
Transcriptome Metabolic Characterization of Tuber borchii SP1—A New Spanish Strain for In Vitro Studies of the Bianchetto Truffle

 ,
, 
 , , and
, , and 
Abstract
1. Introduction
2. Results
2.1. Culture Conditions, Growth Rate, and Biomass Production
2.2. Hyphal Morphology
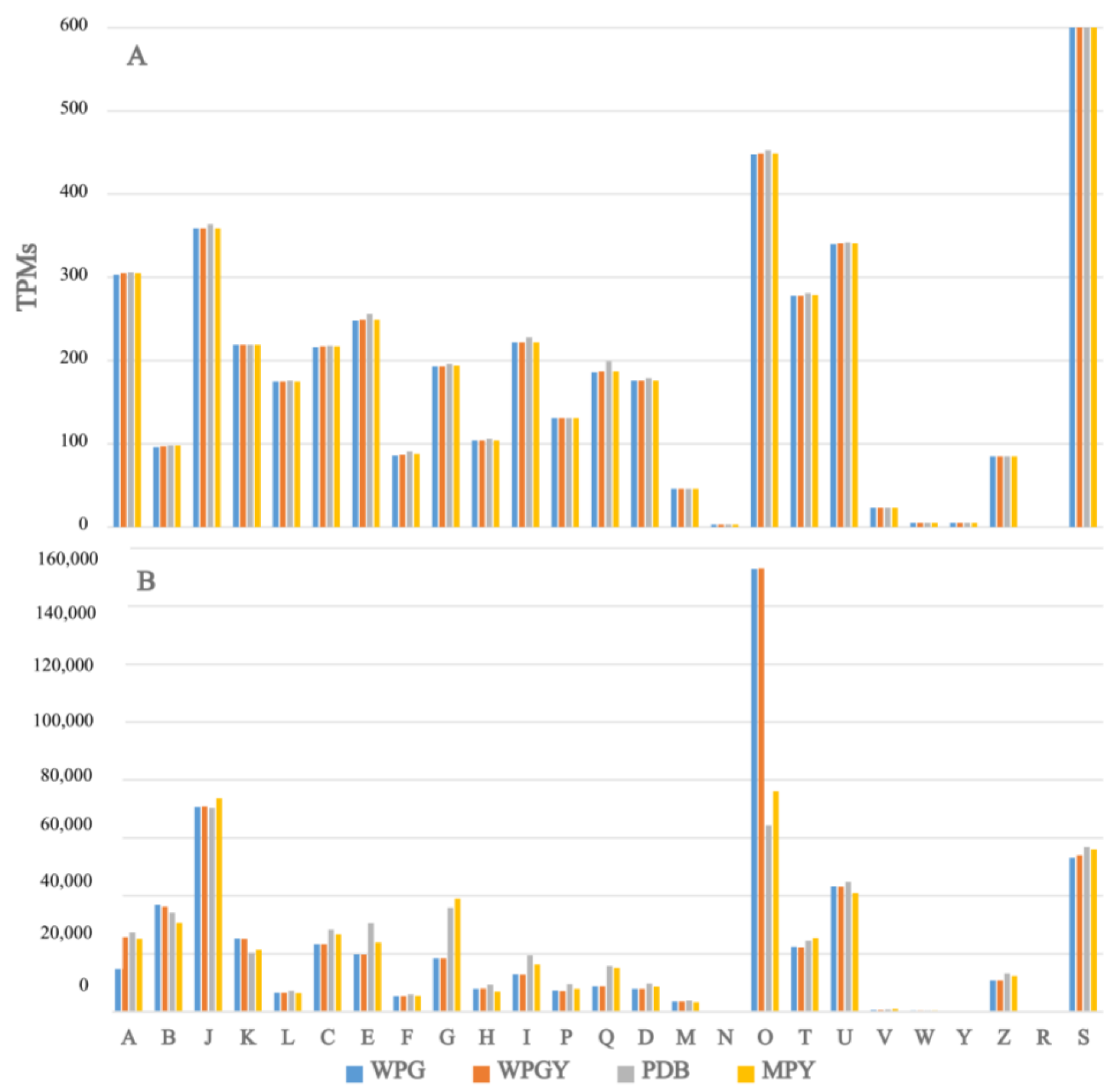
2.3. RNA-Seq and Transcriptome Analysis
2.4. Functional Annotation of the T. borchii SP1 Transcriptome
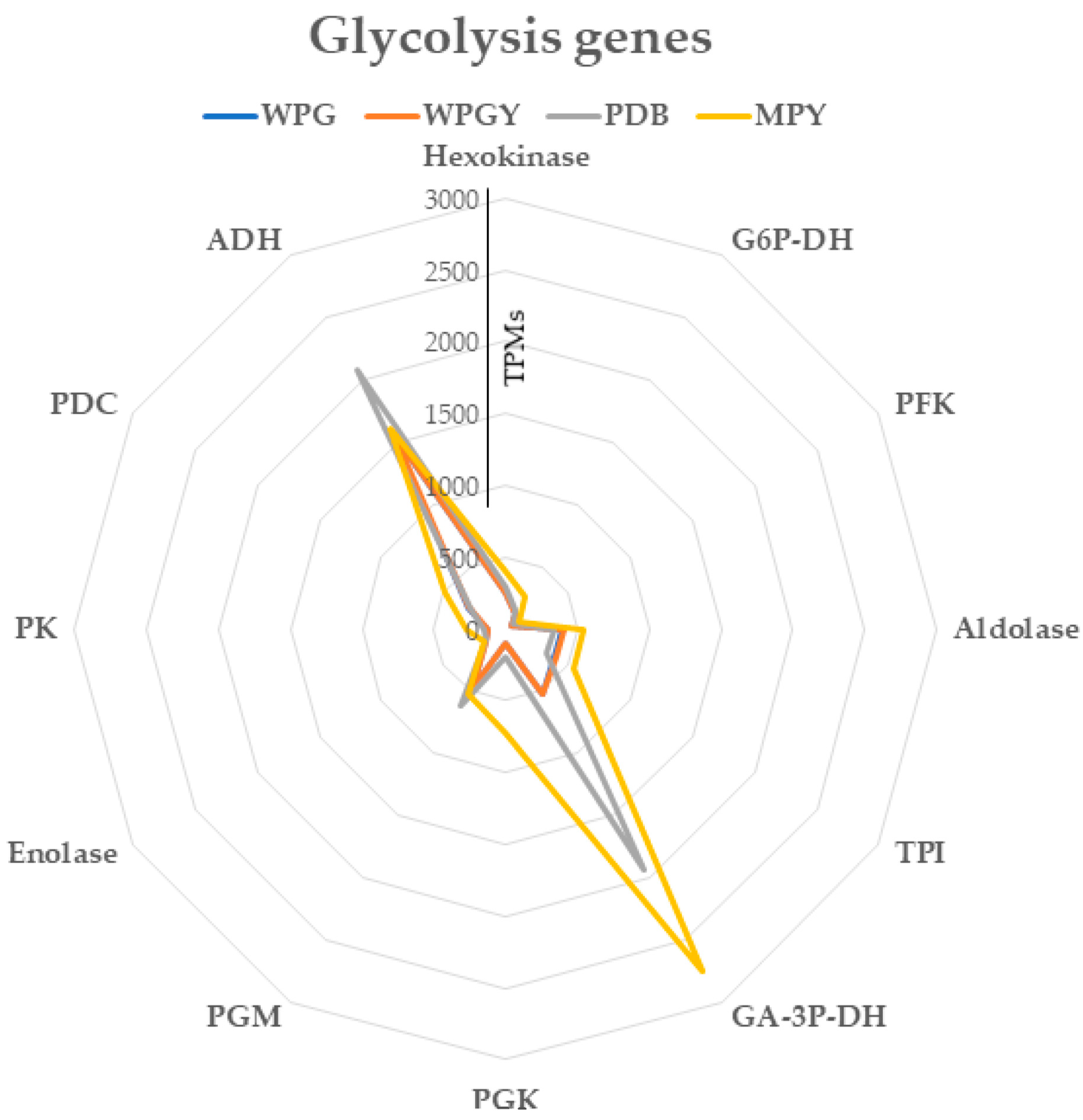
2.5. Pathways of Central Metabolism
2.5.1. Central Metabolism Glycolysis Genes
2.5.2. TCA and Glyoxylate Cycle
2.5.3. Electron Transport Chain
2.6. Genes Involved in Secondary Metabolism Pathways—Clusters
2.7. Genes Related to the Synthesis of Volatile Compounds
3. Discussion
3.1. SP1 Is A New Mycelial Isolate of Tuber Borchii
3.2. RNA-Seq and Transcriptome Analysis
3.3. Comparison of the Transcriptomes from the Different Culture Media
3.4. Pathways of Central Metabolism
3.5. Genes Involved in Secondary Metabolism—Clusters
3.6. Genes Related to the Synthesis of Volatile Compounds
4. Materials and Methods
4.1. Strain and Culture Conditions
4.2. Growth Rate
4.3. Biomass
4.4. Molecular Characterization
4.5. Morphology
4.6. RNA seq
4.7. Transcriptome Data Analysis
4.8. Analysis of Metabolic Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mustafa, A.M.; Angeloni, S.; Nzekoue, F.K.; Abouelenein, D.; Sagratini, G.; Caprioli, G.; Torregiani, E. An Overview on Truffle Aroma and Main Volatile Compounds. Molecules 2020, 25, 5948. [Google Scholar] [CrossRef] [PubMed]
- Mello, A.; Murat, C.; Bonfante, P. Truffles: Much more than a prized and local fungal delicacy. FEMS Microbiol. Lett. 2006, 260, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bonito, G.M.; Smith, M.E. General Systematic Position of the Truffles: Evolutionary Theories. In True Truffle (Tuber spp.) in the World. Soil Ecology, Systematics and Biochemistry; Zambonelli, A., Iotti, M., Murat, C., Eds.; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-31434-1. [Google Scholar]
- Splivallo, R.; Cullere, L. The Smell of Truffles: From Aroma Biosynthesis to Product Quality. In True Truffle (Tuber spp.) in the World. Soil Ecology, Systematics and Biochemistry; Zambonelli, A., Iotti, M., Murat, C., Eds.; Springer International Publishing: Cham, Switzerland, 2016; ISBN 9783319314365. [Google Scholar]
- Bonito, G.; Trappe, J.M.; Rawlinson, P.; Vilgalys, R. Improved resolution of major clades within Tuber and taxonomy of species within the Tuber gibbosum complex. Mycologia 2010, 102, 1042–1057. [Google Scholar] [CrossRef]
- Hall, I.R.; Brown, G.T.; Zambonelli, A. Taming the Truffle: The History Lore and Science of the Ultimate Mushroom, 1st ed.; Timber Press: Portland, OR, USA, 2007; ISBN 9780881928600. [Google Scholar]
- Lacourt, I.; Abba, S.; Bonfante, P.; Martin, F. Isolation and Characterization of Differentially Expressed Genes in the Mycelium and Fruit Body of Tuber borchii. Appl. Environ. Microbiol. 2002, 68, 4574–4582. [Google Scholar] [CrossRef]
- Marathe, S.J.; Hamzi, W.; Bashein, A.M.; Deska, J.; Seppänen-Laakso, T.; Singhal, R.S.; Shamekh, S. Anti-angiogenic and anti-inflammatory activity of the summer truffle (Tuber aestivum Vittad.) extracts and a correlation with the chemical constituents identified therein. Food Res. Int. 2020, 137, 109699. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, X.H.; Li, H.M.; Wang, S.H.; Chen, T.; Yuan, Z.P.; Tang, Y.J. Isolation and characterization of polysaccharides with the antitumor activity from Tuber fruiting bodies and fermentation system. Appl. Microbiol. Biotechnol. 2014, 98, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Wang, G.; Li, H.M.; Zhong, J.J.; Tang, Y.J. Volatile organic compounds from a Tuber melanosporum fermentation system. Food Chem. 2012, 135, 2628–2637. [Google Scholar] [CrossRef]
- Vita, F.; Taiti, C.; Pompeiano, A.; Bazihizina, N.; Lucarotti, V.; Mancuso, S.; Alpi, A. Volatile organic compounds in truffle (Tuber magnatum Pico): Comparison of samples from different regions of Italy and from different seasons. Sci. Rep. 2015, 5, 12629. [Google Scholar] [CrossRef]
- Zambonelli, A.; Iotti, M.; Hall, I. Current status of truffle cultivation: Recent results and future perspectives. Micol. Ital. 2015, 44, 31–40. [Google Scholar] [CrossRef]
- Shah, N.; Marathe, S.J.; Croce, D.; Ciardi, M.; Longo, V.; Juilus, A.; Shamekh, S. An investigation of the antioxidant potential and bioaccumulated minerals in Tuber borchii and Tuber maculatum mycelia obtained by submerged fermentation. Arch. Microbiol. 2022, 204, 64. [Google Scholar] [CrossRef]
- Tang, Y.J.; Liu, R.S.; Li, H.M. Current progress on truffle submerged fermentation: A promising alternative to its fruiting bodies. Appl. Microbiol. Biotechnol. 2015, 99, 2041–2053. [Google Scholar] [CrossRef]
- Splivallo, R.; Bossi, S.; Maffei, M.; Bonfante, P. Discrimination of truffle fruiting body versus mycelial aromas by stir bar sorptive extraction. Phytochemistry 2007, 68, 2584–2598. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Y.J.; Li, H.M.; Tang, Y.Y.J. Comparison of sterol composition between Tuber fermentation mycelia and natural fruiting bodies. Food Chem. 2012, 132, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Tirillini, B.; Verdelli, G.; Paolocci, F.; Ciccioli, P.; Frattoni, M. The volatile organic compounds from the mycelium of Tuber borchii Vitt. Phytochemistry 2000, 55, 983–985. [Google Scholar] [CrossRef]
- Murat, C.; Payen, T.; Noel, B.; Kuo, A.; Morin, E.; Chen, J.; Kohler, A.; Krizsán, K.; Balestrini, R.; Da Silva, C.; et al. Pezizomycetes genomes reveal the molecular basis of ectomycorrhizal truffle lifestyle. Nat. Ecol. Evol. 2018, 2, 1956–1965. [Google Scholar] [CrossRef]
- Wang, S.; Marcone, M.F. The biochemistry and biological properties of the world’s most expensive underground edible mushroom: Truffles. Food Res. Int. 2011, 44, 2567–2581. [Google Scholar] [CrossRef]
- Martin, F.; Kohler, A.; Murat, C.; Balestrini, R.; Coutinho, P.M.; Jaillon, O.; Montanini, B.; Morin, E.; Noel, B.; Percudani, R.; et al. Périgord black truffle genome uncovers evolutionary origins and mechanisms of symbiosis. Nature 2010, 464, 1033–1038. [Google Scholar] [CrossRef]
- Murat, C.; Martin, F. Truffle genomics: Investigating an early diverging lineage of Pezizomycotina. In True Truffle (Tuber spp.) in the world. Soil Ecology, Systematics and Biochemistry; Zambonelli, A., Iotti, M., Murat, C., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 137–149. [Google Scholar]
- Murat, C.; Kuo, A.; Barry, K.W.; Clum, A.; Dockter, R.B.; Fauchery, L.; Iotti, M.; Kohler, A.; Labutti, K.; Lindquist, E.A.; et al. Draft Genome Sequence of Tuber borchii Vittad., a Whitish Edible Truffle. Genome Announc. 2018, 6, e00537-18. [Google Scholar] [CrossRef] [PubMed]
- Ceccaroli, P.; Buffalini, M.; Saltarelli, R.; Barbieri, E.; Polidori, E.; Ottonello, S.; Kohler, A.; Tisserant, E.; Martin, F.; Stocchi, V. Genomic profiling of carbohydrate metabolism in the ectomycorrhizal fungus Tuber melanosporum. New Phytol. 2011, 189, 751–764. [Google Scholar] [CrossRef]
- Alfaro, M.; Castanera, R.; Lavín, J.L.; Grigoriev, I.V.; Oguiza, J.A.; Ramírez, L.; Pisabarro, A.G. Comparative and transcriptional analysis of the predicted secretome in the lignocellulose-degrading basidiomycete fungus Pleurotus ostreatus. Environ. Microbiol. 2016, 18, 4710–4726. [Google Scholar] [CrossRef]
- Aguilar-Pontes, M.V.; Brandl, J.; McDonnell, E.; Strasser, K.; Nguyen, T.T.M.; Riley, R.; Mondo, S.; Salamov, A.; Nybo, J.L.; Vesth, T.C.; et al. The gold-standard genome of Aspergillus niger NRRL 3 enables a detailed view of the diversity of sugar catabolism in fungi. Stud. Mycol. 2018, 91, 61–78. [Google Scholar] [CrossRef]
- Li, J.; Chroumpi, T.; Garrigues, S.; Kun, R.S.; Meng, J.; Salazar-Cerezo, S.; Aguilar-Pontes, M.V.; Zhang, Y.; Tejomurthula, S.; Lipzen, A.; et al. The Sugar Metabolic Model of Aspergillus niger Can Only Be Reliably Transferred to Fungi of Its Phylum. J. Fungi. 2022, 8, 1315. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Smirnov, S.; Nikolskaya, A.N.; et al. The COG database: An updated vesion includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, Z.; Xu, L.; Shi, W.; Liu, X.; Wang, F. Increasing production of truffle polysaccharides in the solid-state fermentation of Tuber melanosporum by diosgenin based on orthogonal matrix and nonlinear regression analysis. Food Sci. Technol. Res. 2020, 26, 487–494. [Google Scholar] [CrossRef]
- Tang, Y.J.; Wang, G.; Li, Y.Y.; Zhong, J.J. Fermentation condition outweighed truffle species in affecting volatile organic compounds analyzed by chromatographic fingerprint system. Anal. Chim. Acta 2009, 647, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.N.; Liu, R.S.; Wang, Y.H.; Mi, Z.Y.; Li, D.S.; Zhong, J.J.; Tang, Y.J. Fed-batch fermentation of Tuber melanosporum for the hyperproduction of mycelia and bioactive Tuber polysaccharides. Bioresour. Technol. 2009, 100, 3644–3649. [Google Scholar] [CrossRef]
- Iotti, M.; Amicucci, A.; Stocchi, V.; Zambonelli, A. Morphological and molecular characterization of mycelia of some Tuber species in pure culture. New Phytol. 2002, 155, 499–505. [Google Scholar] [CrossRef]
- Vahdatzadeh, M.; Deveau, A.; Splivallo, R. The role of the microbiome of truffles in aroma formation: A meta-analysis approach. Appl. Environ. Microbiol. 2015, 81, 6946–6952. [Google Scholar] [CrossRef]
- Barbieri, E.; Gioacchini, A.M.; Zambonelli, A.; Bertini, L.; Stocchi, V. Determination of microbial volatile organic compounds from Staphylococcus pasteuri against Tuber borchii using solid-phase microextraction and gas chromatography/ion trap mass spectrometry. Rapid Commun. Mass Spectrom. 2015, 19, 3411–3415. [Google Scholar] [CrossRef]
- Perlińska-Lenart, U.; Piłsyk, S.; Gryz, E.; Turło, J.; Hilszczańska, D.; Kruszewska, J.S. Identification of bacteria and fungi inhabiting fruiting bodies of Burgundy truffle (Tuber aestivum Vittad.). Arch. Microbiol. 2020, 202, 2727–2738. [Google Scholar] [CrossRef]
- Leonardi, P.; Murat, C.; Puliga, F.; Iotti, M.; Zambonelli, A. Ascoma genotyping and mating type analyses of mycorrhizas and soil mycelia of Tuber borchii in a truffle orchard established by mycelial inoculated plants. Environ. Microbiol. 2020, 22, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Murat, C.; Rubini, A.; Riccioni, C.; De la Varga, H.; Akroume, E.; Belfiori, B.; Guaragno, M.; Le Tacon, F.; Robin, C.; Halkett, F.; et al. Fine-scale spatial genetic structure of the black truffle (Tuber melanosporum) investigated with neutral microsatellites and functional mating type genes. New Phytol. 2013, 199, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Moore, D. Fungal morphogenesis; Cambridge University Press: Cambridge, UK, 1998; ISBN 0-521-55295-8. [Google Scholar]
- Graziosi, S.; Hall, I.R.; Zambonelli, A. The Mysteries of the White Truffle: Its Biology, Ecology and Cultivation. Encyclopedia 2022, 2, 1959–1971. [Google Scholar] [CrossRef]
- Chen, C.C.; Nargotra, P.; Kuo, C.H.; Liu, Y.C. High-Molecular-Weight Exopolysaccharides Production from Tuber brochii Cultivated by Submerged Fermentation. Int. J. Mol. Sci. 2023, 24, 4875. [Google Scholar] [CrossRef] [PubMed]
- Amicucci, A.; Zambonelli, A.; Iotti, M.; Polidori, E.; Menotta, M.; Saltarelli, R.; Potenza, L.; Stocchi, V. Morphological and molecular modifications induced by different carbohydrate sources in Tuber borchii. J. Mol. Microbiol. Biotechnol. 2010, 18, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, E.; Balestrini, R.; Kohler, A.; Abbà, S.; Martin, F.; Bonfante, P. The Perigord black truffle responds to cold temperature with an extensive reprogramming of its transcriptional activity. Fungal Genet. Biol. 2011, 48, 585–591. [Google Scholar] [CrossRef]
- Ling, Y.Y.; Ling, Z.L.; Zhao, R.L. Construction of a heat-resistant strain of Lentinus edodes by fungal Hsp20 protein overexpression and genetic transformation. Front. Microbiol. 2022, 13, 1009885. [Google Scholar] [CrossRef]
- De Maio, A.; Vazquez, D. Extracellular heat shock proteins: A new location, a new function. Shock 2013, 40, 239–246. [Google Scholar] [CrossRef]
- Abba’, S.; Balestrini, R.; Benedetto, A.; Rottensteiner, H.; De Lucas, J.R.; Bonfante, P. The role of the glyoxylate cycle in the symbiotic fungus Tuber borchii: Expression analysis and subcellular localization. Curr. Genet. 2007, 52, 159–170. [Google Scholar] [CrossRef]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Pusztahelyi, T.; Holb, I.J.; Pócsi, I. Secondary metabolites in fungus-plant interactions. Front. Plant Sci. 2015, 6, 573. [Google Scholar] [CrossRef]
- De Angelis, F.; Arcadi, A.; Marinelli, F.; Paci, M.; Botti, D.; Pacioni, G.; Miranda, M. Partial structures of truffle melnins. Phytochemistry 1996, 43, 1103–1106. [Google Scholar] [CrossRef]
- Medina, A.; Schmidt-Heydt, M.; Rodríguez, A.; Parra, R.; Geisen, R.; Magan, N. Impacts of environmental stress on growth, secondary metabolite biosynthetic gene clusters and metabolite production of xerotolerant/xerophilic fungi. Curr. Genet. 2015, 61, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Simon Andrews FastQC. A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 December 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Culture Medium | Fresh Weight (mg) | Dry Weight (mg) |
|---|---|---|
| WPG | 535 ± 103 | 41 ± 7 |
| PDB | 470 ± 89 | 38 ± 1 |
| MPY | 1600 ± 270 | 127 ± 22 |
| Sample | Total Reads | Genes Expressed | Genes KOG |
|---|---|---|---|
| WPG | 116,376,579 | 10,870 | 4645 |
| WPGY | 125,457,058 | 10,941 | 4642 |
| MPY | 47,969,567 | 10,442 | 4582 |
| PDB | 157,335,190 | 10,599 | 4621 |
| Number of genes in the reference genome | 12,346 | 5686 | |
| Protein Id | TPMs-WPG | Function | Protein Id | TPMs-WPGY | Function |
|---|---|---|---|---|---|
| 990338 | 59710.6 | Molecular chaperone | 990338 | 59732.8 | Molecular chaperone |
| 1125059 | 37904.9 | N/A | 1125059 | 38565.1 | N/A |
| 985714 | 19175.7 | N/A | 985714 | 19689.8 | N/A |
| 986569 | 18513.8 | N/A | 986569 | 18720.1 | N/A |
| 907400 | 15291.6 | N/A | 907400 | 15059.6 | N/A |
| 1138063 | 12188.2 | N/A | 1138063 | 12331.1 | N/A |
| 1039918 | 11559.9 | Molecular chaperone | 1039918 | 11506.3 | Molecular chaperone |
| 1067414 | 9526.58 | N/A | 961630 | 9712.31 | Histone H4 |
| 961630 | 9394.31 | Histone H4 | 1067414 | 9622.65 | N/A |
| 1124376 | 9124.24 | Histones H3 and H4 | 1124376 | 9300.14 | Histones H3 and H4 |
| Protein Id | TPMs-PDB | Function | Protein Id | TPMs-MPY | Function |
| 1067414 | 20972.3 | N/A | 957843 | 19500.8 | GAPDH |
| 1138063 | 18989.4 | N/A | 892438 | 18481.5 | N/A |
| 1098806 | 16159.1 | N/A | 1067414 | 13726.7 | N/A |
| 1125059 | 13738.2 | N/A | 1098806 | 11293 | N/A |
| 957843 | 13582.2 | GAPDH | 987363 | 10269.6 | N/A |
| 907400 | 12134 | N/A | 1138063 | 9857.26 | N/A |
| 1124376 | 10231.8 | Histones H3 and H4 | 1041746 | 9720.97 | N/A |
| 1103054 | 9425.23 | N/A | 1124376 | 8576.88 | Histones H3 and H4 |
| 961630 | 9060.03 | Histone H4 | 1127611 | 8393.75 | N/A |
| 981017 | 6404.74 | N/A | 907400 | 8022.94 | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomazeli, E.C.; Alfaro, M.; Zambonelli, A.; Garde, E.; Pérez, G.; Jiménez, I.; Ramírez, L.; Salman, H.; Pisabarro, A.G. Transcriptome Metabolic Characterization of Tuber borchii SP1—A New Spanish Strain for In Vitro Studies of the Bianchetto Truffle. Int. J. Mol. Sci. 2023, 24, 10981. https://doi.org/10.3390/ijms241310981
Tomazeli EC, Alfaro M, Zambonelli A, Garde E, Pérez G, Jiménez I, Ramírez L, Salman H, Pisabarro AG. Transcriptome Metabolic Characterization of Tuber borchii SP1—A New Spanish Strain for In Vitro Studies of the Bianchetto Truffle. International Journal of Molecular Sciences. 2023; 24(13):10981. https://doi.org/10.3390/ijms241310981
Chicago/Turabian StyleTomazeli, Emilia Chuina, Manuel Alfaro, Alessandra Zambonelli, Edurne Garde, Gumer Pérez, Idoia Jiménez, Lucía Ramírez, Hesham Salman, and Antonio G. Pisabarro. 2023. "Transcriptome Metabolic Characterization of Tuber borchii SP1—A New Spanish Strain for In Vitro Studies of the Bianchetto Truffle" International Journal of Molecular Sciences 24, no. 13: 10981. https://doi.org/10.3390/ijms241310981
APA StyleTomazeli, E. C., Alfaro, M., Zambonelli, A., Garde, E., Pérez, G., Jiménez, I., Ramírez, L., Salman, H., & Pisabarro, A. G. (2023). Transcriptome Metabolic Characterization of Tuber borchii SP1—A New Spanish Strain for In Vitro Studies of the Bianchetto Truffle. International Journal of Molecular Sciences, 24(13), 10981. https://doi.org/10.3390/ijms241310981