
Aflatoxin B1 Increases Soluble Epoxide Hydrolase in the Brain and Induces Neuroinflammation and Dopaminergic Neurotoxicity

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
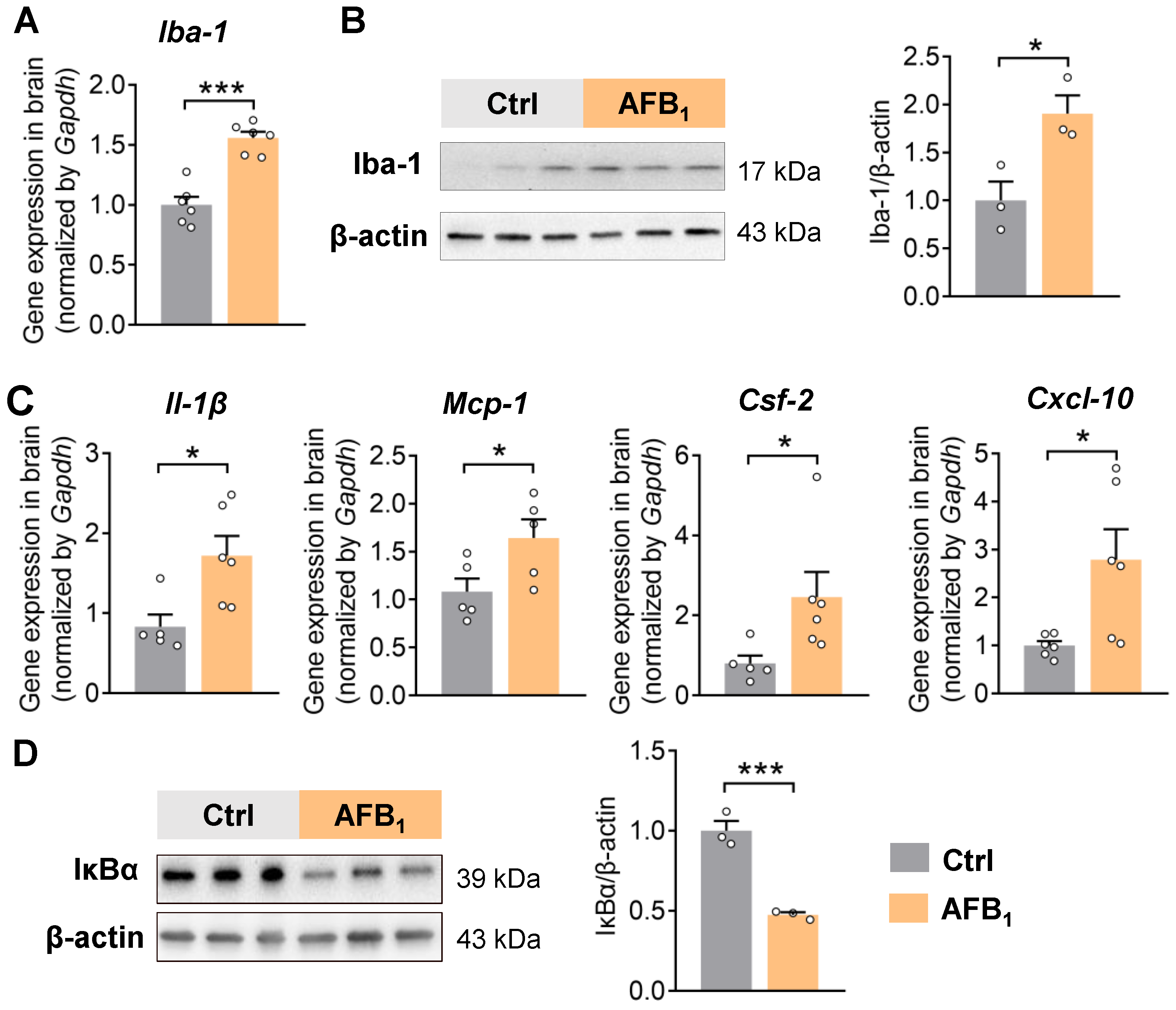
2.1. AFB1 Increases Neuroinflammation
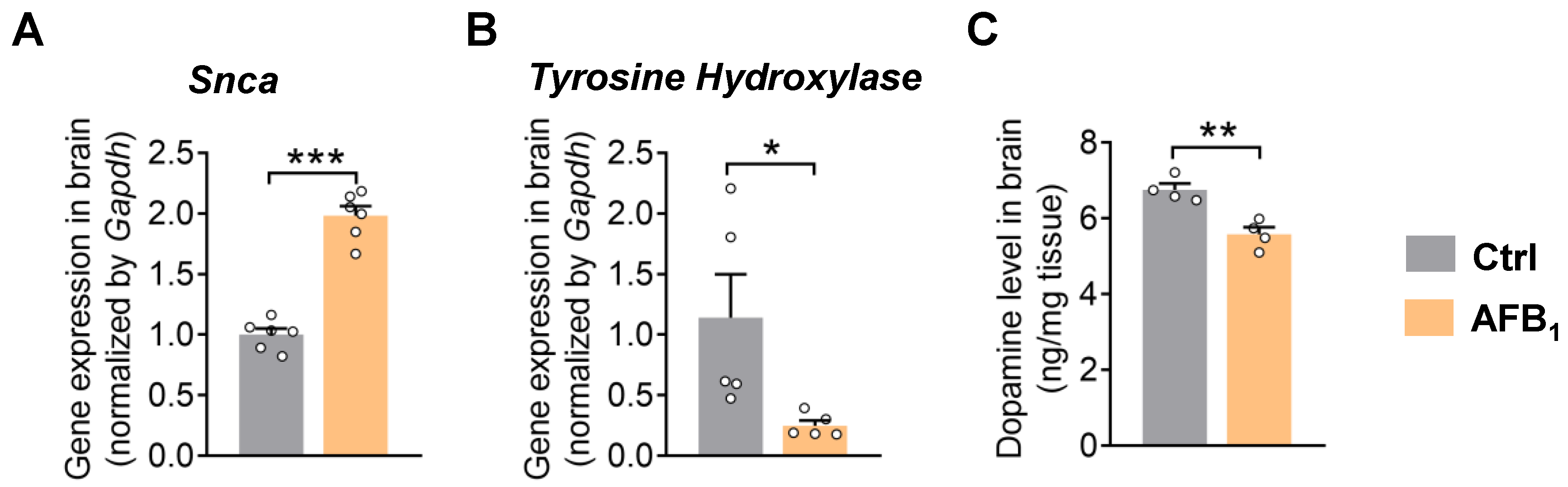
2.2. AFB1 Stimulates α-Synuclein Upregulation and Causes Dopaminergic Neurotoxicity
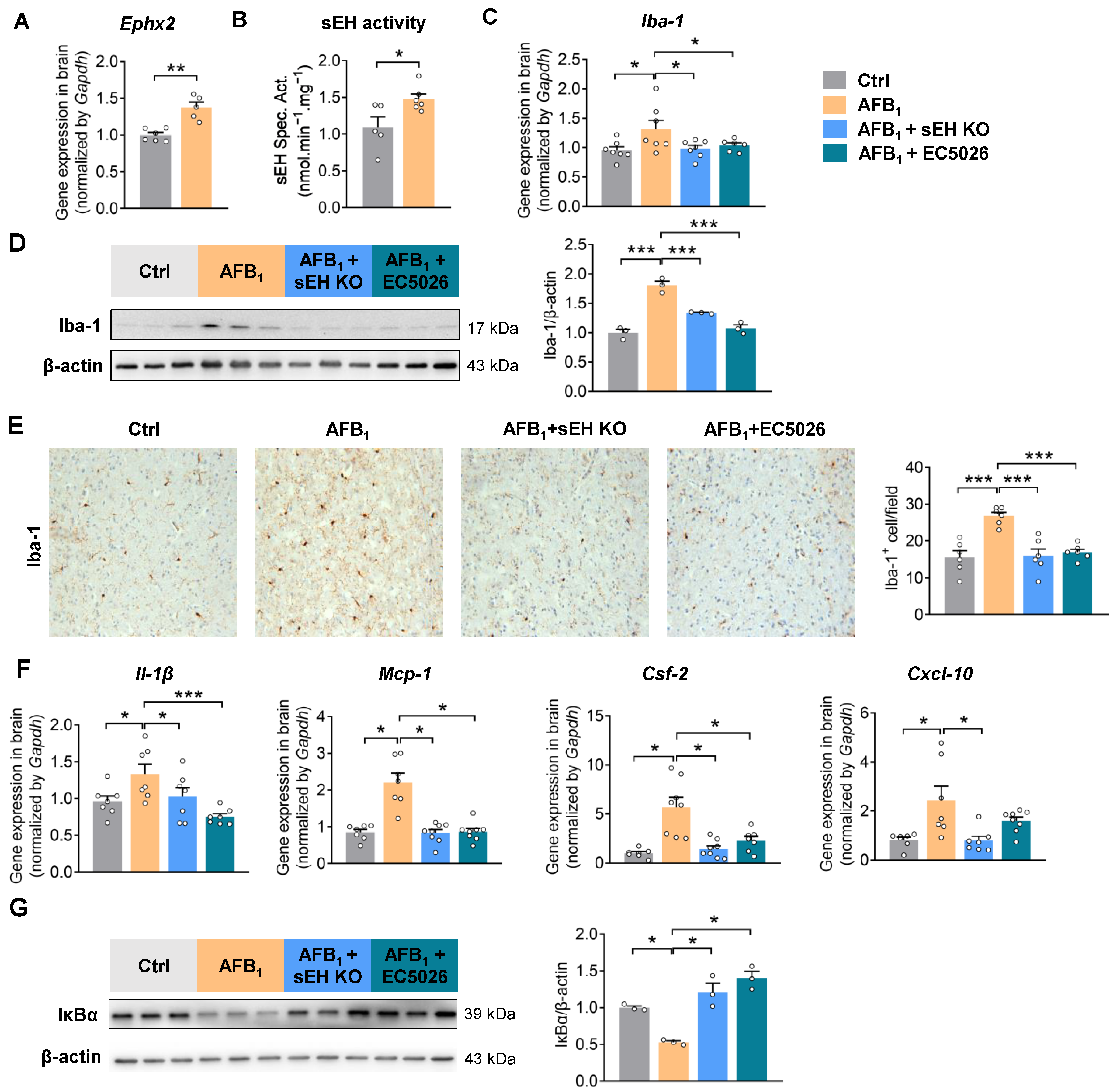
2.3. AFB1 Induces Neuroinflammation in an sEH-Dependent Manner
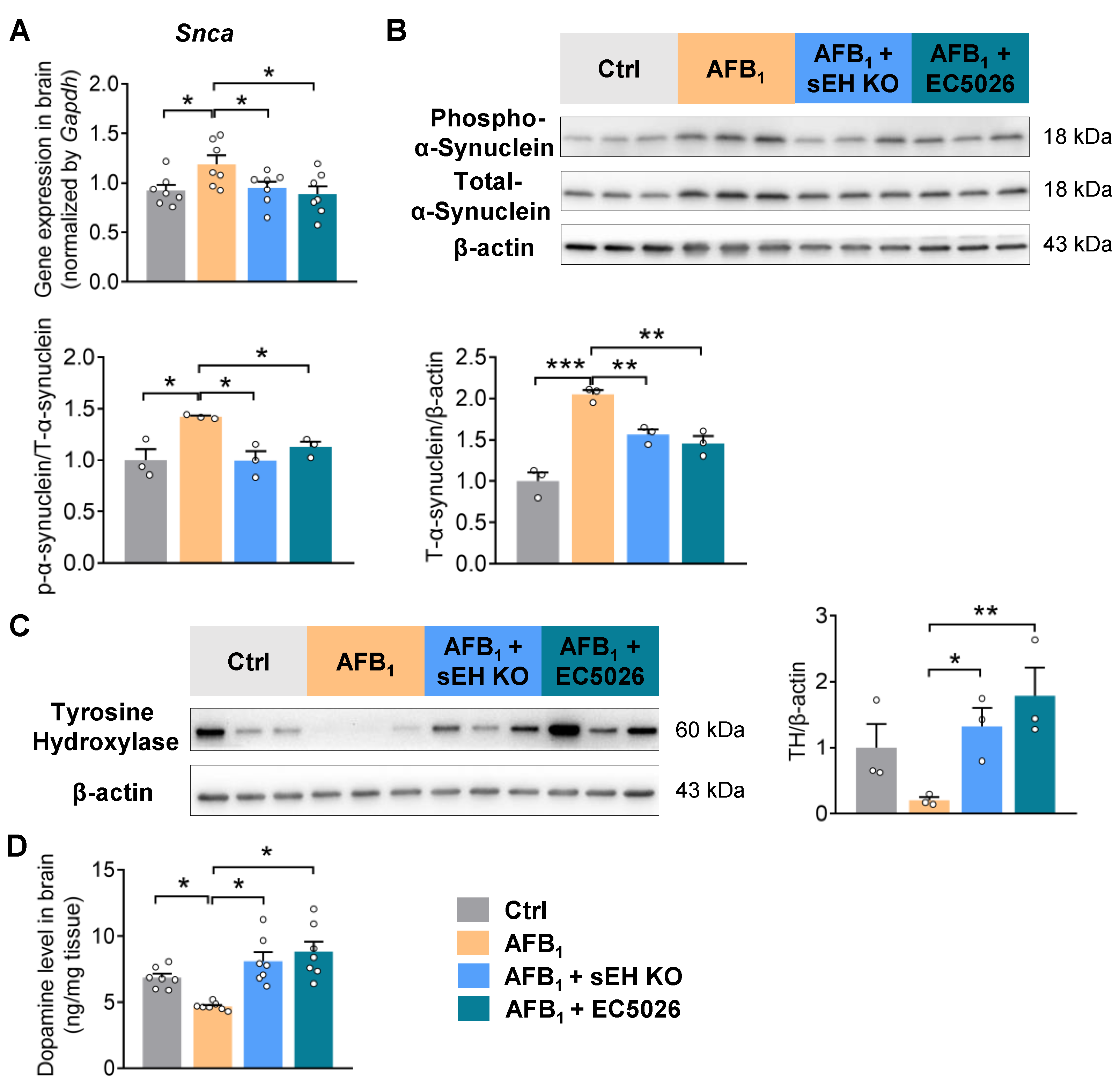
2.4. sEH Deficiency or Inhibition Alleviates AFB1-Induced α-Synuclein Pathology and Dopaminergic Neurotoxicity
3. Discussion
4. Materials and Methods
4.1. Animal Study
4.1.1. Animal Experiment 1: Effects of AFB1 Exposure on Neuroinflammation and Neurotoxicity in Mice
4.1.2. Animal Experiment 2: Effects of sEH Deficiency or Inhibition on AFB1-Induced Neuroinflammation and Neurotoxicity in Mice
4.2. Total RNA Isolation and Quantitative Polymerase Chain Reaction (qPCR) Analysis
4.3. Protein Extraction and Immunoblotting
4.4. Tissue Staining
4.5. Enzyme-Linked Immunosorbent Assay (ELISA) Analysis of Dopamine
4.6. sEH Activity Measurement
4.7. Cell Assays
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Chen, H.; Ritz, B. The Search for Environmental Causes of Parkinson’s Disease: Moving Forward. J. Park. Dis. 2018, 8, S9–S17. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- Anderson, F.L.; Coffey, M.M.; Berwin, B.L.; Havrda, M.C. Inflammasomes: An Emerging Mechanism Translating Environmental Toxicant Exposure Into Neuroinflammation in Parkinson’s Disease. Toxicol. Sci. 2018, 166, 3–15. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM); Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; Del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.R.; Leblanc, J.C.; et al. Risk assessment of aflatoxins in food. EFSA J. 2020, 18, e06040. [Google Scholar] [CrossRef]
- Williams, J.H.; Phillips, T.D.; Jolly, P.E.; Stiles, J.K.; Jolly, C.M.; Aggarwal, D. Human aflatoxicosis in developing countries: A review of toxicology, exposure, potential health consequences, and interventions. Am. J. Clin. Nutr. 2004, 80, 1106–1122. [Google Scholar] [CrossRef]
- Benkerroum, N. Chronic and Acute Toxicities of Aflatoxins: Mechanisms of Action. Int. J. Environ. Res. Public Health 2020, 17, 423. [Google Scholar] [CrossRef]
- Pei, X.; Zhang, W.; Jiang, H.; Liu, D.; Liu, X.; Li, L.; Li, C.; Xiao, X.; Tang, S.; Li, D. Food-Origin Mycotoxin-Induced Neurotoxicity: Intend to Break the Rules of Neuroglia Cells. Oxidative Med. Cell. Longev. 2021, 2021, 9967334. [Google Scholar] [CrossRef]
- Oyelami, O.A.; Maxwell, S.M.; Adelusola, K.A.; Aladekoma, T.A.; Oyelese, A.O. Aflatoxins in the autopsy brain tissue of children in Nigeria. Mycopathologia 1995, 132, 35–38. [Google Scholar] [CrossRef]
- Linardaki, Z.I.; Lamari, F.N.; Margarity, M. Saffron (Crocus sativus L.) Tea Intake Prevents Learning/Memory Defects and Neurobiochemical Alterations Induced by Aflatoxin B1 Exposure in Adult Mice. Neurochem. Res. 2017, 42, 2743–2754. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Peritore, A.F.; D’Amico, R.; Licata, P.; Crupi, R. Evaluation of Neuroprotective Effects of Quercetin against Aflatoxin B1-Intoxicated Mice. Animals 2020, 10, 898. [Google Scholar] [CrossRef]
- Kihara, T.; Matsuo, T.; Sakamoto, M.; Yasuda, Y.; Yamamoto, Y.; Tanimura, T. Effects of prenatal aflatoxin B1 exposure on behaviors of rat offspring. Toxicol. Sci. 2000, 53, 392–399. [Google Scholar] [CrossRef]
- Mehrzad, J.; Malvandi, A.M.; Alipour, M.; Hosseinkhani, S. Environmentally relevant level of aflatoxin B(1) elicits toxic pro-inflammatory response in murine CNS-derived cells. Toxicol. Lett. 2017, 279, 96–106. [Google Scholar] [CrossRef]
- Mehrzad, J.; Hosseinkhani, S.; Malvandi, A.M. Human Microglial Cells Undergo Proapoptotic Induction and Inflammatory Activation upon in vitro Exposure to a Naturally Occurring Level of Aflatoxin B1. Neuroimmunomodulation 2018, 25, 176–183. [Google Scholar] [CrossRef]
- Huang, B.; Chen, Q.; Wang, L.; Gao, X.; Zhu, W.; Mu, P.; Deng, Y. Aflatoxin B1 Induces Neurotoxicity through Reactive Oxygen Species Generation, DNA Damage, Apoptosis, and S-Phase Cell Cycle Arrest. Int. J. Mol. Sci. 2020, 21, 6517. [Google Scholar] [CrossRef]
- Piomelli, D.; Sasso, O. Peripheral gating of pain signals by endogenous lipid mediators. Nat. Neurosci. 2014, 17, 164–174. [Google Scholar] [CrossRef]
- Anand, S.; Ansari, M.A.; Sukrutha, S.K.; Alomary, M.N.; Khan, A.A.; Elderdery, A.Y. Resolvins Lipid Mediators: Potential Therapeutic Targets in Alzheimer and Parkinson Disease. Neuroscience 2022, 507, 139–148. [Google Scholar] [CrossRef]
- Shen, W.; Jiang, L.; Zhao, J.; Wang, H.; Hu, M.; Chen, L.; Chen, Y. Bioactive lipids and their metabolism: New therapeutic opportunities for Parkinson’s disease. Eur. J. Neurosci. 2022, 55, 846–872. [Google Scholar] [CrossRef]
- Warner, J.; Hardesty, J.; Zirnheld, K.; McClain, C.; Warner, D.; Kirpich, I. Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps. Biology 2020, 9, 124. [Google Scholar] [CrossRef]
- Hashimoto, K. Role of Soluble Epoxide Hydrolase in Metabolism of PUFAs in Psychiatric and Neurological Disorders. Front. Pharmacol. 2019, 10, 36. [Google Scholar] [CrossRef]
- Borsini, A. The role of soluble epoxide hydrolase and its inhibitors in depression. Brain Behav. Immun. Health 2021, 16, 100325. [Google Scholar] [CrossRef]
- Ren, Q. Soluble Epoxide Hydrolase Inhibitor: A Novel Potential Therapeutic or Prophylactic Drug for Psychiatric Disorders. Front. Pharmacol. 2019, 10, 420. [Google Scholar] [CrossRef]
- Qin, X.; Wu, Q.; Lin, L.; Sun, A.; Liu, S.; Li, X.; Cao, X.; Gao, T.; Luo, P.; Zhu, X.; et al. Soluble Epoxide Hydrolase Deficiency or Inhibition Attenuates MPTP-Induced Parkinsonism. Mol. Neurobiol. 2015, 52, 187–195. [Google Scholar] [CrossRef]
- Huang, H.J.; Wang, Y.T.; Lin, H.C.; Lee, Y.H.; Lin, A.M. Soluble Epoxide Hydrolase Inhibition Attenuates MPTP-Induced Neurotoxicity in the Nigrostriatal Dopaminergic System: Involvement of alpha-Synuclein Aggregation and ER Stress. Mol. Neurobiol. 2018, 55, 138–144. [Google Scholar] [CrossRef]
- Lakkappa, N.; Krishnamurthy, P.T.; Pandareesh, M.D.; Hammock, B.D.; Hwang, S.H. Soluble epoxide hydrolase inhibitor, APAU, protects dopaminergic neurons against rotenone induced neurotoxicity: Implications for Parkinson’s disease. Neurotoxicology 2019, 70, 135–145. [Google Scholar] [CrossRef]
- Arrus, K.; Blank, G.; Clear, R.; Holley, R.A.; Abramson, D. Microbiological and aflatoxin evaluation of Brazil nut pods and the effects of unit processing operations. J. Food Prot. 2005, 68, 1060–1065. [Google Scholar] [CrossRef]
- Lewis, L.; Onsongo, M.; Njapau, H.; Schurz-Rogers, H.; Luber, G.; Kieszak, S.; Nyamongo, J.; Backer, L.; Dahiye, A.M.; Misore, A.; et al. Aflatoxin contamination of commercial maize products during an outbreak of acute aflatoxicosis in eastern and central Kenya. Environ. Health Perspect. 2005, 113, 1763–1767. [Google Scholar] [CrossRef]
- Johansson, A.S.; Whitaker, T.B.; Hagler, W.M., Jr.; Bowman, D.T.; Slate, A.B.; Payne, G. Predicting aflatoxin and fumonisin in shelled corn lots using poor-quality grade components. J. AOAC Int. 2006, 89, 433–440. [Google Scholar] [CrossRef]
- Desai, S.; Ghewande, M.P.; Nagaraj, G.; Narayan, P.; Chauhan, S.; Singh, H. Screening for resistance to Aspergillus flavus and aflatoxin production in groundnut. Mycotoxin Res. 1991, 7, 79–84. [Google Scholar] [CrossRef]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Deng, Y.; Qing, H. The phosphorylation of alpha-synuclein: Development and implication for the mechanism and therapy of the Parkinson’s disease. J. Neurochem. 2015, 135, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Ghosh, A.; Comerota, M.M.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2020, 12, eabb1206. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Leung, M.C.; Bodhicharla, R.K.; McKeever, M.G.; Arrant, A.E.; Margillo, K.M.; Ryde, I.T.; Cyr, D.D.; Kosmaczewski, S.G.; Hammarlund, M.; et al. Exposure to mitochondrial genotoxins and dopaminergic neurodegeneration in Caenorhabditis elegans. PLoS ONE 2014, 9, e114459. [Google Scholar] [CrossRef]
- Coulombe, R.A., Jr.; Sharma, R.P. Effect of repeated dietary exposure of aflatoxin B1 on brain biogenic amines and metabolites in the rat. Toxicol. Appl. Pharmacol. 1985, 80, 496–501. [Google Scholar] [CrossRef]
- Wangikar, P.B.; Dwivedi, P.; Sharma, A.K.; Sinha, N. Effect in rats of simultaneous prenatal exposure to ochratoxin A and aflatoxin B1. II. Histopathological features of teratological anomalies induced in fetuses. Birth Defects Res. B Dev. Reprod. Toxicol. 2004, 71, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Ma, M.; Yang, J.; Nonaka, R.; Yamaguchi, A.; Ishikawa, K.I.; Kobayashi, K.; Murayama, S.; Hwang, S.H.; Saiki, S.; et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E5815–E5823. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.G.; Beraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-dependent TLR1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal 2015, 8, ra45. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef]
- Huynh, V.A.; Takala, T.M.; Murros, K.E.; Diwedi, B.; Saris, P.E.J. Desulfovibrio bacteria enhance alpha-synuclein aggregation in a Caenorhabditis elegans model of Parkinson’s disease. Front. Cell Infect. Microbiol. 2023, 13, 1181315. [Google Scholar] [CrossRef]
- Pu, Y.; Yang, J.; Chang, L.; Qu, Y.; Wang, S.; Zhang, K.; Xiong, Z.; Zhang, J.; Tan, Y.; Wang, X.; et al. Maternal glyphosate exposure causes autism-like behaviors in offspring through increased expression of soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2020, 117, 11753–11759. [Google Scholar] [CrossRef]
- Terashvili, M.; Sarkar, P.; Nostrand, M.V.; Falck, J.R.; Harder, D.R. The protective effect of astrocyte-derived 14,15-epoxyeicosatrienoic acid on hydrogen peroxide-induced cell injury in astrocyte-dopaminergic neuronal cell line co-culture. Neuroscience 2012, 223, 68–76. [Google Scholar] [CrossRef]
- Hammock, B.D.; McReynolds, C.B.; Wagner, K.; Buckpitt, A.; Cortes-Puch, I.; Croston, G.; Lee, K.S.S.; Yang, J.; Schmidt, W.K.; Hwang, S.H. Movement to the Clinic of Soluble Epoxide Hydrolase Inhibitor EC5026 as an Analgesic for Neuropathic Pain and for Use as a Nonaddictive Opioid Alternative. J. Med. Chem. 2021, 64, 1856–1872. [Google Scholar] [CrossRef]
- Gilda, J.E.; Ghosh, R.; Cheah, J.X.; West, T.M.; Bodine, S.C.; Gomes, A.V. Western Blotting Inaccuracies with Unverified Antibodies: Need for a Western Blotting Minimal Reporting Standard (WBMRS). PLoS ONE 2015, 10, e0135392. [Google Scholar] [CrossRef] [PubMed]
- Borhan, B.; Mebrahtu, T.; Nazarian, S.; Kurth, M.J.; Hammock, B.D. Improved radiolabeled substrates for soluble epoxide hydrolase. Anal. Biochem. 1995, 231, 188–200. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wang, Y.; Wagner, K.M.; Lee, R.D.; Hwang, S.H.; Morisseau, C.; Wulff, H.; Hammock, B.D. Aflatoxin B1 Increases Soluble Epoxide Hydrolase in the Brain and Induces Neuroinflammation and Dopaminergic Neurotoxicity. Int. J. Mol. Sci. 2023, 24, 9938. https://doi.org/10.3390/ijms24129938
Wang W, Wang Y, Wagner KM, Lee RD, Hwang SH, Morisseau C, Wulff H, Hammock BD. Aflatoxin B1 Increases Soluble Epoxide Hydrolase in the Brain and Induces Neuroinflammation and Dopaminergic Neurotoxicity. International Journal of Molecular Sciences. 2023; 24(12):9938. https://doi.org/10.3390/ijms24129938
Chicago/Turabian StyleWang, Weicang, Yuxin Wang, Karen M. Wagner, Ruth Diana Lee, Sung Hee Hwang, Christophe Morisseau, Heike Wulff, and Bruce D. Hammock. 2023. "Aflatoxin B1 Increases Soluble Epoxide Hydrolase in the Brain and Induces Neuroinflammation and Dopaminergic Neurotoxicity" International Journal of Molecular Sciences 24, no. 12: 9938. https://doi.org/10.3390/ijms24129938
APA StyleWang, W., Wang, Y., Wagner, K. M., Lee, R. D., Hwang, S. H., Morisseau, C., Wulff, H., & Hammock, B. D. (2023). Aflatoxin B1 Increases Soluble Epoxide Hydrolase in the Brain and Induces Neuroinflammation and Dopaminergic Neurotoxicity. International Journal of Molecular Sciences, 24(12), 9938. https://doi.org/10.3390/ijms24129938