
S-Nitrosylation of Tissue Transglutaminase in Modulating Glycolysis, Oxidative Stress, and Inflammatory Responses in Normal and Indoxyl-Sulfate-Induced Endothelial Cells

 , ,
, ,
Abstract
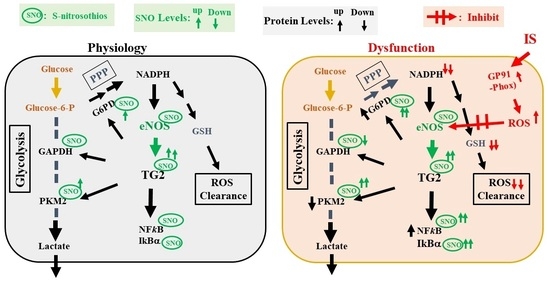
1. Introduction
2. Results
2.1. Intracellular Cellular NADP+/NADPH and GSH/GSSG Ratios
2.2. Effect of ZDON on Extracellular Acidification Rate (ECAR) upon IS Exposure
2.3. Effects of ZDON on the Expression of eNOS and Phospho-eNOS upon IS Exposure
2.4. In Situ TGase Assay to Identify Potential TG2/TGase Substrates
2.5. SNO-RAC on Purified TG2, Total SNO Proteins and S-nitrosylation of TG2/TGase Substrate Proteins during IS-Induced Injury
2.6. Time Courses Studies on the Expression of TG2/TGase Substrates, Redox, and Inflammatory Proteins upon IS Exposure Either in the Presence or Absence of ZDON
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Antibodies
4.3. SDS-PAGE and Immunoblotting
4.4. Cells
4.5. Cell Treatments and Total Lysates Preparation
4.6. Cellular GSH/GSSG Assay
4.7. Cellular NADP+/NADPH Assay
4.8. XFe24 Seahorse Glycolysis Stress Assays
4.9. Resin Assisted Capture for S-nitrosothiols (SNO-RAC)
4.10. SNO-RAC of Purified TG2
4.11. Total SNO Proteins Content
4.12. In Situ TG2/TGase Assay
4.13. Intracellular ROS Generation Assay
4.14. Molecular Modeling
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Taki, K.; Nakamura, S.; Miglinas, M.; Enomoto, A.; Niwa, T. Accumulation of indoxyl sulfate in OAT1/3-positive tubular cells in kidneys of patients with chronic renal failure. J. Ren. Nutr. 2006, 16, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.L.; Zhang, R.; Anand, P.; Stomberski, C.T.; Qian, Z.; Hausladen, A.; Wang, L.; Rhee, E.P.; Parikh, S.M.; Karumanchi, S.A.; et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 2019, 565, 96–100. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef]
- Zhou, H.L.; Stomberski, C.T.; Stamler, J.S. Cross Talk Between S-Nitrosylation and Phosphorylation Involving Kinases and Nitrosylases. Circ. Res. 2018, 122, 1485–1487. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- Lai, T.S.; Hausladen, A.; Slaughter, T.F.; Eu, J.P.; Stamler, J.S.; Greenberg, C.S. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry 2001, 40, 4904–4910. [Google Scholar] [CrossRef]
- Lai, T.S.; Lindberg, R.A.; Zhou, H.L.; Haroon, Z.A.; Dewhirst, M.W.; Hausladen, A.; Juang, Y.L.; Stamler, J.S.; Greenberg, C.S. Endothelial cell-surface tissue transglutaminase inhibits neutrophil adhesion by binding and releasing nitric oxide. Sci. Rep. 2017, 7, 16163. [Google Scholar] [CrossRef]
- Mastroberardino, P.G.; Farrace, M.G.; Viti, I.; Pavone, F.; Fimia, G.M.; Melino, G.; Rodolfo, C.; Piacentini, M. “Tissue” transglutaminase contributes to the formation of disulphide bridges in proteins of mitochondrial respiratory complexes. Biochim. Biophys. Acta 2006, 1757, 1357–1365. [Google Scholar] [CrossRef]
- Lai, T.S.; Lin, C.J.; Greenberg, C.S. Role of tissue transglutaminase-2 (TG2)-mediated aminylation in biological processes. Amino Acids 2017, 49, 501–515. [Google Scholar] [CrossRef]
- Stamnaes, J.; Pinkas, D.M.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [Google Scholar] [CrossRef]
- Izquierdo-Alvarez, A.; Ramos, E.; Villanueva, J.; Hernansanz-Agustin, P.; Fernandez-Rodriguez, R.; Tello, D.; Carrascal, M.; Martinez-Ruiz, A. Differential redox proteomics allows identification of proteins reversibly oxidized at cysteine residues in endothelial cells in response to acute hypoxia. J. Proteom. 2012, 75, 5449–5462. [Google Scholar] [CrossRef]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef]
- Lai, T.S.; Liu, Y.; Li, W.; Greenberg, C.S. Identification of two GTP-independent alternatively spliced forms of tissue transglutaminase in human leukocytes, vascular smooth muscle, and endothelial cells. FASEB J. 2007, 21, 4131–4143. [Google Scholar] [CrossRef]
- Johnson, T.S.; Fisher, M.; Haylor, J.L.; Hau, Z.; Skill, N.J.; Jones, R.; Saint, R.; Coutts, I.; Vickers, M.E.; El Nahas, A.M.; et al. Transglutaminase inhibition reduces fibrosis and preserves function in experimental chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 3078–3088. [Google Scholar] [CrossRef]
- Burhan, I.; Furini, G.; Lortat-Jacob, H.; Atobatele, A.G.; Scarpellini, A.; Schroeder, N.; Atkinson, J.; Maamra, M.; Nutter, F.H.; Watson, P.; et al. Interplay between transglutaminases and heparan sulphate in progressive renal scarring. Sci. Rep. 2016, 6, 31343. [Google Scholar] [CrossRef]
- Balajthy, Z.; Csomos, K.; Vamosi, G.; Szanto, A.; Lanotte, M.; Fesus, L. Tissue-transglutaminase contributes to neutrophil granulocyte differentiation and functions. Blood 2006, 108, 2045–2054. [Google Scholar] [CrossRef]
- Liu, C.; Luo, R.; Wang, W.; Peng, Z.; Johnson, G.V.W.; Kellems, R.E.; Xia, Y. Tissue Transglutaminase-Mediated AT1 Receptor Sensitization Underlies Pro-inflammatory Cytokine LIGHT-Induced Hypertension. Am. J. Hypertens. 2019, 32, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, L.; Tuday, E.C.; Webb, A.K.; Dowzicky, P.; Kim, J.H.; Oh, Y.J.; Sikka, G.; Kuo, M.; Halushka, M.K.; Macgregor, A.M.; et al. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ. Res. 2010, 107, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Jandu, S.; Steppan, J.; Belkin, A.; An, S.S.; Pak, A.; Choi, E.Y.; Nyhan, D.; Butlin, M.; Viegas, K.; et al. Increased tissue transglutaminase activity contributes to central vascular stiffness in eNOS knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H803–H810. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, M.; Thole, J.; Bibli, S.I.; Luck, B.; Loot, A.E.; de Silva, K.; Wittig, I.; Heidler, J.; Stingl, H.; Randriamboavonjy, V.; et al. Nitric oxide maintains endothelial redox homeostasis through PKM2 inhibition. EMBO J. 2019, 38, e100938. [Google Scholar] [CrossRef]
- Kim, B.; Jang, C.; Dharaneeswaran, H.; Li, J.; Bhide, M.; Yang, S.; Li, K.; Arany, Z. Endothelial pyruvate kinase M2 maintains vascular integrity. J. Clin. Investig. 2018, 128, 4543–4556. [Google Scholar] [CrossRef]
- Sun, J.; Steenbergen, C.; Murphy, E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid. Redox Signal. 2006, 8, 1693–1705. [Google Scholar] [CrossRef]
- Keillor, J.W.; Apperley, K.Y.; Akbar, A. Inhibitors of tissue transglutaminase. Trends Pharmacol. Sci. 2015, 36, 32–40. [Google Scholar] [CrossRef]
- Kumar, A.; Xu, J.; Sung, B.; Kumar, S.; Yu, D.; Aggarwal, B.B.; Mehta, K. Evidence that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2012, 14, R4. [Google Scholar] [CrossRef]
- Slaughter, T.F.; Achyuthan, K.E.; Lai, T.S.; Greenberg, C.S. A microtiter plate transglutaminase assay utilizing 5-(biotinamido)pentylamine as substrate. Anal. Biochem. 1992, 205, 166–171. [Google Scholar] [CrossRef]
- Staffler, R.; Pasternack, R.; Hils, M.; Kaiser, W.; Moller, F.M. Nucleotide binding kinetics and conformational change analysis of tissue transglutaminase with switchSENSE. Anal. Biochem. 2020, 605, 113719. [Google Scholar] [CrossRef]
- Forrester, M.T.; Thompson, J.W.; Foster, M.W.; Nogueira, L.; Moseley, M.A.; Stamler, J.S. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 2009, 27, 557–559. [Google Scholar] [CrossRef]
- Mizuno, Y.; Jacob, R.F.; Mason, R.P. Effects of calcium channel and renin-angiotensin system blockade on intravascular and neurohormonal mechanisms of hypertensive vascular disease. Am. J. Hypertens. 2008, 21, 1076–1085. [Google Scholar] [CrossRef]
- Arenas, I.A.; Xu, Y.; Lopez-Jaramillo, P.; Davidge, S.T. Angiotensin II-induced MMP-2 release from endothelial cells is mediated by TNF-alpha. Am. J. Physiol. Cell Physiol. 2004, 286, C779–C784. [Google Scholar] [CrossRef]
- Shimizu, H.; Saito, S.; Higashiyama, Y.; Nishijima, F.; Niwa, T. CREB, NF-kappaB, and NADPH oxidase coordinately upregulate indoxyl sulfate-induced angiotensinogen expression in proximal tubular cells. Am. J. Physiol. Cell Physiol. 2013, 304, C685–C692. [Google Scholar] [CrossRef]
- Shimizu, H.; Bolati, D.; Adijiang, A.; Muteliefu, G.; Enomoto, A.; Nishijima, F.; Dateki, M.; Niwa, T. NF-kappaB plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am. J. Physiol. Cell Physiol. 2011, 301, C1201–C1212. [Google Scholar] [CrossRef]
- Hilfiker, A.; Hilfiker-Kleiner, D.; Fuchs, M.; Kaminski, K.; Lichtenberg, A.; Rothkotter, H.J.; Schieffer, B.; Drexler, H. Expression of CYR61, an angiogenic immediate early gene, in arteriosclerosis and its regulation by angiotensin II. Circulation 2002, 106, 254–260. [Google Scholar] [CrossRef]
- Qin, Z.; Fisher, G.J.; Quan, T. Cysteine-rich protein 61 (CCN1) domain-specific stimulation of matrix metalloproteinase-1 expression through alphaVbeta3 integrin in human skin fibroblasts. J. Biol. Chem. 2013, 288, 12386–12394. [Google Scholar] [CrossRef]
- Seth, D.; Stamler, J.S. The SNO-proteome: Causation and classifications. Curr. Opin. Chem. Biol. 2011, 15, 129–136. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid. Redox Signal. 2013, 18, 239–249. [Google Scholar] [CrossRef]
- Furini, G.; Schroeder, N.; Huang, L.; Boocock, D.; Scarpellini, A.; Coveney, C.; Tonoli, E.; Ramaswamy, R.; Ball, G.; Verderio, C.; et al. Proteomic Profiling Reveals the Transglutaminase-2 Externalization Pathway in Kidneys after Unilateral Ureteric Obstruction. J. Am. Soc. Nephrol. 2018, 29, 880–905. [Google Scholar] [CrossRef]
- Ravi, K.; Brennan, L.A.; Levic, S.; Ross, P.A.; Black, S.M. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc. Natl. Acad. Sci. USA 2004, 101, 2619–2624. [Google Scholar] [CrossRef] [PubMed]
- Pinilla, E.; Comerma-Steffensen, S.; Prat-Duran, J.; Rivera, L.; Matchkov, V.V.; Buus, N.H.; Simonsen, U. Transglutaminase 2 Inhibitor LDN 27219 Age-Dependently Lowers Blood Pressure and Improves Endothelium-Dependent Vasodilation in Resistance Arteries. Hypertension 2021, 77, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, M.D.; Sen, N.; Hara, M.R.; Juluri, K.R.; Nguyen, J.V.; Snowman, A.M.; Law, L.; Hester, L.D.; Snyder, S.H. GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 2010, 12, 1094–1100. [Google Scholar] [CrossRef]
- Reynaert, N.L.; Ckless, K.; Korn, S.H.; Vos, N.; Guala, A.S.; Wouters, E.F.; van der Vliet, A.; Janssen-Heininger, Y.M. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. USA 2004, 101, 8945–8950. [Google Scholar] [CrossRef]
- Kelleher, Z.T.; Matsumoto, A.; Stamler, J.S.; Marshall, H.E. NOS2 regulation of NF-kappaB by S-nitrosylation of p65. J. Biol. Chem. 2007, 282, 30667–30672. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.S.; Choi, D.H.; Bang, M.S.; Han, T.R.; Joh, T.H.; Kim, S.Y. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J. Biol. Chem. 2004, 279, 53725–53735. [Google Scholar] [CrossRef]
- Kumar, S.; Mehta, K. Tissue transglutaminase constitutively activates HIF-1alpha promoter and nuclear factor-kappaB via a non-canonical pathway. PLoS ONE 2012, 7, e49321. [Google Scholar] [CrossRef]
- Tatsukawa, H.; Hitomi, K. Role of Transglutaminase 2 in Cell Death, Survival, and Fibrosis. Cells 2021, 10, 1842. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | PDB Code | Cysteine Number |
|---|---|---|
| GDP | 1KV3 | 98, 545, 554 |
| GTP | 4PYG | 10, 98, 269, 545, 554 |
| Ac-P(DON)LPF-NH2 | 2Q3Z | 10, 98, 269, 545, 554 |
| Z-DON-VPL-Ome (ZDON) | 3S3J | 10, 98, 545 |
| Ca2+ | 6KZB | 10, 27, 98, 269, 545, 554 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-J.; Chiu, C.Y.; Liao, E.-C.; Wu, C.-J.; Chung, C.-H.; Greenberg, C.S.; Lai, T.-S. S-Nitrosylation of Tissue Transglutaminase in Modulating Glycolysis, Oxidative Stress, and Inflammatory Responses in Normal and Indoxyl-Sulfate-Induced Endothelial Cells. Int. J. Mol. Sci. 2023, 24, 10935. https://doi.org/10.3390/ijms241310935
Lin C-J, Chiu CY, Liao E-C, Wu C-J, Chung C-H, Greenberg CS, Lai T-S. S-Nitrosylation of Tissue Transglutaminase in Modulating Glycolysis, Oxidative Stress, and Inflammatory Responses in Normal and Indoxyl-Sulfate-Induced Endothelial Cells. International Journal of Molecular Sciences. 2023; 24(13):10935. https://doi.org/10.3390/ijms241310935
Chicago/Turabian StyleLin, Cheng-Jui, Chun Yu Chiu, En-Chih Liao, Chih-Jen Wu, Ching-Hu Chung, Charles S. Greenberg, and Thung-S. Lai. 2023. "S-Nitrosylation of Tissue Transglutaminase in Modulating Glycolysis, Oxidative Stress, and Inflammatory Responses in Normal and Indoxyl-Sulfate-Induced Endothelial Cells" International Journal of Molecular Sciences 24, no. 13: 10935. https://doi.org/10.3390/ijms241310935
APA StyleLin, C.-J., Chiu, C. Y., Liao, E.-C., Wu, C.-J., Chung, C.-H., Greenberg, C. S., & Lai, T.-S. (2023). S-Nitrosylation of Tissue Transglutaminase in Modulating Glycolysis, Oxidative Stress, and Inflammatory Responses in Normal and Indoxyl-Sulfate-Induced Endothelial Cells. International Journal of Molecular Sciences, 24(13), 10935. https://doi.org/10.3390/ijms241310935