1. Introduction
Identifying novel anticancer compounds has long been a major focus of medical research. Conventional cancer treatments, such as surgery, radiotherapy, and chemotherapy, often adversely affect healthy cells and tissues and can lead to the development of treatment resistance. Therefore, it is essential to identify additional effective therapeutic options. Anticancer peptides (ACPs) are short peptide sequences that typically contain fewer than 50 amino acids. They exert their anticancer effects via a variety of mechanisms, for example, by inhibiting the proliferation and migration of cancer cells, inducing apoptosis, changing the pH in the internal and external cellular environment, or by damaging the cell membrane of cancer cells without affecting healthy tissues [
1]. Compared to conventional therapeutic compounds, ACPs offer several advantages, such as high specificity, low intrinsic toxicity, high tissue permeability, and the convenience of sequence modification [
2,
3,
4,
5]. These features make ACPs a promising new therapeutic option in the management of cancer.
Historically, the identification of ACPs involved conventional laboratory experiments that were time-consuming and costly. However, with the accumulation of considerable ACP sequence data and the establishment of experimentally validated ACP databases, such as CancerPPD [
2], DADP [
6], CAMP [
7], and APD [
8], the rapid identification of novel ACP sequences using machine learning algorithms is becoming increasingly feasible. For example, in 2018, Wei et al. [
9] used amino acid binary profile, amino acid type group, composition–transition–distribution, and twenty-one-bit features to represent peptides and then adopted a support vector machine learning method named ACPred_FL. In 2020, Rao et al. [
10] developed another model named ACPred_Fuse, which applied 114 type features to represent peptides. In the same year, Agrawal et al. [
11] used amino acid composition, dipeptide composition, terminus composition, and binary profile to develop an extra-tree-based model named AntiCP-2.0. With the rise in deep learning and protein representation learning, many new anticancer peptide recognition methods [
11,
12,
13,
14,
15,
16] (e.g., iACP-DRLF and TriNet) continue to emerge, and the performances of the methods are becoming increasingly better.
Nonetheless, current machine-learning-based ACP prediction models are limited. Analyzing standard datasets with alternative sequence feature extraction methods often results in greatly variable outcomes, and the causes of this divergence are currently unclear. In addition, studies to date have paid little attention to the key physicochemical features that differentiate ACPs from peptides with no ACP activity, resulting in an insufficient understanding of what features determine whether an amino acid sequence can act as an ACP. To overcome these shortcomings, we used the AAindex database [
17,
18,
19,
20] as the feature database of peptide sequences. This database consists of two subdatabases, AAindex1 and AAindex2. AAindex1 is a database representing the different physical, chemical, and biological properties of amino acids, currently containing 566 amino acid features. AAindex2 is an amino acid mutation matrix database that represents the similarity between amino acids, currently containing 94 matrices. The AAindex database is primarily used in protein-related research fields and as a machine learning database in protein prediction applications. However, the information contained within this database has not been previously utilized for extracting the key features of ACP sequences. Thus, no information is available on the physicochemical properties of key amino acids that affect the function of ACPs from a holistic perspective.
To address the problems of current ACP prediction models, we explored the physicochemical characteristics of amino acids predicting ACP activity. We combined multiple feature selection techniques and a random forest (RF) model to construct a single-feature model based on the physicochemical characteristics of ACP sequences [
21,
22,
23]. The resulting model performed well in predicting ACPs in the calculation-based analysis of the physicochemical properties of key amino acids within the peptide sequences. This approach allowed us to identify 19 key amino acid properties that were useful in detecting ACP sequences in various benchmark datasets. A user-friendly webserver (
https://www.aibiochem.net/servers/RFaaindexACP, accessed on 18 June 2023) is provided.
3. Materials and Methods
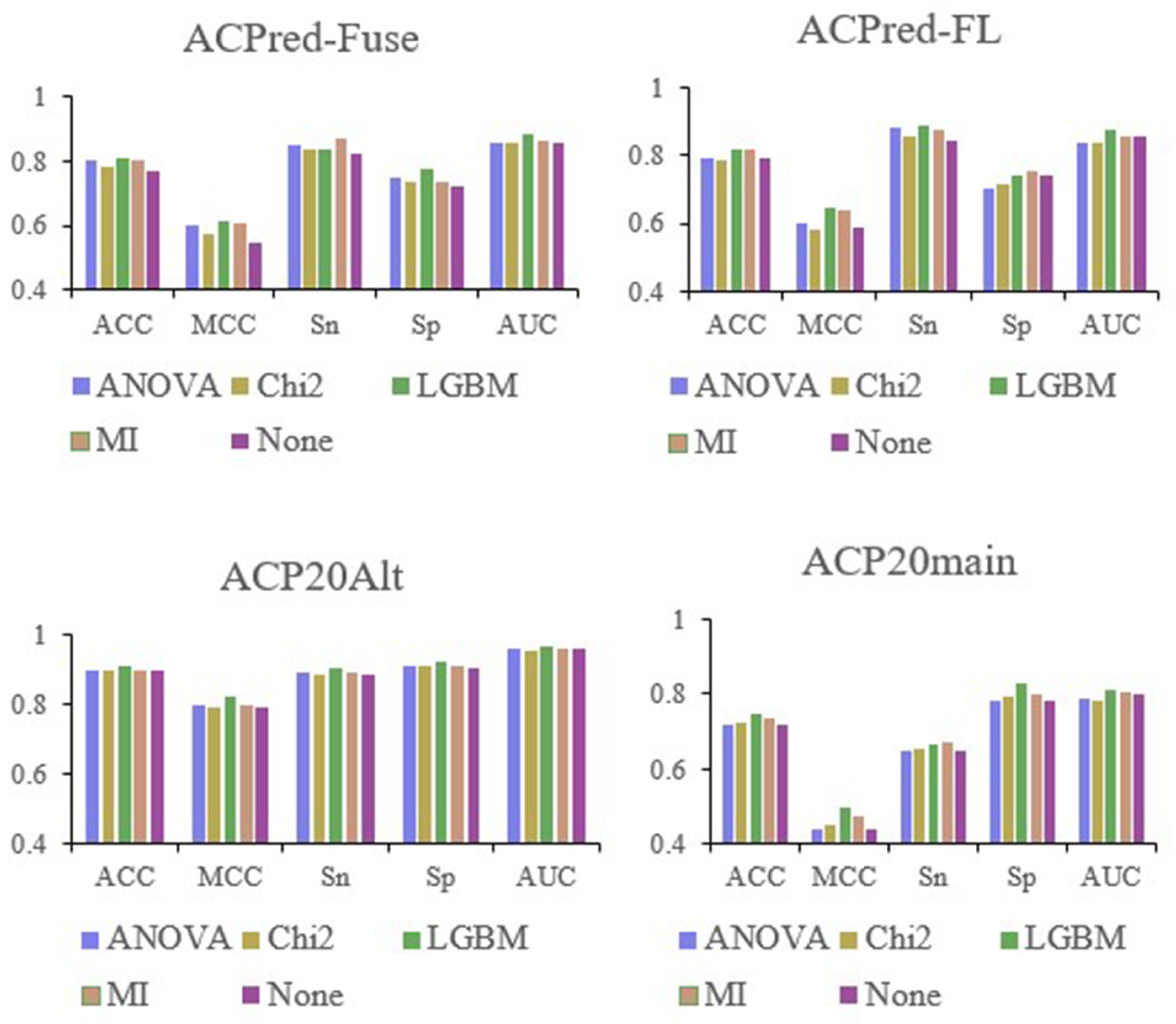
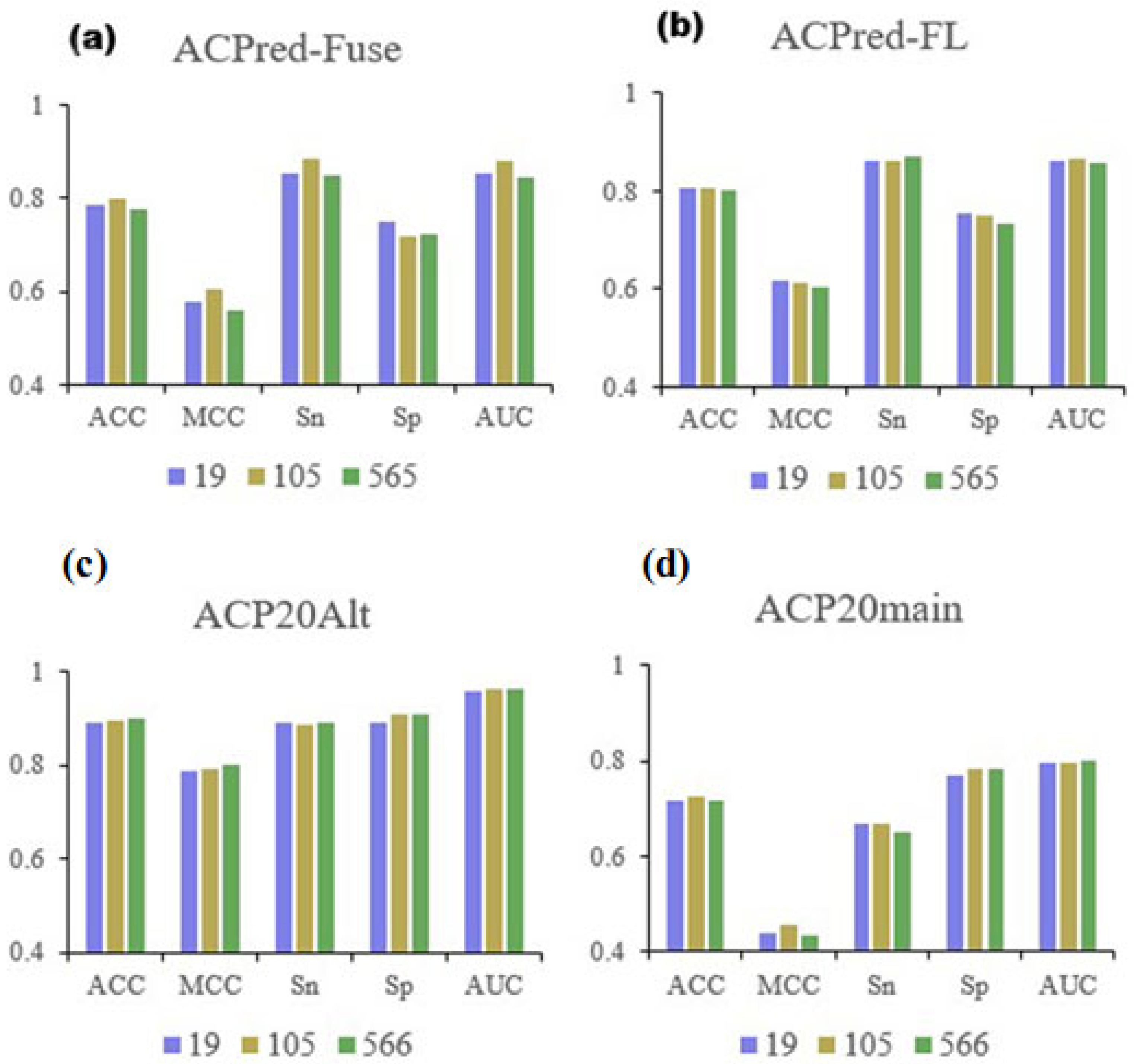
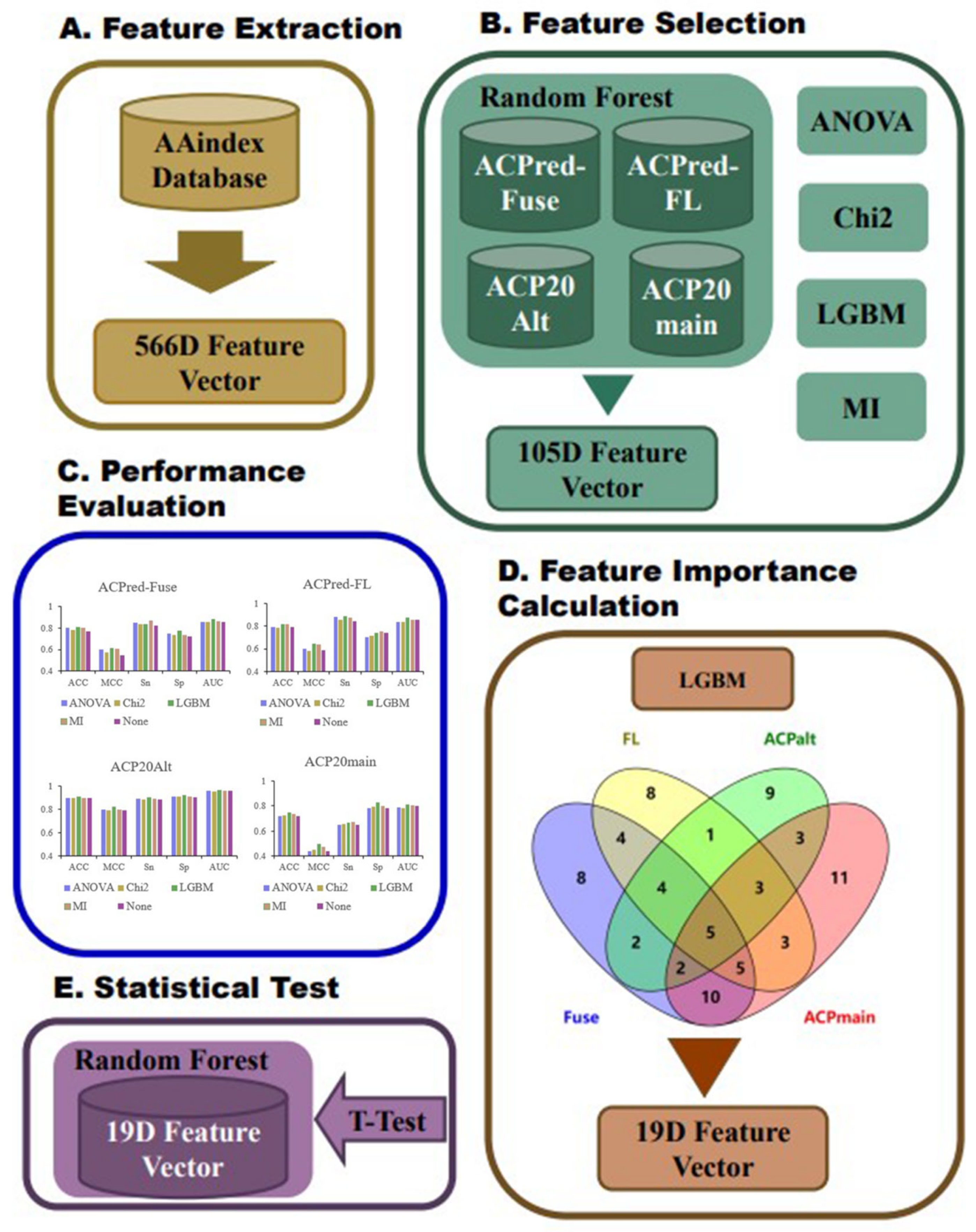
We constructed an amino acid sequence feature extraction tool based on the AAindex database, converting peptide amino acid sequences into 566-dimensional feature vectors, where each dimension represents a physicochemical property of an amino acid [
25,
26,
27]. First, the ACP datasets were divided into training and test datasets, and we analyzed them by a random forest (RF) model based on the full 566-dimensional features of the AAindex database to select the most informative features. Next, we used light gradient-boosting machine (LGBM), analysis of variance (ANOVA), chi-squared test (Chi
2), and mutual information (MI) analysis for feature selection. By examining the performance of the RF model based on the best top n features under the four methods, we initially selected 105 features. RF modeling was performed again based on these 105 features on the same benchmark datasets, and the best features were selected based on hyperparameter optimization and feature importance analysis. As a result, we identified the best performing top 19 features. Finally, the RF model was trained based on the 19 features using all four datasets. The best performing models were compared with previously reported ACP prediction algorithms described in the literature, and the statistical significance of the differences in prediction performance indices was calculated. The overall flowchart of the conducted work is summarized in
Figure 5.
3.1. Benchmark Datasets
Throughout the work presented here, we used the ACP20Alt [
28], ACP20main [
28], ACPred-FL [
9], and ACPred-Fuse [
10] datasets. Of these, the positive samples of ACPred-FL and ACPred-Fuse were primarily published by Chen et al. [
29], and Tyagi et al. [
2] in their CancerPPD datasets. Peptides with no ACP activity were represented by antimicrobial peptides (AMPs) and by a collection of peptides that had no anticancer effect during experimental testing. We randomly selected 250 ACP and 250 non-ACP sequences from the ACPred-FL dataset to act as the training dataset, and 82 ACPs and 92 non-ACPs as the test dataset. The 250 positive samples in the ACPred-Fuse training dataset were selected from the work of Wei et al. [
9] Half of the 250 negative samples were also derived from Wei et al., while the other half was collected from the AMP dataset. The test dataset contained all the remaining ACPred-Fuse data (82 ACPs and 2628 non-ACPs) as positive and negative samples. The ACP20main and ACP20Alt databases were compiled by Lv et al. [
28]. ACP20main contains 861 experimentally verified ACPs and 861 peptides with no documented ACP activity. Peptides from this dataset were divided into two subsets for 5-fold cross-validation and independent testing. Finally, the ACP20Alt database contains 970 ACPs and 970 non-ACPs. These were also subdivided into a training set and an independent test subset. The main difference between the ACP20main and ACP20Alt databases is that the negative samples in the former are represented by AMPs, while the negative samples in ACP20Alt are randomly selected peptides, assumed to have no antitumor activity. The main details of the used benchmark datasets are summarized in
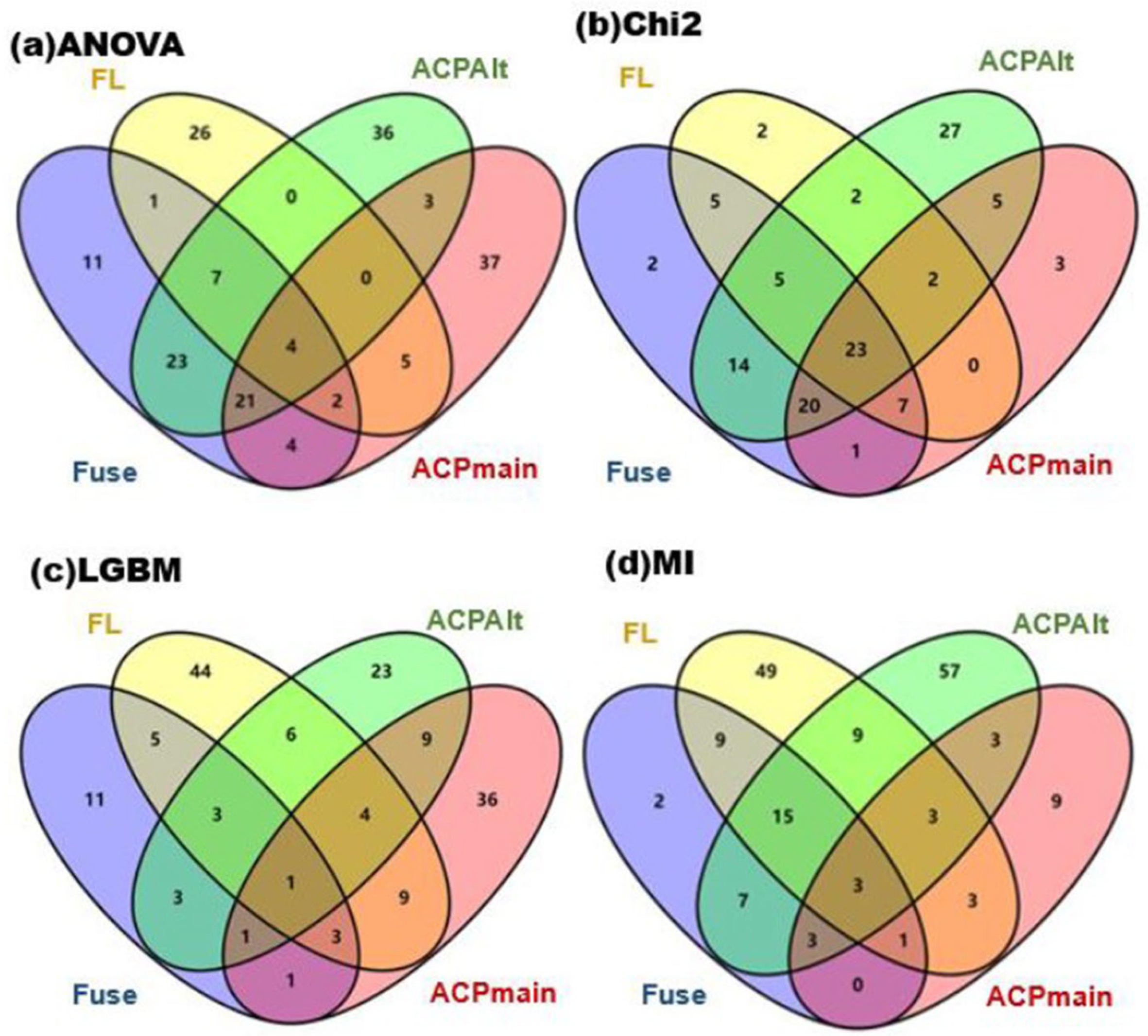
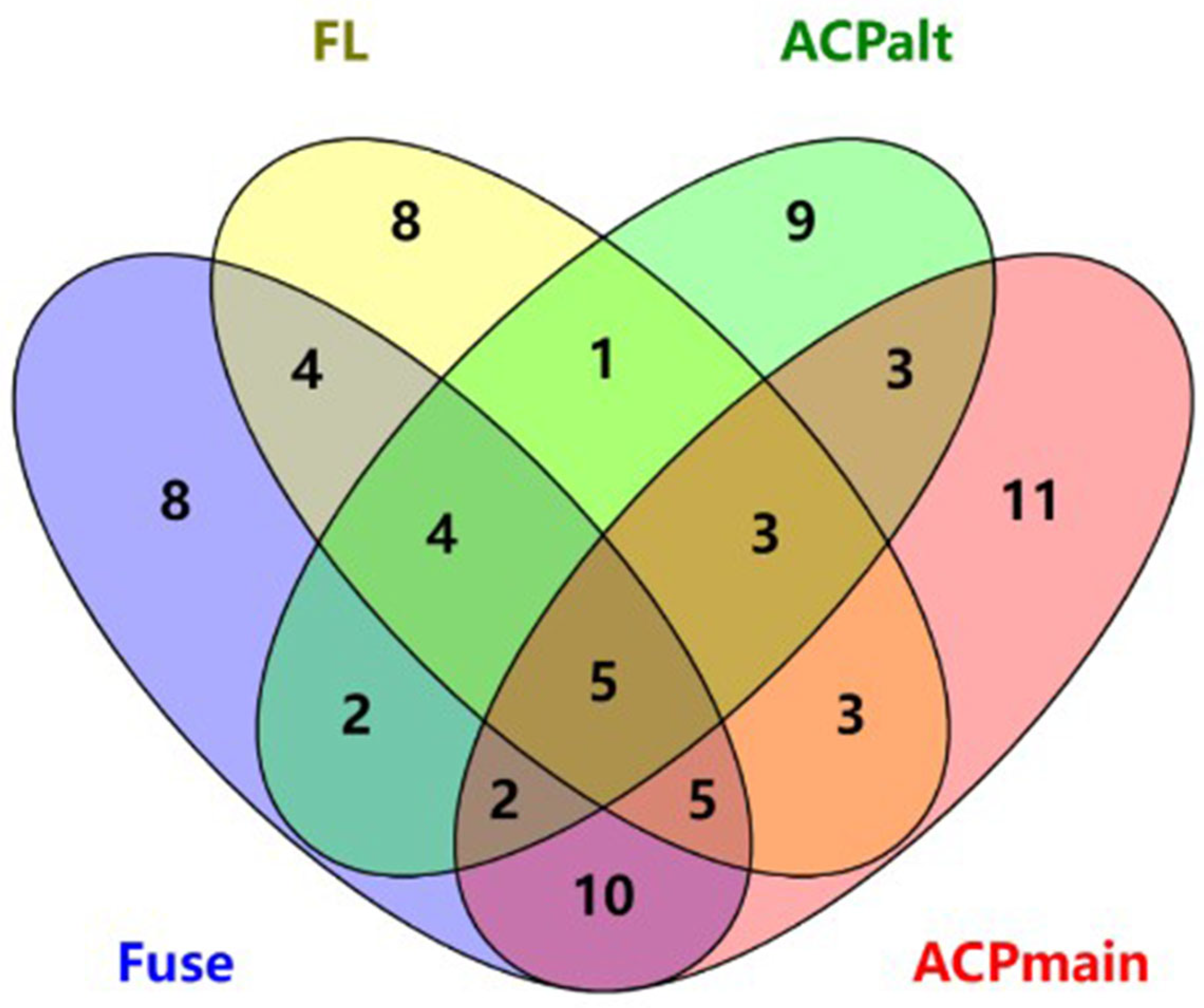
Table 9. The shared sequences numbers are shown in
Figure S6 in the supporting information.
3.2. Feature Extraction
To investigate the physicochemical properties of key amino acids, we first had to extract the information contained in the ACP sequences and convert raw sequences into interpretable features [
30,
31]. The data for this feature extraction were obtained from the AAindex databases. The AAindex1 database, available at
https://www.genome.jp/aaindex/ [
32] (accessed on 1 January 2020), describes the physicochemical properties of amino acids using 566 indices.
The physicochemical properties of a peptide sequence with a length of
L were extracted from the amino acid index database as numerical values for each amino acid [
33,
34]. It means that each amino acid in the peptide sequence is represented by a 566-dimensional vector, and then a peptide sequence of
L amino acids will be transformed into an
L × 566 matrix. Next, an average pooling of the matrix is conducted, resulting in a 566-dimensional vector representing the peptide sequence. This approach transformed each peptide sequence into a 566-dimensional feature vector, where each dimension represents a particular physicochemical property of amino acids [
21]. The source code for peptides from the AAindex feature extraction can be downloaded from
https://github.com/zhibinlv/RFaaindexACP (accessed on 19 June 2023).
3.3. Feature Selection Methods
Feature selection is the process of filtering the most relevant features by machine learning [
35]. The main purpose of feature selection is to identify irrelevant or redundant features, reducing the runtime of machine learning algorithms and obtaining more accurate results. Four feature selection methods were used in our research: light gradient-boosting machine (LGBM), analysis of variance (ANOVA), chi-squared test (Chi
2), and mutual information (MI). The LGBM feature selection code can be found at
https://github.com/zhibinlv/iACP-DRLF/tree/main/feature_selection (accessed on 1 January 2020). For Chi
2, ANOVA, and MI, it can be found using the scikit-learn toolkits at
https://scikit-learn.org/stable/index.html (accessed on 1 January 2020).
3.3.1. LGBM
LGBM adopts the histogram algorithm [
36], in which continuous features are turned into
k discrete values in order to construct a histogram with a width of
k. The algorithm is trained to count the value of each discrete value in the histogram. Based on these discrete values, the optimal feature segmentation points can be determined, and the exact number of key features can be obtained.
3.3.2. ANOVA
ANOVA analyzes the relationship between independent variables and dependent variables by studying whether the variance of multiple samples is equal to the overall mean value [
37,
38]. It can perform feature extraction before the data enter classifier training, thus reducing data dimensionality. Taking binary classification as an example, using ANOVA for feature selection divides the value of a certain feature into two groups, a positive and a negative group. The greater the difference between these groups in ANOVA, the greater their impact on the sample.
3.3.3. Chi-Squared Test
A chi-squared test can be used to determine whether two variables are correlated and to calculate the extent of this correlation [
39], by performing a test between the feature and the real label. Assuming that the number of independent variables is
A, and the number of dependent variables is
B, the following equation
χ can be constructed:
The result indicates the degree of dependence between the independent variables and dependent variables.
3.3.4. MI
Mutual information [
40,
41] can also be used to test the correlation between two variables, which can be defined as follows:
If
x and
y are random variables that are independent of each other, then
Thus, the result of
I(
X,
Y) is 0. Therefore, a larger
I(
X,
Y) indicates a greater correlation between the two variables, allowing feature filtering.
3.4. Random Forest Algorithm
In this study, we used a random forest (RF) [
42,
43,
44,
45] machine learning algorithm to analyze the importance of features. By determining the contribution of each feature to each tree in the models, the importance of key features can be ranked, identifying key amino acid physicochemical properties that determine the likelihood of a given peptide having ACP activity.
During this process, the RF model selects several samples from the sample set to construct a training dataset with replacement. It then uses the obtained training dataset to generate a decision tree and randomly selects multiple non-repetitive features at each node, using these features to divide the sample set [
46]. After the optimal division features are found, the process is repeated until all the decision trees in a random forest are generated [
47]. Finally, the model trained by the above steps is used to predict the sample set, and the prediction result is determined by the number of classifications.
3.5. Feature Importance Analysis
Common evaluation metrics for calculating feature importance include the
Gini index [
48] and the out-of-bag (OOB) error. The
Gini index is calculated according to the following equation:
where
indicates the number of features divided into
categories, and
represents the importance of category
.
The OOB dataset refers to the data that are not chosen in the sampling process. For a tree in the RF model, the error
is obtained by the out-of-bag data sample, and then, error
is derived by randomly permuting the
i-th column of the out-of-bag data matrix. This way, the importance of feature
i can be represented by
:
where
n represents the number of decision trees in the model.
3.6. Evaluation of Model Performance
To evaluate the performance of each model, the sensitivity (
Sn), specificity (
Sp), accuracy (
ACC), Matthews correlation coefficient (
MCC), and area under receiver-operating characteristic curve (AUC) [
49,
50] were calculated as follows:
TP,
TN,
FP, and
FN represent the number of true positive, true negative, false positive, and false negative samples, respectively. In addition, we also analyzed the ROC curve and AUC area, where a larger AUC value indicates a stronger predictive performance.
3.7. Websever
After the model was developed and optimized, we developed a simple and easy-to-use website for interested readers to use. The reader only needs to enter the peptide sequence in FASTA format and after a few minutes can find out whether those peptides have anticancer activity. The website can be found at
https://www.aibiochem.net/servers/RFaaindexACP (accessed on 18 June 2023). A simple screenshot of the application is shown in
Figures S1–S4.
4. Conclusions
The work presented in this paper proposed to identify the key physicochemical properties of ACPs based on existing machine learning algorithms, using existing ACP datasets available in the literature and amino acid features collated in the AAindex database. We analyzed the influence of four feature selection methods, ANOVA, Chi2, LGBM, and MI, on identifying key features. The comparison of these methods revealed that LGBM was the best approach for selecting features that led to the creation of the best fitting RF model and resulted in the best performance indices. Ultimately, this work identified 19 key amino acid features, which compared favorably with machine learning models reported previously in the literature. Furthermore, statistical tests revealed that the 19 key identified features provided as much information as predictions based on a much larger feature dimensionality or alternative machine learning algorithms, enhancing the credibility of our approach. Based on these 19 key features, we can develop new machine learning models with better effects or further refine existing models. In addition, based on these key properties, investigators will be able to design prospective ACPs with an improved probability of therapeutic effectiveness, thereby increasing the speed of transitioning peptides into clinical ACP research.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}