lemos, A.P.S.; Vadivelu, K.; Pizza, M.; Rappuoli, R.; Bekkat-Berkani, R. Looking beyond meningococcal B with the 4CMenB vaccine: The Neisseria effect. npj Vaccines 2021, 6, 130. [Google Scholar] [CrossRef]
- Micoli, F.; Nakakana, U.N.; Scorza, F.B. Towards a Four-Component GMMA-Based Vaccine against Shigella. Vaccines 2022, 10, 328. [Google Scholar] [CrossRef] [PubMed]
- Micoli, F.; Rossi, O.; Conti, V.; Launay, O.; Sciré, A.S.; Aruta, M.G.; Nakakana, U.N.; Marchetti, E.; Rappuoli, R.; Saul, A.; et al. Antibodies Elicited by the Shigella sonnei GMMA Vaccine in Adults Trigger Complement-Mediated Serum Bactericidal Activity: Results From a Phase 1 Dose Escalation Trial Followed by a Booster Extension. Front. Immunol. 2021, 12, 671325. [Google Scholar] [CrossRef]
- Morelli, L.; Poletti, L.; Lay, L. Carbohydrates and Immunology: Synthetic Oligosaccharide Antigens for Vaccine Formulation. Eur. J. Org. Chem. 2011, 2011, 5723–5777. [Google Scholar] [CrossRef]
- MacCalman, T.E.; Phillips-Jones, M.K.; Harding, S.E. Glycoconjugate vaccines: Some observations on carrier and production methods. Biotechnol. Genet. Eng. Rev. 2019, 35, 93–125. [Google Scholar] [CrossRef]
- Rose, F.; Wern, J.E.; Ingvarsson, P.T.; van de Weert, M.; Andersen, P.; Follmann, F.; Foged, C. Engineering of a novel adjuvant based on lipid-polymer hybrid nanoparticles: A quality-by-design approach. J. Control. Release 2015, 210, 48–57. [Google Scholar] [CrossRef]
- Zhang, F.; Lu, Y.-J.; Malley, R. Multiple antigen-presenting system (MAPS) to induce comprehensive B- and T-cell immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 13564–13569. [Google Scholar] [CrossRef]
- Jiang, X.; Bai, J.; Zhang, H.; Yuan, J.; Lu, G.; Wang, Y.; Jiang, L.; Liu, B.; Huang, D.; Feng, L. Development of an O-polysaccharide based recombinant glycoconjugate vaccine in engineered E. coli against ExPEC O1. Carbohydr. Polym. 2022, 277, 118796. [Google Scholar] [CrossRef]
- Martin, P.; Alaimo, C. The Ongoing Journey of a Shigella Bioconjugate Vaccine. Vaccines 2022, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Trotter, C.L.; Maiden, M.C. Meningococcal vaccines and herd immunity: Lessons learned from serogroup C conjugate vaccination programs. Expert Rev. Vaccines 2009, 8, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Pneumococcal Vaccine Overview. Available online: https://www.nhs.uk/conditions/vaccinations/pneumococcal-vaccination/ (accessed on 15 May 2023).
- U.S. FDA Approves PREVNAR 20TM, Pfizer’s Pneumococcal 20-Valent Conjugate Vaccine for Adults Ages 18 Years or Older. Available online: https://www.pfizer.com/news/press-release/press-release-detail/us-fda-approves-prevnar-20tm-pfizers-pneumococcal-20-valent (accessed on 15 May 2023).
- Zhang, F.; Boerth, E.M.; Gong, J.; Ma, N.; Lucas, K.; Ledue, O.; Malley, R.; Lu, Y.-J. A Bivalent MAPS Vaccine Induces Protective Antibody Responses against Salmonella Typhi and Paratyphi A. Vaccines 2022, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Chichili, G.R.; Smulders, R.; Santos, V.; Cywin, B.; Kovanda, L.; Van Sant, C.; Malinoski, F.; Sebastian, S.; Siber, G.; Malley, R. Phase 1/2 study of a novel 24-valent pneumococcal vaccine in healthy adults aged 18 to 64 years and in older adults aged 65 to 85 years. Vaccine 2022, 40, 4190–4198. [Google Scholar] [CrossRef]
- Inoue, M.; Ogawa, T.; Tamura, H.; Hagiwara, Y.; Saito, Y.; Abbanat, D.; Dobbelsteen, G.V.D.; Hermans, P.; Thoelen, S.; Poolman, J.; et al. Safety, tolerability and immunogenicity of the ExPEC4V (JNJ-63871860) vaccine for prevention of invasive extraintestinal pathogenic Escherichia coli disease: A phase 1, randomized, double-blind, placebo-controlled study in healthy Japanese participants. Hum. Vaccines Immunother. 2018, 14, 2150–2157. [Google Scholar] [CrossRef]
- Huttner, A.; Hatz, C.; Dobbelsteen, G.V.D.; Abbanat, D.; Hornacek, A.; Frölich, R.; Dreyer, A.M.; Martin, P.; Davies, T.; Fae, K.; et al. Safety, immunogenicity, and preliminary clinical efficacy of a vaccine against extraintestinal pathogenic Escherichia coli in women with a history of recurrent urinary tract infection: A randomised, single-blind, placebo-controlled phase 1b trial. Lancet Infect. Dis. 2017, 17, 528–537. [Google Scholar] [CrossRef]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Kon, E.; Levy, Y.; Elia, U.; Cohen, H.; Hazan-Halevy, I.; Aftalion, M.; Ezra, A.; Bar-Haim, E.; Naidu, G.S.; Diesendruck, Y.; et al. Applied Sciences and Engineering A Single-Dose F1-Based mRNA-LNP Vaccine Provides Protection Against the Lethal Plague Bacterium. 2023. Available online: https://www.science.org (accessed on 22 March 2023).
- Maruggi, G.; Chiarot, E.; Giovani, C.; Buccato, S.; Bonacci, S.; Frigimelica, E.; Margarit, I.; Geall, A.; Bensi, G.; Maione, D. Immunogenicity and protective efficacy induced by self-amplifying mRNA vaccines encoding bacterial antigens. Vaccine 2017, 35, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, C.; Rcheulishvili, N.; Papukashvili, D.; Xie, F.; Zhao, J.; Hu, X.; Yu, K.; Yang, N.; Pan, X.; et al. Strong immune responses and protection of PcrV and OprF-I mRNA vaccine candidates against Pseudomonas aeruginosa. npj Vaccines 2023, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.E.; Erasmus, J.H.; Reese, V.A.; Pecor, T.; Archer, J.; Kandahar, A.; Hsu, F.-C.; Nicholes, K.; Reed, S.G.; Baldwin, S.L.; et al. An RNA-Based Vaccine Platform for Use against Mycobacterium tuberculosis. Vaccines 2023, 11, 130. [Google Scholar] [CrossRef]
- Jawalagatti, V.; Kirthika, P.; Lee, J.H. Oral mRNA Vaccines Against Infectious Diseases- A Bacterial Perspective [Invited]. Front. Immunol. 2022, 13, 884862. [Google Scholar] [CrossRef]
- Jawalagatti, V.; Kirthika, P.; Hewawaduge, C.; Yang, M.-S.; Park, J.-Y.; Oh, B.; Lee, J.H. Bacteria-enabled oral delivery of a replicon-based mRNA vaccine candidate protects against ancestral and delta variant SARS-CoV-2. Mol. Ther. 2022, 30, 1926–1940. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Qu, L.; Yi, Z.; Shen, Y.; Lin, L.; Chen, F.; Xu, Y.; Wu, Z.; Tang, H.; Zhang, X.; Tian, F.; et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 2022, 185, 1728–1744.e16. [Google Scholar] [CrossRef]
- Study of Investigational Pneumococcal Vaccine in Healthy Adults, Toddlers and Infants. Available online: https://clinicaltrials.gov/ct2/show/NCT01446926 (accessed on 22 February 2023).
- Safety and Immunogenicity Study of GSK’s Clostridium Difficile Vaccine 2904545A When Administered in Healthy Adults Aged 18–45 Years and 50–70 Years. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04026009 (accessed on 22 February 2023).
- A Clinical Trial to Evaluate a Recombinant Staphylococcus Aureus Vaccine (Escherichia coli) in Healthy Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT02804711 (accessed on 22 February 2023).
- A Study to Investigate the Safety and Immunogenicity of the SF2a-TT15 Synthetic Carbohydrate-Based Conjugate Vaccine Against Shigella Flexneri 2a (GlycoShig3). Available online: https://clinicaltrials.gov/ct2/show/NCT04602975 (accessed on 22 February 2023).
- Phase 2b Challenge Study With the Bioconjugate Vaccine Flexyn2a. Available online: https://clinicaltrials.gov/ct2/show/NCT02646371 (accessed on 22 February 2023).
- Phase I to Test a New Pneumococcal Vaccine. Available online: https://www.bing.com/search?q=NCT03303976&qs=n&form=QBRE&sp=-1&pq=nct03303976&sc=1-11&sk=&cvid=1A3703AB7D384D64A109369EC68B2BBE&ghsh=0&ghacc=0&ghpl= (accessed on 22 February 2023).
- A Study to Evaluate the Different Doses of VAC52416 (ExPEC10V) in Japanese Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT04306302 (accessed on 22 February 2023).
- Preventing UTIs in Chronic Neurogenic Bladder Dysfunction (Mix Methods) (PReSuTINeB). Available online: https://clinicaltrials.gov/ct2/show/NCT02591901 (accessed on 22 February 2023).
- A Study to Evaluate the Efficacy, Safety and Immunogenicity of a Vaccine Designed to Protect Against Infection With Shigella Sonnei in Healthy Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT03527173 (accessed on 22 February 2023).
- A Study on the Safety and Immune Responses to the GVGH altSonflex1-2-3 Vaccine Against Shigellosis in Adults, Children, and Infants. Available online: https://clinicaltrials.gov/ct2/show/NCT05073003 (accessed on 22 February 2023).
- Singh, R.; Lillard, J.W., Jr. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef]
- Campos, E.; Branquinho, J.; Carreira, A.S.; Carvalho, A.; Coimbra, P.; Ferreira, P.; Gil, M. Designing polymeric microparticles for biomedical and industrial applications. Eur. Polym. J. 2013, 49, 2005–2021. [Google Scholar] [CrossRef]
- Nguyen, B.; Tolia, N.H. Protein-based antigen presentation platforms for nanoparticle vaccines. npj Vaccines 2021, 6, 70. [Google Scholar] [CrossRef]
- Singh, H.; Richard, G.; Compans, W. (Eds.) Current Topics in Microbiology and Immunology Nanoparticles for Rational Vaccine Design. Available online: http://www.springer.com/series/82 (accessed on 28 October 2022).
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Leleux, J.; Roy, K. Micro and Nanoparticle-Based Delivery Systems for Vaccine Immunotherapy: An Immunological and Materials Perspective. Adv. Health Mater. 2012, 2, 72–94. [Google Scholar] [CrossRef]
- Zong, G.; Toonstra, C.; Yang, Q.; Zhang, R.; Wang, L.-X. Chemoenzymatic Synthesis and Antibody Binding of HIV-1 V1/V2 Glycopeptide-Bacteriophage Qβ Conjugates as a Vaccine Candidate. Int. J. Mol. Sci. 2021, 22, 12538. [Google Scholar] [CrossRef]
- López-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-assembling protein nanoparticles in the design of vaccines. Comput. Struct. Biotechnol. J. 2016, 14, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: Preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 2021, 19, 59. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus–like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Cappelli, L.; Cinelli, P.; Giusti, F.; Ferlenghi, I.; Utrio-Lanfaloni, S.; Wahome, N.; Bottomley, M.J.; Maione, D.; Cozzi, R. Self-assembling protein nanoparticles and virus like particles correctly display β-barrel from meningococcal factor H-binding protein through genetic fusion. PLoS ONE 2022, 17, e0273322. [Google Scholar] [CrossRef]
- Wang, L.; Xing, D.; Le Van, A.; Jerse, A.E.; Wang, S. Structure-based design of ferritin nanoparticle immunogens displaying antigenic loops of Neisseria gonorrhoeae. FEBS Open Bio 2017, 7, 1196–1207. [Google Scholar] [CrossRef]
- Joyner, J.A.; Daly, S.M.; Peabody, J.; Triplett, K.D.; Pokhrel, S.; Elmore, B.O.; Adebanjo, D.; Peabody, D.S.; Chackerian, B.; Hall, P.R. Vaccination with VLPs Presenting a Linear Neutralizing Domain of S. aureus Hla Elicits Protective Immunity. Toxins 2020, 12, 450. [Google Scholar] [CrossRef]
- Huo, C.-X.; Dhara, D.; Baliban, S.M.; Nick, S.T.; Tan, Z.; Simon, R.; Misra, A.K.; Huang, X. Synthetic and immunological studies of Salmonella Enteritidis O-antigen tetrasaccharides as potential anti-Salmonella vaccines. Chem. Commun. 2019, 55, 4519–4522. [Google Scholar] [CrossRef] [PubMed]
- McHugh, K.J.; Guarecuco, R.; Langer, R.; Jaklenec, A. Single-injection vaccines: Progress, challenges, and opportunities. J. Control. Release 2015, 219, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Cirelli, K.M.; Carnathan, D.G.; Nogal, B.; Martin, J.T.; Rodriguez, O.L.; Upadhyay, A.A.; Enemuo, C.A.; Gebru, E.H.; Choe, Y.; Viviano, F.; et al. Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses via Modulation of Immunodominance. Cell 2019, 177, 1153–1171.e28. [Google Scholar] [CrossRef]
- Washington, M.A.; Balmert, S.; Fedorchak, M.V.; Little, S.R.; Watkins, S.; Meyer, T.Y. Monomer sequence in PLGA microparticles: Effects on acidic microclimates and in vivo inflammatory response. Acta Biomater. 2018, 65, 259–271. [Google Scholar] [CrossRef]
- Zhu, G.; Mallery, S.R.; Schwendeman, S.P. Stabilization of Proteins Encapsulated in Injectable Poly (Lactide-co-Glycolide). 2000. Available online: http://biotech.nature.com (accessed on 13 January 2023).
- Desai, K.-G.H.; Kadous, S.; Schwendeman, S.P. Gamma Irradiation of Active Self-Healing PLGA Microspheres for Efficient Aqueous Encapsulation of Vaccine Antigens. Pharm. Res. 2013, 30, 1768–1778. [Google Scholar] [CrossRef]
- Xie, X.; Hu, Y.; Ye, T.; Chen, Y.; Zhou, L.; Li, F.; Xi, X.; Wang, S.; He, Y.; Gao, X.; et al. Therapeutic vaccination against leukaemia via the sustained release of co-encapsulated anti-PD-1 and a leukaemia-associated antigen. Nat. Biomed. Eng. 2020, 5, 414–428. [Google Scholar] [CrossRef]
- Desai, K.-G.H.; Schwendeman, S.P. Active self-healing encapsulation of vaccine antigens in PLGA microspheres. J. Control. Release 2013, 165, 62–74. [Google Scholar] [CrossRef]
- Jiang, W.; Gupta, R.K.; Deshpande, M.C.; Schwendeman, S.P. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv. Drug Deliv. Rev. 2005, 57, 391–410. [Google Scholar] [CrossRef]
- Khademi, F.; Derakhshan, M.; Yousefi-Avarvand, A.; Najafi, A.; Tafaghodi, M. A novel antigen of Mycobacterium tuberculosis and MPLA adjuvant co-entrapped into PLGA:DDA hybrid nanoparticles stimulates mucosal and systemic immunity. Microb. Pathog. 2018, 125, 507–513. [Google Scholar] [CrossRef]
- de Figueiredo, D.B.; Kaneko, K.; Rodrigues, T.d.C.; MacLoughlin, R.; Miyaji, E.N.; Saleem, I.; Gonçalves, V.M. Pneumococcal Surface Protein A-Hybrid Nanoparticles Protect Mice from Lethal Challenge after Mucosal Immunization Targeting the Lungs. Pharmaceutics 2022, 14, 1238. [Google Scholar] [CrossRef]
- Evans, J.T.; Ward, J.R.; Kern, J.; Johnson, M.E. A single vaccination with protein-microspheres elicits a strong CD8 T-cell-mediated immune response against Mycobacterium tuberculosis antigen Mtb8.4. Vaccine 2004, 22, 1964–1972. [Google Scholar] [CrossRef]
- Manriquez, G.G.G.; Tuero, I. Adjuvants: Friends in vaccine formulations against infectious diseases. Hum. Vaccines Immunother. 2021, 17, 3539–3550. [Google Scholar] [CrossRef]
- de Souza Apostólico, J.; Lunardelli, V.A.S.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, Modus Operandi, and Licensing. J. Immunol. Res. 2016, 2016, 1459394. [Google Scholar] [CrossRef]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine Adjuvants: Putting Innate Immunity to Work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef]
- Tan, K.; Li, R.; Huang, X.; Liu, Q. Outer Membrane Vesicles: Current Status and Future Direction of These Novel Vaccine Adjuvants. Front. Microbiol. 2018, 9, 783. [Google Scholar] [CrossRef]
- Kool, M.; Fierens, K.; Lambrecht, B.N. Alum adjuvant: Some of the tricks of the oldest adjuvant. J. Med. Microbiol. 2012, 61, 927–934. [Google Scholar] [CrossRef]
- Raponi, A.; Brewer, J.M.; Garside, P.; Laera, D. Nanoalum adjuvanted vaccines: Small details make a big difference. Semin. Immunol. 2021, 56, 101544. [Google Scholar] [CrossRef]
- Meppen, M.; Stranges, D. QbD approach to formulation development for protein-based vaccines. In Practical Aspects of Vaccine Development; Academic Press: Cambridge, MA, USA, 2021; pp. 137–156. [Google Scholar] [CrossRef]
- Clapp, T.; Munks, M.W.; Trivedi, R.; Kompella, U.B.; Braun, L.J. Freeze-thaw stress of Alhydrogel® alone is sufficient to reduce the immunogenicity of a recombinant hepatitis B vaccine containing native antigen. Vaccine 2014, 32, 3765–3771. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Aldayel, A.M.; Cui, Z. Aluminum hydroxide nanoparticles show a stronger vaccine adjuvant activity than traditional aluminum hydroxide microparticles. J. Control. Release 2013, 173, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Ruwona, T.B.; Xu, H.; Li, X.; Taylor, A.N.; Shi, Y.-C.; Cui, Z. Toward understanding the mechanism underlying the strong adjuvant activity of aluminum salt nanoparticles. Vaccine 2016, 34, 3059–3067. [Google Scholar] [CrossRef]
- Rinaldi, M.; Fioretti, D.; Iurescia, S. (Eds.) DNA Vaccines; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1143. [Google Scholar] [CrossRef]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef]
- Muslehiddinoglu, J.; Simler, R.; Hill, M.L.; Mueller, C.; Amery, J.H.A.; Dixon, L.; Watson, A.; Storch, K.; Gazziola, C.; Gielen, F.; et al. Technical Considerations for Use of Oligonucleotide Solution API. Nucleic Acid Ther. 2020, 30, 189–197. [Google Scholar] [CrossRef]
- Scheiermann, J.; Klinman, D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef]
- Shi, S.; Zhu, H.; Xia, X.; Liang, Z.; Ma, X.; Sun, B. Vaccine adjuvants: Understanding the structure and mechanism of adjuvanticity. Vaccine 2019, 37, 3167–3178. [Google Scholar] [CrossRef]
- Søgaard, O.S.; Lohse, N.; Harboe, Z.B.; Offersen, R.; Bukh, A.R.; Davis, H.L.; Schønheyder, H.C.; Østergaard, L. Improving the Immunogenicity of Pneumococcal Conjugate Vaccine in HIV-Infected Adults with a Toll-Like Receptor 9 Agonist Adjuvant: A Randomized, Controlled Trial. Clin. Infect. Dis. 2010, 51, 42–50. [Google Scholar] [CrossRef]
- Chu, R.S.; McCool, T.; Greenspan, N.S.; Schreiber, J.R.; Harding, C.V. CpG Oligodeoxynucleotides Act as Adjuvants for Pneumococcal Polysaccharide-Protein Conjugate Vaccines and Enhance Antipolysaccharide Immunoglobulin G2a (IgG2a) and IgG3 Antibodies. Infect. Immun. 2000, 68, 1450–1456. [Google Scholar] [CrossRef]
- Akbari, S.; Nour, A.H. Emulsion Types, Stability Mechanisms and Rheology: A Review. 2018. Available online: www.ijirss.com (accessed on 13 March 2023).
- O’hagan, D.T.; van der Most, R.; Lodaya, R.N.; Coccia, M.; Lofano, G. “World in motion”—Emulsion adjuvants rising to meet the pandemic challenges. npj Vaccines 2021, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Schultze, V.; D’agosto, V.; Wack, A.; Novicki, D.; Zorn, J.; Hennig, R. Safety of MF59™ adjuvant. Vaccine 2008, 26, 3209–3222. [Google Scholar] [CrossRef]
- Garçon, N.; Vaughn, D.W.; Didierlaurent, A.M. Development and evaluation of AS03, an Adjuvant System containing α-tocopherol and squalene in an oil-in-water emulsion. Expert Rev. Vaccines 2012, 11, 349–366. [Google Scholar] [CrossRef]
- Pellegrino, P.; Clementi, E.; Radice, S. On vaccine’s adjuvants and autoimmunity: Current evidence and future perspectives. Autoimmun. Rev. 2015, 14, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Morel, S.; Didierlaurent, A.; Bourguignon, P.; Delhaye, S.; Baras, B.; Jacob, V.; Planty, C.; Elouahabi, A.; Harvengt, P.; Carlsen, H.; et al. Adjuvant System AS03 containing α-tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine 2011, 29, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Haensler, J. Manufacture of Oil-in-Water Emulsion Adjuvants. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2016; Volume 1494, pp. 165–180. [Google Scholar] [CrossRef]
- Ott, G.; Radhakrishnan, R.; Fang, J.-H.; Hora, M. The Adjuvant MF59: A 10-Year Perspective. Vaccine Adjuv. 2003, 42, 211–228. [Google Scholar] [CrossRef]
- Ko, E.-J.; Kang, S.-M. Immunology and efficacy of MF59-adjuvanted vaccines. Hum. Vaccines Immunother. 2018, 14, 3041–3045. [Google Scholar] [CrossRef]
- Song, J.Y.; Cheong, H.J.; Hyun, H.J.; Bin Seo, Y.; Lee, J.; Wie, S.-H.; Choi, M.J.; Choi, W.S.; Noh, J.Y.; Yun, J.W.; et al. Immunogenicity and safety of a 13-valent pneumococcal conjugate vaccine and an MF59-adjuvanted influenza vaccine after concomitant vaccination in ≥60-year-old adults. Vaccine 2017, 35, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Roman, F.; Vaman, T.; Kafeja, F.; Hanon, E.; Van Damme, P. AS03A-Adjuvanted Influenza A (H1N1) 2009 Vaccine for Adults up to 85 Years of Age. Clin. Infect. Dis. 2010, 51, 668–677. [Google Scholar] [CrossRef]
- Safety and Immunogenicity of a Klebsiella Pneumoniae Tetravalent Bioconjugate Vaccine (Kleb4V). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04959344 (accessed on 18 January 2023).
- Chatzikleanthous, D.; O’hagan, D.T.; Adamo, R. Lipid-Based Nanoparticles for Delivery of Vaccine Adjuvants and Antigens: Toward Multicomponent Vaccines. Mol. Pharm. 2021, 18, 2867–2888. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef]
- Marasini, N.; Ghaffar, K.A.; Skwarczynski, M.; Toth, I. Liposomes as a Vaccine Delivery System. In Micro- and Nanotechnology in Vaccine Development; Skwarczynski, M., Toth, I., Eds.; William Andrew Inc: Norwich, UK, 2017. [Google Scholar] [CrossRef]
- Webb, C.; Khadke, S.; Schmidt, S.T.; Roces, C.B.; Forbes, N.; Berrie, G.; Perrie, Y. The Impact of Solvent Selection: Strategies to Guide the Manufacturing of Liposomes Using Microfluidics. Pharmaceutics 2019, 11, 653. [Google Scholar] [CrossRef]
- Forbes, N.; Hussain, M.T.; Briuglia, M.L.; Edwards, D.P.; ter Horst, J.H.; Szita, N.; Perrie, Y. Rapid and scale-independent microfluidic manufacture of liposomes entrapping protein incorporating in-line purification and at-line size monitoring. Int. J. Pharm. 2018, 556, 68–81. [Google Scholar] [CrossRef]
- Filipczak, N.; Pan, J.; Yalamarty, S.S.K.; Torchilin, V.P. Recent advancements in liposome technology. Adv. Drug Deliv. Rev. 2020, 156, 4–22. [Google Scholar] [CrossRef]
- Jahn, A.; Vreeland, W.N.; DeVoe, D.L.; Locascio, L.E.; Gaitan, M. Microfluidic Directed Formation of Liposomes of Controlled Size. Langmuir 2007, 23, 6289–6293. [Google Scholar] [CrossRef] [PubMed]
- Didierlaurent, A.M.; Laupeze, B.; Di Pasquale, A.; Hergli, N.; Collignon, C.; Garçon, N. Adjuvant system AS01: Helping to overcome the challenges of modern vaccines. Expert Rev. Vaccines 2017, 16, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Casella, C.R.; Mitchell, T.C. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. 2008, 65, 3231–3240. [Google Scholar] [CrossRef]
- Marty-Roix, R.; Vladimer, G.I.; Pouliot, K.; Weng, D.; Buglione-Corbett, R.; West, K.; MacMicking, J.D.; Chee, J.D.; Wang, S.; Lu, S.; et al. Identification of QS-21 as an Inflammasome-activating Molecular Component of Saponin Adjuvants. J. Biol. Chem. 2016, 291, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Alving, C.R.; Rao, M.; Matyas, G.R. Similarities and differences of chemical compositions and physical and functional properties of adjuvant system 01 and army liposome formulation with QS21. Front. Immunol. 2023, 14, 1102524. [Google Scholar] [CrossRef] [PubMed]
- Alving, C.R.; Peachman, K.K.; Matyas, G.R.; Rao, M.; Beck, Z. Army Liposome Formulation (ALF) family of vaccine adjuvants. Expert Rev. Vaccines 2020, 19, 279–292. [Google Scholar] [CrossRef]
- Beck, Z.; Matyas, G.R.; Jalah, R.; Rao, M.; Polonis, V.R.; Alving, C.R. Differential immune responses to HIV-1 envelope protein induced by liposomal adjuvant formulations containing monophosphoryl lipid A with or without QS21. Vaccine 2015, 33, 5578–5587. [Google Scholar] [CrossRef]
- Study to Evaluate the Efficacy of GlaxoSmithKline (GSK) Biologicals’ Candidate Tuberculosis (TB) Vaccine in Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT01755598 (accessed on 24 May 2023).
- Eldridge, G.R.; Hughey, H.; Rosenberger, L.; Martin, S.M.; Shapiro, A.M.; D’antonio, E.; Krejci, K.G.; Shore, N.; Peterson, J.; Lukes, A.S.; et al. Safety and immunogenicity of an adjuvanted Escherichia coli adhesin vaccine in healthy women with and without histories of recurrent urinary tract infections: Results from a first-in-human phase 1 study. Hum. Vaccines Immunother. 2020, 17, 1262–1270. [Google Scholar] [CrossRef]
- A Study of a Vaccine for Pneumonia in Adults and Toddlers in Kenya. Available online: https://clinicaltrials.gov/ct2/show/NCT02543892 (accessed on 26 February 2023).
- Safety Study of a Capsule-Conjugate Vaccine to Prevent Campylobacter-Caused Diarrhea (CJCV1-01). Available online: https://clinicaltrials.gov/ct2/show/NCT02067676 (accessed on 22 February 2023).
- Study of the Safety and Efficacy of the Subunit Recombinant Tuberculosis Vaccine GamTBvac. Available online: https://clinicaltrials.gov/ct2/history/NCT04975737?V_4=View#StudyPageTop (accessed on 23 February 2023).
- Pulendran, B.; Arunachalam, P.S.; O’hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- O’Hagan, D.T.; Singh, M. MF59: A Safe and Potent Oil-in-Water Emulsion Adjuvant; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 115–129. [Google Scholar] [CrossRef]
- Song, J.Y.; Cheong, H.J.; Tsai, T.; Chang, H.-A.; Choi, M.J.; Jeon, J.H.; Kang, S.H.; Jeong, E.J.; Noh, J.Y.; Kim, W.J. Immunogenicity and safety of concomitant MF59-adjuvanted influenza vaccine and 23-valent pneumococcal polysaccharide vaccine administration in older adults. Vaccine 2015, 33, 4647–4652. [Google Scholar] [CrossRef]
- Criscuolo, E.; Caputo, V.; Diotti, R.A.; Sautto, G.A.; Kirchenbaum, G.A.; Clementi, N. Alternative Methods of Vaccine Delivery: An Overview of Edible and Intradermal Vaccines. J. Immunol. Res. 2019, 2019, 8303648. [Google Scholar] [CrossRef] [PubMed]
- Kis, E.E.; Winter, G.; Myschik, J. Devices for intradermal vaccination. Vaccine 2011, 30, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Sun, Y.; Cui, H.; Zhu, S.J.; Qiu, H.-J. Mucosal vaccines: Strategies and challenges. Immunol. Lett. 2019, 217, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Lin, S.-J.; Yang, Y.-C.; Wang, D.-Y.; Cheng, H.-F.; Yeh, M.-K. Biodegradable polymeric microsphere-based vaccines and their applications in infectious diseases. Hum. Vaccines Immunother. 2015, 11, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Corthésy, B.; Bioley, G. Lipid-Based Particles: Versatile Delivery Systems for Mucosal Vaccination against Infection. Front. Immunol. 2018, 9, 431. [Google Scholar] [CrossRef]
- Vivotif Typhoid Vaccine Live Oral Ty21a. Available online: https://www.fda.gov/media/75988/download (accessed on 6 April 2023).
- Lycke, N. Recent progress in mucosal vaccine development: Potential and limitations. Nat. Rev. Immunol. 2012, 12, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Tribble, D.; Kaminski, R.; Cantrell, J.; Nelson, M.; Porter, C.; Baqar, S.; Williams, C.; Arora, R.; Saunders, J.; Ananthakrishan, M.; et al. Safety and immunogenicity of a Shigella flexneri 2a Invaplex 50 intranasal vaccine in adult volunteers. Vaccine 2010, 28, 6076–6085. [Google Scholar] [CrossRef]
- Combadiere, B.; Liard, C. Transcutaneous and intradermal vaccination. Hum. Vaccines 2011, 7, 811–827. [Google Scholar] [CrossRef]
- Menon, I.; Bagwe, P.; Gomes, K.B.; Bajaj, L.; Gala, R.; Uddin, M.N.; D’souza, M.J.; Zughaier, S.M. Microneedles: A New Generation Vaccine Delivery System. Micromachines 2021, 12, 435. [Google Scholar] [CrossRef] [PubMed]
- Frech, S.A.; DuPont, H.L.; Bourgeois, A.L.; McKenzie, R.; Belkind-Gerson, J.; Figueroa, J.F.; Okhuysen, P.C.; Guerrero, N.H.; Martinez-Sandoval, F.G.; Meléndez-Romero, J.H.; et al. Use of a patch containing heat-labile toxin from Escherichia coli against travellers’ diarrhoea: A phase II, randomised, double-blind, placebo-controlled field trial. Lancet 2008, 371, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, H.; Yang, C.; Wu, Y.; Zhou, X.; Liu, H.; Wang, Y. Bacterial outer membrane vesicles as a platform for biomedical applications: An update. J. Control. Release 2020, 323, 253–268. [Google Scholar] [CrossRef]
- ICH Official Web Site: Q2(R2)/Q14 EWG, Analytical Procedure Development and Revision of Q2(R1) Analytical Validation. Available online: https://www.ich.org/page/quality-guidelines (accessed on 19 February 2023).
- ICH Official Web Site: ICH Implementation Working Group (IWG) on ICH Q8, Q9 and Q10. Available online: https://database.ich.org/sites/default/files/Q8_Q9_Q10_Q%26As_R4_Q%26As_0.pdf (accessed on 18 February 2023).
- ICH Official Web Site: Q2A and Q2B, “Validation of Analytical Procedures: Text and Methodology” and Q14, ‘Analytical Procedure Development’. Proposed Revision Realeas in May 2023. Available online: https://database.ich.org/sites/default/files/Q2_Q14%20ICH_Step_2_Presentation_2022_0325.pdf (accessed on 18 February 2023).
- Rogers, R.S.; Abernathy, M.; Richardson, D.D.; Rouse, J.C.; Sperry, J.B.; Swann, P.; Wypych, J.; Yu, C.; Zang, L.; Deshpande, R. A View on the Importance of “Multi-Attribute Method” for Measuring Purity of Biopharmaceuticals and Improving Overall Control Strategy. AAPS J. 2017, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Eris, T.; Zhang, L.; Ren, D.; Ricci, M.S.; Thiel, T.; Goudar, C.T. A high-resolution multi-attribute method for product characterization, process characterization, and quality control of therapeutic proteins. Anal. Biochem. 2022, 643, 114575. [Google Scholar] [CrossRef] [PubMed]
- Millán-Martín, S.; Jakes, C.; Carillo, S.; Rogers, R.; Da Ren, D.; Bones, J. Comprehensive multi-attribute method workflow for biotherapeutic characterization and current good manufacturing practices testing. Nat. Protoc. 2022, 18, 1056–1089. [Google Scholar] [CrossRef]
- Sharma, V.K.; Sharma, I.; Glick, J. The expanding role of mass spectrometry in the field of vaccine development. Mass Spectrom. Rev. 2018, 39, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Gunawardena, H.P.; Li, X.; Kim, Y.-I.; Dewald, H.D.; Chen, H. Standard-Free Absolute Quantitation of Antibody Deamidation Degradation and Host Cell Proteins by Coulometric Mass Spectrometry. Anal. Chem. 2022, 94, 12490–12499. [Google Scholar] [CrossRef]
- Li, C.; Chu, S.; Tan, S.; Yin, X.; Jiang, Y.; Dai, X.; Gong, X.; Fang, X.; Tian, D. Towards Higher Sensitivity of Mass Spectrometry: A Perspective From the Mass Analyzers. Front. Chem. 2021, 9, 813359. [Google Scholar] [CrossRef] [PubMed]
- Voeten, R.L.C.; Ventouri, I.K.; Haselberg, R.; Somsen, G.W. Capillary Electrophoresis: Trends and Recent Advances. Anal. Chem. 2018, 90, 1464–1481. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Ma, L.; Wang, L.; Xiao, P.; Li, H.; Zhou, M.; Song, D. Research advances in hydrogen–deuterium exchange mass spectrometry for protein epitope mapping. Anal. Bioanal. Chem. 2021, 413, 2345–2359. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Liuni, P.; Chen, T.; Houy, C.; Wilson, D.J.; James, D.A. Epitope screening using Hydrogen/Deuterium Exchange Mass Spectrometry (HDX-MS): An accelerated workflow for evaluation of lead monoclonal antibodies. Biotechnol. J. 2021, 17, 2100358. [Google Scholar] [CrossRef] [PubMed]
- Ständer, S.; Grauslund, L.R.; Scarselli, M.; Norais, N.; Rand, K. Epitope Mapping of Polyclonal Antibodies by Hydrogen–Deuterium Exchange Mass Spectrometry (HDX-MS). Anal. Chem. 2021, 93, 11669–11678. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Z.; Lancaster, C.; Winters, M.A.; Phillips, K.M.; Zhuang, P.; Ha, S. Multi-attribute characterization of pneumococcal conjugate vaccine by Size-exclusion chromatography coupled with UV-MALS-RI detections. Vaccine 2022, 40, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, N.; Berti, F. NMR characterization of bacterial glycans and glycoconjugate vaccines. In Recent Trends in Carbohydrate Chemistry; Elsevier: Amsterdam, The Netherlands, 2020; pp. 239–281. [Google Scholar] [CrossRef]
- Berti, F.; Ravenscroft, N. Characterization of Carbohydrate Vaccines by NMR Spectroscopy. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1331, pp. 189–209. [Google Scholar] [CrossRef]
- Berti, F. NMR characterization of a multi-valent conjugate vaccine against Neisseria meningitidis A, C, W, Y and Haemophilus influenzae b infections. J. Pharm. Biomed. Anal. 2021, 205, 114302. [Google Scholar] [CrossRef] [PubMed]
- Martini, S.; Aggravi, M.; Cianetti, S.; Egan, W.; Meppen, M.; Moriconi, A.; Simeone, L.; Berti, F. NMR Assays for Estimating the O-Acetyl Content of Meningococcal Polysaccharide Serogroup A in Quadrivalent Conjugate Vaccine Formulation. ACS Omega 2019, 4, 12827–12832. [Google Scholar] [CrossRef] [PubMed]
- Beri, S.; Gandhi, D. Quantification of residual cetyltrimethylammonium bromide (CTAB) and sodium deoxycholate (DOC) in Haemophilus influenzae type b (Hib) polysaccharide using NMR. Biologicals 2021, 70, 22–27. [Google Scholar] [CrossRef]
- Khatun, R.; Hunter, H.; Magcalas, W.; Sheng, Y.; Carpick, B.; Kirkitadze, M. Nuclear Magnetic Resonance (NMR) Study for the Detection and Quantitation of Cholesterol in HSV529 Therapeutic Vaccine Candidate. Comput. Struct. Biotechnol. J. 2017, 15, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Garrido, R.; Baró, B.; Soubal, J.P.; Santana, D. Quantitative assessment of C-polysaccharide in capsular polysaccharides of Streptococcus pneumoniae by 31PNMR. J. Pharm. Biomed. Anal. 2020, 192, 113670. [Google Scholar] [CrossRef] [PubMed]
- Vialle, S.; Sepulcri, P.; Dubayle, J.; Talaga, P. The teichoic acid (C-polysaccharide) synthesized by Streptococcus pneumoniae serotype 5 has a specific structure. Carbohydr. Res. 2005, 340, 91–96. [Google Scholar] [CrossRef]
- Carboni, F.; Adamo, R.; Fabbrini, M.; De Ricco, R.; Cattaneo, V.; Brogioni, B.; Veggi, D.; Pinto, V.; Passalacqua, I.; Oldrini, D.; et al. Structure of a protective epitope of group B Streptococcus type III capsular polysaccharide. Proc. Natl. Acad. Sci. USA 2017, 114, 5017–5022. [Google Scholar] [CrossRef]
- Ardenkjaer-Larsen, J.-H.; Boebinger, G.S.; Comment, A.; Duckett, S.; Edison, A.S.; Engelke, F.; Griesinger, C.; Griffin, R.G.; Hilty, C.; Maeda, H.; et al. Facing and Overcoming Sensitivity Challenges in Biomolecular NMR Spectroscopy. Angew. Chem. Int. Ed. 2015, 54, 9162–9185. [Google Scholar] [CrossRef]
- Kiss, R.; Fizil, Á.; Szántay, C. What NMR can do in the biopharmaceutical industry. J. Pharm. Biomed. Anal. 2018, 147, 367–377. [Google Scholar] [CrossRef]
- Beger, R.D.; Dunn, W.B.; Bandukwala, A.; Bethan, B.; Broadhurst, D.; Clish, C.B.; Dasari, S.; Derr, L.; Evans, A.; Fischer, S.; et al. Towards quality assurance and quality control in untargeted metabolomics studies. Metabolomics 2019, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Puranik, A.; Dandekar, P.; Jain, R. Exploring the potential of machine learning for more efficient development and production of biopharmaceuticals. Biotechnol. Prog. 2022, 38, e3291. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Abraham, A.; Baldwin, J.; Piplani, S.; Petrovsky, N. Artificial Intelligence in Vaccine and Drug Design. Vaccines Hum. Dis. 2021, 1, 131–146. [Google Scholar] [CrossRef]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- CHMP Position on Bexsero. Available online: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-bexsero_en.pdf (accessed on 14 February 2023).
- Chaudhury, S.; Duncan, E.H.; Atre, T.; Storme, C.K.; Beck, K.; Kaba, S.A.; Lanar, D.E.; Bergmann-Leitner, E.S. Identification of Immune Signatures of Novel Adjuvant Formulations Using Machine Learning. Sci. Rep. 2018, 8, 17508. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Duncan, E.H.; Atre, T.; Dutta, S.; Spring, M.D.; Leitner, W.W.; Bergmann-Leitner, E.S. Combining immunoprofiling with machine learning to assess the effects of adjuvant formulation on human vaccine-induced immunity. Hum. Vaccines Immunother. 2019, 16, 400–411. [Google Scholar] [CrossRef]
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 2021, 11, 3238. [Google Scholar] [CrossRef]
- Bahrami, A.A.; Payandeh, Z.; Khalili, S.; Zakeri, A.; Bandehpour, M. Immunoinformatics: In Silico Approaches and Computational Design of a Multi-epitope, Immunogenic Protein. Int. Rev. Immunol. 2019, 38, 307–322. [Google Scholar] [CrossRef]
- Verch, T.; Trausch, J.J.; Shank-Retzlaff, M. Principles of vaccine potency assays. Bioanalysis 2018, 10, 163–180. [Google Scholar] [CrossRef]
- Sanyal, G. Development of functionally relevant potency assays for monovalent and multivalent vaccines delivered by evolving technologies. npj Vaccines 2022, 7, 50. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Platform | Pathogen | Clinical Trial |
|---|---|---|
| Recombinant protein antigens | Streptococcus pneumoniae | phase 2 [91] |
| Clostridioides difficile | phase 1 [92] | |
| Staphylococcus aureus | phase 1 [93] | |
| Glycoconjugate/bioconjugate | Shigella flexneri | phase2-phase 2b [94,95] |
| Streptococcus pneumoniae | phase 1 [96] | |
| Escherichia coli | phase 1/2 [97] | |
| OMV/GMMA | Escherichia coli | phase 2 [98] |
| Shigella sonneii | phase 2 [99] | |
| Shigella flexneri | phase 1/2 [100] | |
| RNA | Yersinia pestis | preclinical [83] |
| P. aeruginosa | preclinical [85] | |
| GAS/GBS | preclinical [84] | |
| Mycobacterium tuberculosis | preclinical [86] |
| Adjuvant | Mechanism of Action | Pathogen | Antigen | Clinical Trial |
|---|---|---|---|---|
| Alum | Activation of dendritic cells via Nlrp3 inflammasome and induction of innate immune response via interaction between CD11c+ dendritic cells and lymphocytes T [130]. | Staphylococcus aureus | Recombinant protein | phase 1 [93] |
| Streptococcus pneumoniae | Recombinant protein | phase 1\2 [170] | ||
| Campylobacter spp. | glycoconjugate | phase 1 [171] | ||
| Shigella flexneri | bioconjugate | phase 2b [95] | ||
| CpG | Stimulation of TLR9 causing the induction of MyD88 pathway and type I interferon [137]. | Mycobacterium tuberculosis | Recombinant protein | Phase 3 [172] |
| AS01 | Stimulation of T-cell and TLR4, activation of IL-1β/IL-18 after Nlrp3 inflammasome interaction [164]. | Mycobacterium tuberculosis Clostridioides difficiles | Recombinant protein Recombinant protein | phase 2b [168] phase 1 [92] |
| MF59/AS03 | Induction of CD4+ T-cells, T-follicular helper cells (TFHs), and germinal centres (GCs); TLR9-independent activation of MyD88 [173,174]. | Streptococcus pneumoniae Klebsiella pneumoniae | Polysaccharide bioconjugate | phase 1 [175] phase 1 [154] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tognetti, F.; Biagini, M.; Denis, M.; Berti, F.; Maione, D.; Stranges, D. Evolution of Vaccines Formulation to Tackle the Challenge of Anti-Microbial Resistant Pathogens. Int. J. Mol. Sci. 2023, 24, 12054. https://doi.org/10.3390/ijms241512054
Tognetti F, Biagini M, Denis M, Berti F, Maione D, Stranges D. Evolution of Vaccines Formulation to Tackle the Challenge of Anti-Microbial Resistant Pathogens. International Journal of Molecular Sciences. 2023; 24(15):12054. https://doi.org/10.3390/ijms241512054
Chicago/Turabian StyleTognetti, Francesco, Massimiliano Biagini, Maxime Denis, Francesco Berti, Domenico Maione, and Daniela Stranges. 2023. "Evolution of Vaccines Formulation to Tackle the Challenge of Anti-Microbial Resistant Pathogens" International Journal of Molecular Sciences 24, no. 15: 12054. https://doi.org/10.3390/ijms241512054
APA StyleTognetti, F., Biagini, M., Denis, M., Berti, F., Maione, D., & Stranges, D. (2023). Evolution of Vaccines Formulation to Tackle the Challenge of Anti-Microbial Resistant Pathogens. International Journal of Molecular Sciences, 24(15), 12054. https://doi.org/10.3390/ijms241512054