
Soluble Epoxide Hydrolase Contributes to Cell Senescence and ER Stress in Aging Mice Colon

 , ,
, ,  and
and 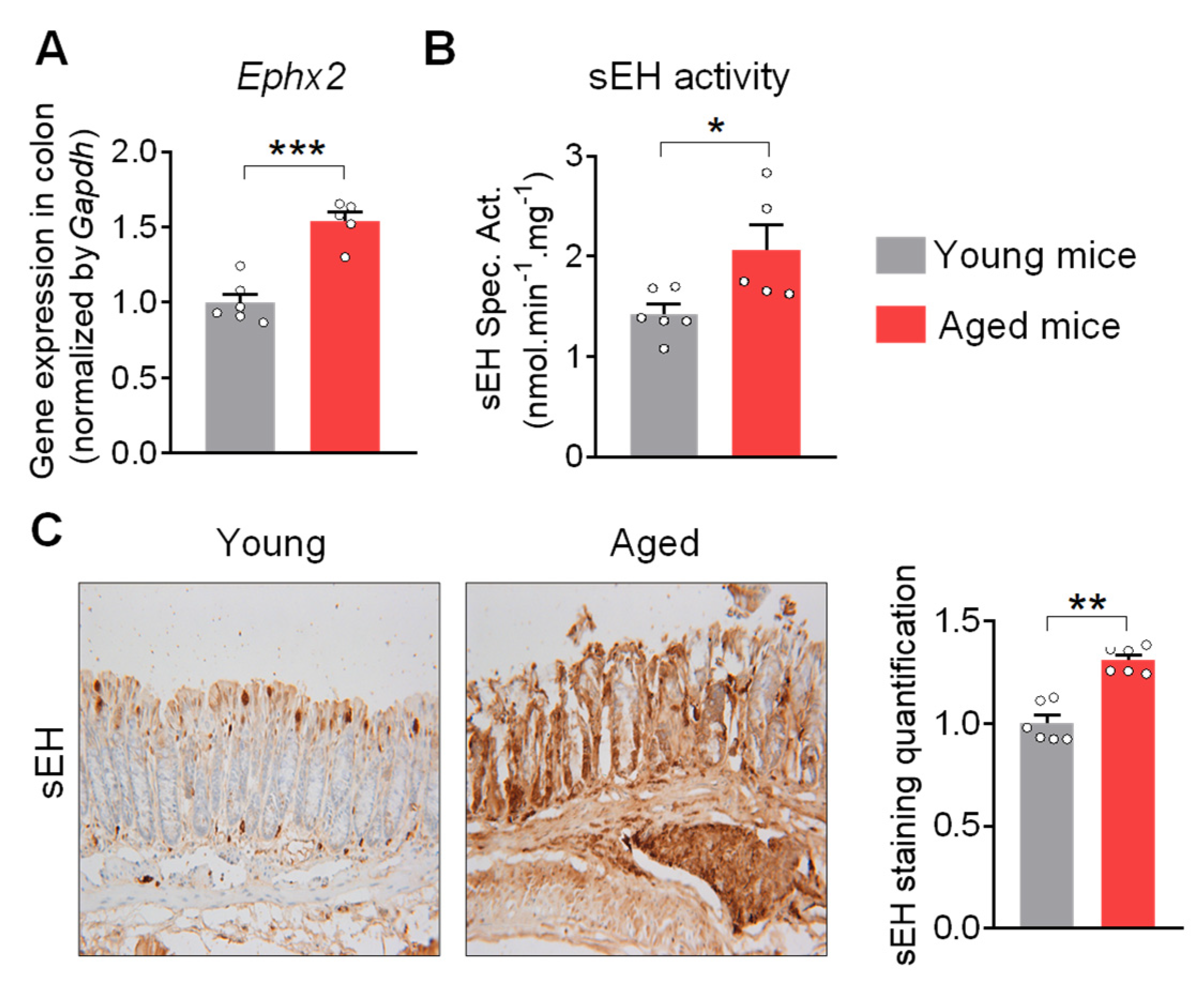{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. sEH Is Increased in the Colon of Aged Mice
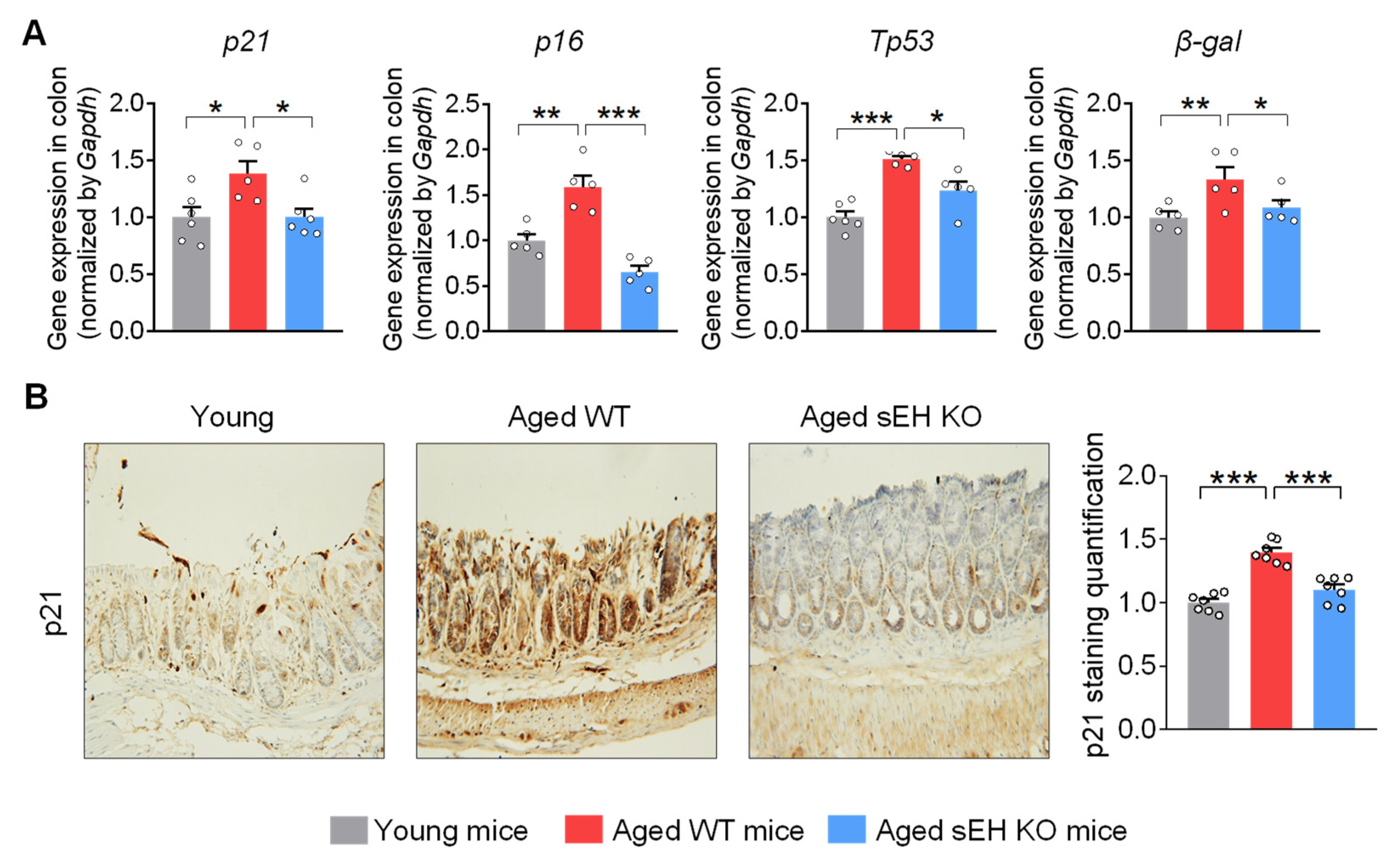
2.2. Genetic Ablation of sEH Attenuates Senescent Markers in the Colon of Aged Mice
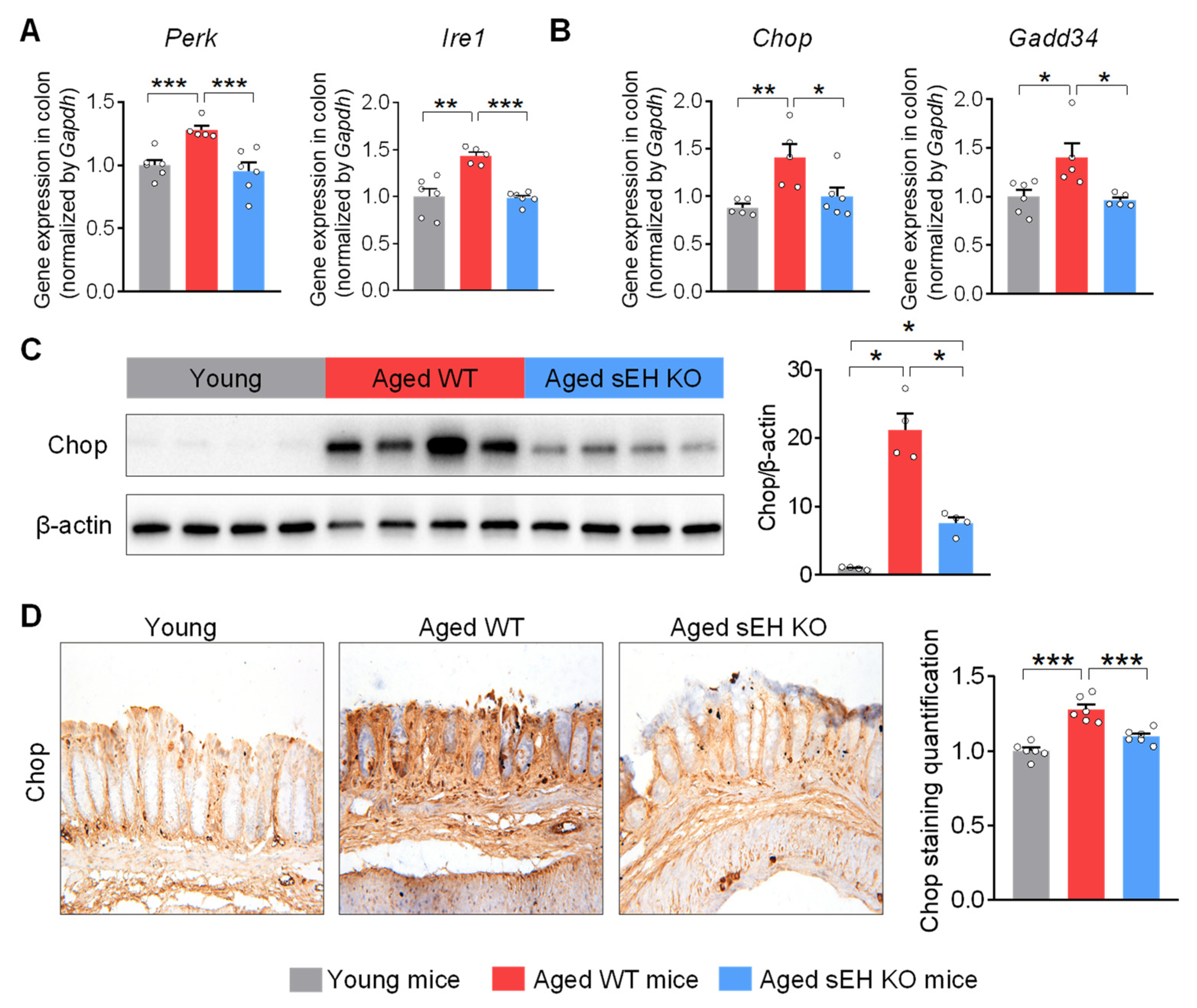
2.3. Genetic Ablation of sEH Abolishes Age-Related ER Stress in the Colon of Aged Mice
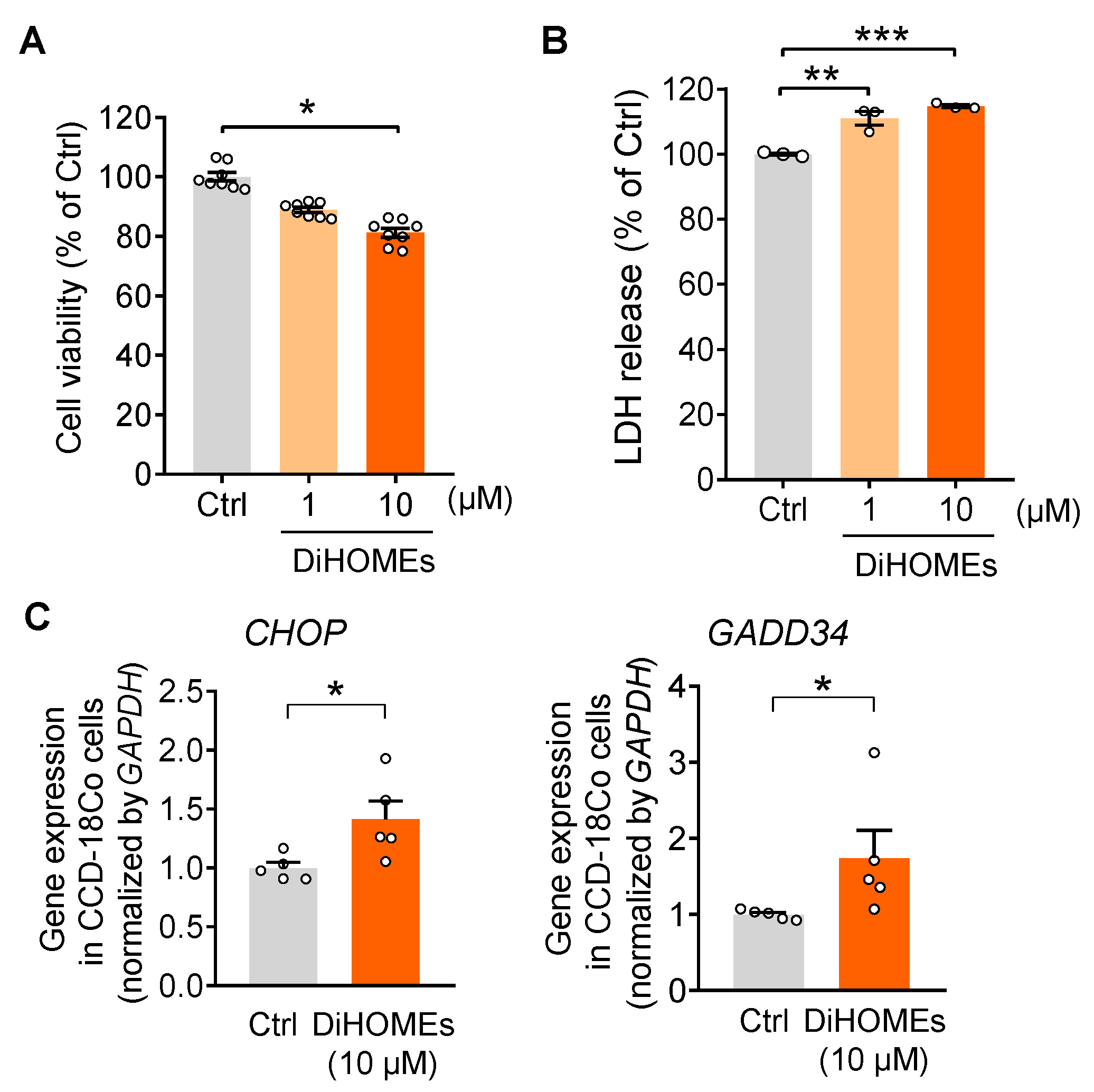
2.4. sEH-Produced DiHOMEs Reduce Cell Viability and Enhance ER Stress In Vitro
3. Discussion
4. Methods and Materials
4.1. Animal Study
4.2. sEH Activity Measurement
4.3. Total RNA Isolation and Quantitative Reverse-Transcriptase DNA Polymerase Chain Reaction (qRT-PCR) Analysis
4.4. Tissue Staining
4.5. Protein Extraction and Immunoblotting
4.6. Cell Assays
4.7. Enzyme-Linked Immunosorbent Assay (ELISA) Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ling, Z.; Liu, X.; Cheng, Y.; Yan, X.; Wu, S. Gut microbiota and aging. Crit. Rev. Food Sci. Nutr. 2022, 62, 3509–3534. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.H.; Lim, S.Y.; Lang, A.E. The microbiome-gut-brain axis in Parkinson disease—From basic research to the clinic. Nat. Rev. Neurol. 2022, 18, 476–495. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Frey, N.; Venturelli, S.; Zender, L.; Bitzer, M. Cellular senescence in gastrointestinal diseases: From pathogenesis to therapeutics. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Idda, M.L.; McClusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of senescent cell markers with age in human tissues. Aging 2020, 12, 4052–4066. [Google Scholar] [CrossRef]
- Jeong, J.J.; Woo, J.Y.; Ahn, Y.T.; Shim, J.H.; Huh, C.S.; Im, S.H.; Han, M.J.; Kim, D.H. The probiotic mixture IRT5 ameliorates age-dependent colitis in rats. Int. Immunopharmacol. 2015, 26, 416–422. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Frakes, A.E.; Dillin, A. The UPR(ER): Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.G.; Ramaiah, K.V. Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 2007, 355, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Paz Gavilan, M.; Vela, J.; Castano, A.; Ramos, B.; del Rio, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Ikeyama, S.; Wang, X.T.; Li, J.; Podlutsky, A.; Martindale, J.L.; Kokkonen, G.; van Huizen, R.; Gorospe, M.; Holbrook, N.J. Expression of the pro-apoptotic gene gadd153/chop is elevated in liver with aging and sensitizes cells to oxidant injury. J. Biol. Chem. 2003, 278, 16726–16731. [Google Scholar] [CrossRef]
- Li, J.; Holbrook, N.J. Elevated gadd153/chop expression and enhanced c-Jun N-terminal protein kinase activation sensitizes aged cells to ER stress. Exp. Gerontol. 2004, 39, 735–744. [Google Scholar] [CrossRef]
- Wang, L.; Ryoo, H.D.; Qi, Y.; Jasper, H. PERK Limits Drosophila Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress. PLoS Genet. 2015, 11, e1005220. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Rasool, R.U.; Kumar, S.; Nayak, D.; Rah, B.; Katoch, A.; Amin, H.; Ali, A.; Goswami, A. Cristacarpin promotes ER stress-mediated ROS generation leading to premature senescence by activation of p21(waf-1). Age 2016, 38, 62. [Google Scholar] [CrossRef]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef]
- Morisseau, C.; Hammock, B.D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. [Google Scholar] [CrossRef]
- McReynolds, C.; Morisseau, C.; Wagner, K.; Hammock, B. Epoxy Fatty Acids Are Promising Targets for Treatment of Pain, Cardiovascular Disease and Other Indications Characterized by Mitochondrial Dysfunction, Endoplasmic Stress and Inflammation. Adv. Exp. Med. Biol. 2020, 1274, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, K.L.; Keshavarz-Bahaghighat, H.; Darwesh, A.M.; Sosnowski, D.K.; Seubert, J.M. Age and Sex Differences in Hearts of Soluble Epoxide Hydrolase Null Mice. Front. Physiol. 2020, 11, 48. [Google Scholar] [CrossRef]
- Zuloaga, K.L.; Zhang, W.; Roese, N.E.; Alkayed, N.J. Soluble epoxide hydrolase gene deletion improves blood flow and reduces infarct size after cerebral ischemia in reproductively senescent female mice. Front. Pharmacol. 2014, 5, 290. [Google Scholar] [CrossRef]
- Sun, C.; Simon, S.I.; Foster, G.A.; Radecke, C.E.; Hwang, H.V.; Zhang, X.; Hammock, B.D.; Chiamvimonvat, N.; Knowlton, A.A. 11,12-Epoxyecosatrienoic acids mitigate endothelial dysfunction associated with estrogen loss and aging: Role of membrane depolarization. J. Mol. Cell Cardiol. 2016, 94, 180–188. [Google Scholar] [CrossRef]
- McReynolds, C.B.; Hwang, S.H.; Yang, J.; Wan, D.; Wagner, K.; Morisseau, C.; Li, D.; Schmidt, W.K.; Hammock, B.D. Pharmaceutical Effects of Inhibiting the Soluble Epoxide Hydrolase in Canine Osteoarthritis. Front. Pharmacol. 2019, 10, 533. [Google Scholar] [CrossRef]
- Ren, Q.; Ma, M.; Yang, J.; Nonaka, R.; Yamaguchi, A.; Ishikawa, K.I.; Kobayashi, K.; Murayama, S.; Hwang, S.H.; Saiki, S.; et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E5815–E5823. [Google Scholar] [CrossRef]
- Ghosh, A.; Comerota, M.M.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2020, 12, eabb1206. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.P.; Zhang, X.Y.; Zhou, J.J.; Huo, X.K.; Yu, Z.L.; Morisseau, C.; Hammock, B.D.; Ma, X.C. Inhibition of sEH via stabilizing the level of EETs alleviated Alzheimer’s disease through GSK3beta signaling pathway. Food Chem. Toxicol. 2021, 156, 112516. [Google Scholar] [CrossRef]
- Bettaieb, A.; Koike, S.; Hsu, M.F.; Ito, Y.; Chahed, S.; Bachaalany, S.; Gruzdev, A.; Calvo-Rubio, M.; Lee, K.S.S.; Inceoglu, B.; et al. Soluble epoxide hydrolase in podocytes is a significant contributor to renal function under hyperglycemia. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2758–2765. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.R.; Bettaieb, A.; Kodani, S.; Dong, H.; Myers, R.; Chiamvimonvat, N.; Haj, F.G.; Hammock, B.D. Inhibition of soluble epoxide hydrolase attenuates hepatic fibrosis and endoplasmic reticulum stress induced by carbon tetrachloride in mice. Toxicol. Appl. Pharmacol. 2015, 286, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Trindade-da-Silva, C.A.; Bettaieb, A.; Napimoga, M.H.; Lee, K.S.S.; Inceoglu, B.; Ueira-Vieira, C.; Bruun, D.; Goswami, S.K.; Haj, F.G.; Hammock, B.D. Soluble Epoxide Hydrolase Pharmacological Inhibition Decreases Alveolar Bone Loss by Modulating Host Inflammatory Response, RANK-Related Signaling, Endoplasmic Reticulum Stress, and Apoptosis. J. Pharmacol. Exp. Ther. 2017, 361, 408–416. [Google Scholar] [CrossRef]
- United Nations Department of Economic and Social Affairs. Population Division; United Nations Department of Economic and Social Affairs: New York, NY, USA, 2022. [Google Scholar]
- Jamieson, K.L.; Darwesh, A.M.; Sosnowski, D.K.; Zhang, H.; Shah, S.; Zhabyeyev, P.; Yang, J.; Hammock, B.D.; Edin, M.L.; Zeldin, D.C.; et al. Soluble Epoxide Hydrolase in Aged Female Mice and Human Explanted Hearts Following Ischemic Injury. Int. J. Mol. Sci. 2021, 22, 1691. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, L.; Gasek, N.S.; Zhou, Y.; Kim, T.; Guo, C.; Jellison, E.R.; Haynes, L.; Yadav, S.; Tchkonia, T.; et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat. Aging 2021, 1, 962–973. [Google Scholar] [CrossRef]
- Zhou, J.; Hou, C.; Chen, H.; Qin, Z.; Miao, Z.; Zhao, J.; Wang, Q.; Cui, M.; Xie, C.; Wang, R.; et al. P16 (I NK 4a) Deletion Ameliorates Damage of Intestinal Epithelial Barrier and Microbial Dysbiosis in a Stress-Induced Premature Senescence Model of Bmi-1 Deficiency. Front. Cell Dev. Biol. 2021, 9, 671564. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.F.; Grant, D.F.; Cheek, J.M.; Greene, J.F.; Williamson, K.C.; Hammock, B.D. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997, 3, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.B.; McReynolds, C.B.; Wan, D.; Singh, N.; Goetzman, H.; Caldwell, C.C.; Supp, D.M.; Hammock, B.D. sEH-derived metabolites of linoleic acid drive pathologic inflammation while impairing key innate immune cell function in burn injury. Proc. Natl. Acad. Sci. USA 2022, 119, e2120691119. [Google Scholar] [CrossRef]
- Sisemore, M.F.; Zheng, J.; Yang, J.C.; Thompson, D.A.; Plopper, C.G.; Cortopassi, G.A.; Hammock, B.D. Cellular characterization of leukotoxin diol-induced mitochondrial dysfunction. Arch. Biochem. Biophys. 2001, 392, 32–37. [Google Scholar] [CrossRef]
- Markaverich, B.M.; Crowley, J.R.; Alejandro, M.A.; Shoulars, K.; Casajuna, N.; Mani, S.; Reyna, A.; Sharp, J. Leukotoxin diols from ground corncob bedding disrupt estrous cyclicity in rats and stimulate MCF-7 breast cancer cell proliferation. Environ. Health Perspect. 2005, 113, 1698–1704. [Google Scholar] [CrossRef]
- McReynolds, C.B.; Cortes-Puch, I.; Ravindran, R.; Khan, I.H.; Hammock, B.G.; Shih, P.B.; Hammock, B.D.; Yang, J. Plasma Linoleate Diols Are Potential Biomarkers for Severe COVID-19 Infections. Front. Physiol. 2021, 12, 663869. [Google Scholar] [CrossRef]
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef]
- Hammock, B.D.; McReynolds, C.B.; Wagner, K.; Buckpitt, A.; Cortes-Puch, I.; Croston, G.; Lee, K.S.S.; Yang, J.; Schmidt, W.K.; Hwang, S.H. Movement to the Clinic of Soluble Epoxide Hydrolase Inhibitor EC5026 as an Analgesic for Neuropathic Pain and for Use as a Nonaddictive Opioid Alternative. J. Med. Chem. 2021, 64, 1856–1872. [Google Scholar] [CrossRef]
- Chen, D.; Whitcomb, R.; MacIntyre, E.; Tran, V.; Do, Z.N.; Sabry, J.; Patel, D.V.; Anandan, S.K.; Gless, R.; Webb, H.K. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol. 2012, 52, 319–328. [Google Scholar] [CrossRef]
- Lazaar, A.L.; Yang, L.; Boardley, R.L.; Goyal, N.S.; Robertson, J.; Baldwin, S.J.; Newby, D.E.; Wilkinson, I.B.; Tal-Singer, R.; Mayer, R.J.; et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol. 2016, 81, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Luria, A.; Morisseau, C.; Tsai, H.J.; Yang, J.; Inceoglu, B.; De Taeye, B.; Watkins, S.M.; Wiest, M.M.; German, J.B.; Hammock, B.D. Alteration in plasma testosterone levels in male mice lacking soluble epoxide hydrolase. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E375–E383. [Google Scholar] [CrossRef]
- Borhan, B.; Mebrahtu, T.; Nazarian, S.; Kurth, M.J.; Hammock, B.D. Improved radiolabeled substrates for soluble epoxide hydrolase. Anal. Biochem. 1995, 231, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Spandidos, A.; Wang, X.; Wang, H.; Seed, B. PrimerBank: A resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic. Acids Res. 2010, 38, D792–D799. [Google Scholar] [CrossRef] [PubMed]
- Spandidos, A.; Wang, X.; Wang, H.; Dragnev, S.; Thurber, T.; Seed, B. A comprehensive collection of experimentally validated primers for Polymerase Chain Reaction quantitation of murine transcript abundance. BMC Genomics. 2008, 9, 633. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Seed, B. A PCR primer bank for quantitative gene expression analysis. Nucleic. Acids Res. 2003, 31, e154. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Lan, Y.; Wu, X.; Han, Y.; Wang, M.; Zheng, J.; Li, Z.; Li, F.; Zhou, J.; Xiao, J.; et al. The chemopreventive effect of 5-demethylnobiletin, a unique citrus flavonoid, on colitis-driven colorectal carcinogenesis in mice is associated with its colonic metabolites. Food Funct. 2020, 11, 4940–4952. [Google Scholar] [CrossRef]
- Gilda, J.E.; Ghosh, R.; Cheah, J.X.; West, T.M.; Bodine, S.C.; Gomes, A.V. Western Blotting Inaccuracies with Unverified Antibodies: Need for a Western Blotting Minimal Reporting Standard (WBMRS). PLoS ONE 2015, 10, e0135392. [Google Scholar] [CrossRef]
- Xiao, H.; Yang, C.S.; Li, S.; Jin, H.; Ho, C.T.; Patel, T. Monodemethylated polymethoxyflavones from sweet orange (Citrus sinensis) peel inhibit growth of human lung cancer cells by apoptosis. Mol. Nutr. Food Res. 2009, 53, 398–406. [Google Scholar] [CrossRef]
- Singh, N.; Li, D.; McReynolds, C.B.; Morisseau, C.; Hammock, B.D. Improved ELISA for linoleate-derived diols in human plasma utilizing a polyHRP-based secondary tracer. Anal. Methods 2022, 14, 1810–1819. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wagner, K.M.; Wang, Y.; Singh, N.; Yang, J.; He, Q.; Morisseau, C.; Hammock, B.D. Soluble Epoxide Hydrolase Contributes to Cell Senescence and ER Stress in Aging Mice Colon. Int. J. Mol. Sci. 2023, 24, 4570. https://doi.org/10.3390/ijms24054570
Wang W, Wagner KM, Wang Y, Singh N, Yang J, He Q, Morisseau C, Hammock BD. Soluble Epoxide Hydrolase Contributes to Cell Senescence and ER Stress in Aging Mice Colon. International Journal of Molecular Sciences. 2023; 24(5):4570. https://doi.org/10.3390/ijms24054570
Chicago/Turabian StyleWang, Weicang, Karen M. Wagner, Yuxin Wang, Nalin Singh, Jun Yang, Qiyi He, Christophe Morisseau, and Bruce D. Hammock. 2023. "Soluble Epoxide Hydrolase Contributes to Cell Senescence and ER Stress in Aging Mice Colon" International Journal of Molecular Sciences 24, no. 5: 4570. https://doi.org/10.3390/ijms24054570
APA StyleWang, W., Wagner, K. M., Wang, Y., Singh, N., Yang, J., He, Q., Morisseau, C., & Hammock, B. D. (2023). Soluble Epoxide Hydrolase Contributes to Cell Senescence and ER Stress in Aging Mice Colon. International Journal of Molecular Sciences, 24(5), 4570. https://doi.org/10.3390/ijms24054570