
Targeting MET in Non-Small Cell Lung Cancer (NSCLC): A New Old Story?

 , ,
, ,  ,
,  , ,
, ,
Abstract
1. Introduction
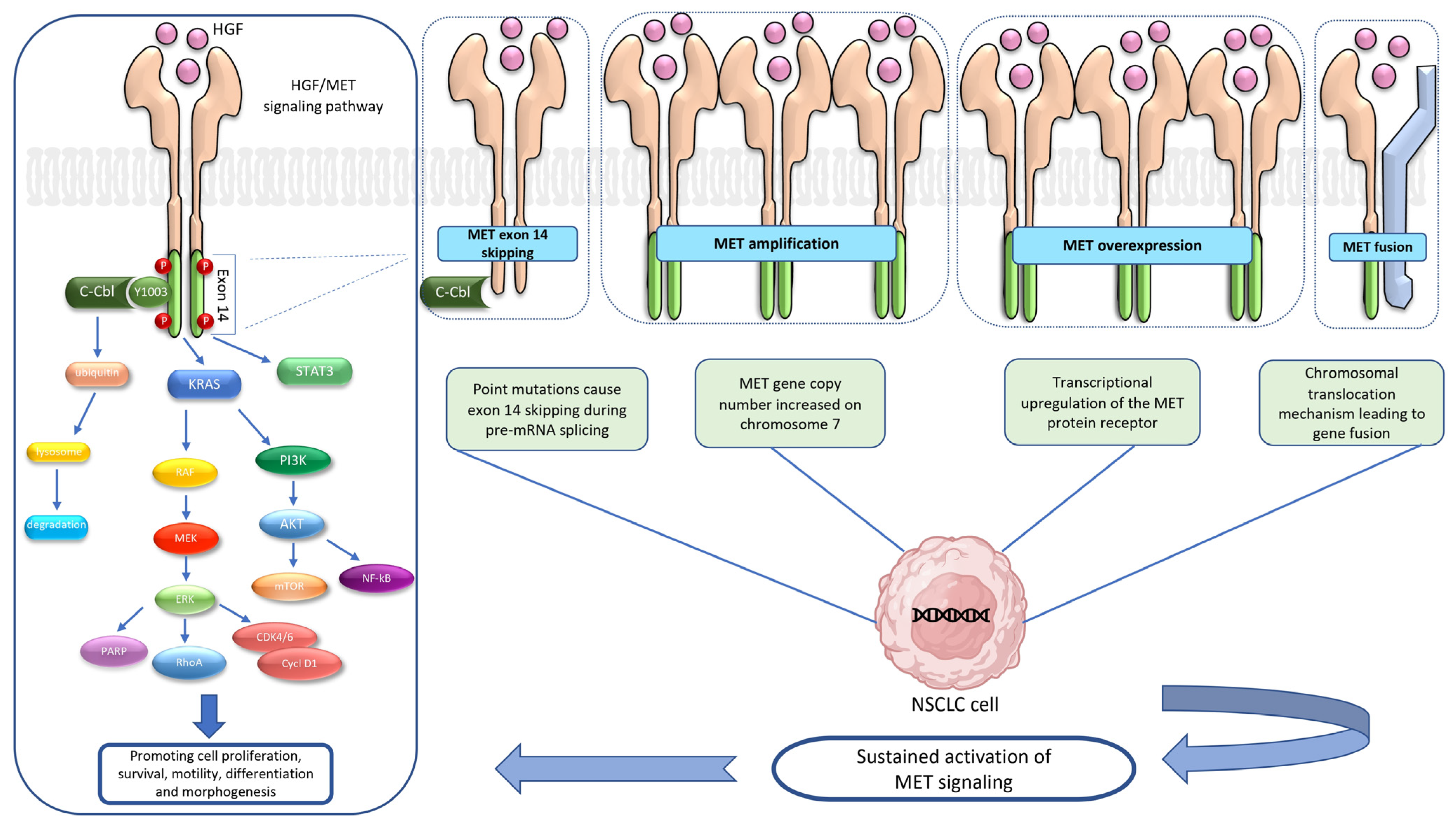
2. HGF/MET Signaling Pathway
3. MET Alterations
3.1. MET Exon 14 Skipping Mutation
3.2. MET Amplification
3.3. MET Overexpression
3.4. MET Fusion
4. Targeting the HGF/MET Axis
4.1. MET TKIs
4.1.1. Multitargeted or Non-Selective MET TKIs
Crizotinib
Cabozantinib
Merestinib
Glesatinib
4.1.2. Selective MET TKIs
Capmatinib
Tepotinib
Savolitinib
4.2. MET Antibodies
4.2.1. Anti-MET/HGF Antibodies
4.2.2. Anti-MET Antibody–Drug Conjugates
5. Immunotherapy for Patients with MET Alterations
6. Mechanism of Resistance to MET Inhibitors
Acquired MET Alterations as a Mechanism of Resistance in EGFR NSCLC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, N.; Temin, S.; Baker, S., Jr.; Blanchard, E.; Brahmer, J.R.; Celano, P.; Duma, N.; Ellis, P.M.; Elkins, I.B.; Haddad, R.Y.; et al. Therapy for Stage IV Non-Small-Cell Lung Cancer With Driver Alterations: ASCO Living Guideline. J. Clin. Oncol. 2022, 40, 3310–3322. [Google Scholar] [CrossRef]
- Singh, N.; Temin, S.; Baker, S., Jr.; Blanchard, E.; Brahmer, J.R.; Celano, P.; Duma, N.; Ellis, P.M.; Elkins, I.B.; Haddad, R.Y.; et al. Therapy for Stage IV Non-Small-Cell Lung Cancer Without Driver Alterations: ASCO Living Guideline. J. Clin. Oncol. 2022, 40, 3323–3343. [Google Scholar] [CrossRef]
- Santarpia, M.; Ciappina, G.; Spagnolo, C.C.; Squeri, A.; Passalacqua, M.I.; Aguilar, A.; Gonzalez-Cao, M.; Giovannetti, E.; Silvestris, N.; Rosell, R. Targeted therapies for KRAS-mutant non-small cell lung cancer: From preclinical studies to clinical development-a narrative review. Transl. Lung Cancer Res. 2023, 12, 346–368. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.E.; Kerr, K.M.; Menis, J.; Mok, T.S.; Nestle, U.; Passaro, A.; Peters, S.; Planchard, D.; Smit, E.F.; Solomon, B.J.; et al. Oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef]
- Santarpia, M.; Massafra, M.; Gebbia, V.; D’Aquino, A.; Garipoli, C.; Altavilla, G.; Rosell, R. A narrative review of MET inhibitors in non-small cell lung cancer with MET exon 14 skipping mutations. Transl. Lung Cancer Res. 2021, 10, 1536–1556. [Google Scholar] [CrossRef] [PubMed]
- Michaels, E.; Bestvina, C.M. Meeting an un-MET need: Targeting MET in non-small cell lung cancer. Front. Oncol. 2022, 12, 1004198. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Skead, G.; Govender, D. Gene of the month: MET. J. Clin. Pathol. 2015, 68, 405–409. [Google Scholar] [CrossRef]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef]
- Linossi, E.M.; Estevam, G.O.; Oshima, M.; Fraser, J.S.; Collisson, E.A.; Jura, N. State of the structure address on MET receptor activation by HGF. Biochem. Soc. Trans. 2021, 49, 645–661. [Google Scholar] [CrossRef]
- Recondo, G.; Che, J.; Janne, P.A.; Awad, M.M. Targeting MET Dysregulation in Cancer. Cancer Discov. 2020, 10, 922–934. [Google Scholar] [CrossRef]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 2013, 1833, 122–139. [Google Scholar] [CrossRef]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ye, W.; Wang, Y.D.; Chen, W.D. HGF/c-Met: A Key Promoter in Liver Regeneration. Front. Pharmacol. 2022, 13, 808855. [Google Scholar] [CrossRef]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. The Hepatocyte Growth Factor Receptor: Structure, Function and Pharmacological Targeting in Cancer. Curr. Signal Transduct. Ther. 2011, 6, 146–151. [Google Scholar] [CrossRef]
- Raj, S.; Kesari, K.K.; Kumar, A.; Rathi, B.; Sharma, A.; Gupta, P.K.; Jha, S.K.; Jha, N.K.; Slama, P.; Roychoudhury, S.; et al. Molecular mechanism(s) of regulation(s) of c-MET/HGF signaling in head and neck cancer. Mol. Cancer 2022, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Hervieu, A.; Kermorgant, S. The Role of PI3K in Met Driven Cancer: A Recap. Front. Mol. Biosci. 2018, 5, 86. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Sakamoto, M.; Patil, T. MET alterations in advanced non-small cell lung cancer. Lung Cancer 2023, 178, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Suda, K.; Mitsudomi, T. Lung Cancer with MET exon 14 Skipping Mutation: Genetic Feature, Current Treatments, and Future Challenges. Lung Cancer 2021, 12, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yin, J.; Bohlman, S.; Walker, P.; Dacic, S.; Kim, C.; Khan, H.; Liu, S.V.; Ma, P.C.; Nagasaka, M.; et al. Characterization of MET Exon 14 Skipping Alterations (in NSCLC) and Identification of Potential Therapeutic Targets Using Whole Transcriptome Sequencing. JTO Clin. Res. Rep. 2022, 3, 100381. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Liang, Y.; Zhu, V.; Ou, S.I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The Why, the How, the Who, the Unknown, and the Inevitable. Lung Cancer 2017, 103, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Malapelle, U.; Russo, A. Skipping or Not Skipping? That’s the Question! An Algorithm to Classify Novel MET Exon 14 Variants in Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2023, 7, e2200674. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Lomboy, A.; Lawrence, C.A.; Yourshaw, M.; Bocsi, G.T.; Camidge, D.R.; Aisner, D.L. DNA-Based versus RNA-Based Detection of MET Exon 14 Skipping Events in Lung Cancer. J. Thorac. Oncol. 2019, 14, 737–741. [Google Scholar] [CrossRef]
- Moore, D.A.; Benafif, S.; Poskitt, B.; Argue, S.; Lee, S.M.; Ahmad, T.; Papadatos-Pastos, D.; Jamal-Hanjani, M.; Bennett, P.; Forster, M.D. Optimising fusion detection through sequential DNA and RNA molecular profiling of non-small cell lung cancer. Lung Cancer 2021, 161, 55–59. [Google Scholar] [CrossRef]
- Lee, J.K.; Madison, R.; Classon, A.; Gjoerup, O.; Rosenzweig, M.; Frampton, G.M.; Alexander, B.M.; Oxnard, G.R.; Venstrom, J.M.; Awad, M.M.; et al. Characterization of Non-Small-Cell Lung Cancers with MET Exon 14 Skipping Alterations Detected in Tissue or Liquid: Clinicogenomics and Real-World Treatment Patterns. JCO Precis. Oncol. 2021, 5, PO.21.00122. [Google Scholar] [CrossRef]
- Sun, R.; Wang, Z.; Zhao, J.; Ren, P.; Ma, J.; Guo, Y. Optimized Detection of Unknown MET Exon 14 Skipping Mutations in Routine Testing for Patients With Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2023, 7, e2200482. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Alex, D.; Bosdet, I.; Hughesman, C.; Karsan, A.; Yip, S.; Ho, C. MET exon 14 skipping mutation positive non-small cell lung cancer: Response to systemic therapy. Lung Cancer 2021, 154, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Hendriks, L.E.L.; Mountzios, G.; Garcia-Campelo, R.; Saw, S.P.L.; Uprety, D.; Recondo, G.; Villacampa, G.; Reck, M. MET alterations in NSCLC-Current Perspectives and Future Challenges. J Thorac. Oncol. 2023, 18, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-dependent solid tumours-molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Duncan, D.J.; Vandenberghe, M.E.; Scott, M.L.J.; Barker, C. Fast fluorescence in situ hybridisation for the enhanced detection of MET in non-small cell lung cancer. PLoS ONE 2019, 14, e0223926. [Google Scholar] [CrossRef]
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Baron, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number-Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304. [Google Scholar] [CrossRef]
- Yin, W.; Guo, M.; Tang, Z.; Toruner, G.A.; Cheng, J.; Medeiros, L.J.; Tang, G. MET Expression Level in Lung Adenocarcinoma Loosely Correlates with MET Copy Number Gain/Amplification and Is a Poor Predictor of Patient Outcome. Cancers 2022, 14, 2433. [Google Scholar] [CrossRef]
- Passaro, A.; Janne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Qin, K.; Hong, L.; Zhang, J.; Le, X. MET Amplification as a Resistance Driver to TKI Therapies in Lung Cancer: Clinical Challenges and Opportunities. Cancers 2023, 15, 612. [Google Scholar] [CrossRef]
- Gong, C.; Xiong, H.; Qin, K.; Wang, J.; Cheng, Y.; Zhao, J.; Zhang, J. MET alterations in advanced pulmonary sarcomatoid carcinoma. Front. Oncol. 2022, 12, 1017026. [Google Scholar] [CrossRef]
- Kron, A.; Scheffler, M.; Heydt, C.; Ruge, L.; Schaepers, C.; Eisert, A.K.; Merkelbach-Bruse, S.; Riedel, R.; Nogova, L.; Fischer, R.N.; et al. Genetic Heterogeneity of MET-Aberrant NSCLC and Its Impact on the Outcome of Immunotherapy. J. Thorac. Oncol. 2021, 16, 572–582. [Google Scholar] [CrossRef]
- Li, J.W.; Cao, S.H.; Xu, J.L.; Zhong, H. De novo MET amplification promotes intrinsic resistance to first-generation EGFR tyrosine kinase inhibitors. Cancer Biol. Ther. 2019, 20, 1183–1186. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.S.; Kim, B.J. Prognostic value of MET copy number gain in non-small-cell lung cancer: An updated meta-analysis. J. Cancer 2018, 9, 1836–1845. [Google Scholar] [CrossRef]
- Watermann, I.; Schmitt, B.; Stellmacher, F.; Muller, J.; Gaber, R.; Kugler, C.; Reinmuth, N.; Huber, R.M.; Thomas, M.; Zabel, P.; et al. Improved diagnostics targeting c-MET in non-small cell lung cancer: Expression, amplification and activation? Diagn. Pathol. 2015, 10, 130. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Toschi, L.; Gianoncelli, L.; Baretti, M.; Santoro, A. Prognostic and predictive value of MET deregulation in non-small cell lung cancer. Ann. Transl. Med. 2015, 3, 83. [Google Scholar] [CrossRef]
- Scagliotti, G.; von Pawel, J.; Novello, S.; Ramlau, R.; Favaretto, A.; Barlesi, F.; Akerley, W.; Orlov, S.; Santoro, A.; Spigel, D.; et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2667–2674. [Google Scholar] [CrossRef]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results From the Phase III Randomized Trial of Onartuzumab Plus Erlotinib Versus Erlotinib in Previously Treated Stage IIIB or IV Non-Small-Cell Lung Cancer: METLung. J. Clin. Oncol. 2017, 35, 412–420. [Google Scholar] [CrossRef]
- Kang, J.; Deng, Q.M.; Feng, W.; Chen, Z.H.; Su, J.W.; Chen, H.J.; Wang, W.X.; Zhang, S.; Wang, Q.; Chen, Z.; et al. Response and acquired resistance to MET inhibitors in de novo MET fusion-positive advanced non-small cell lung cancer. Lung Cancer 2023, 178, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Ng, T.L.; Estrada-Bernal, A.; Le, A.T.; Ennever, P.R.; Camidge, D.R.; Doebele, R.C.; Aisner, D.L. Dramatic Response to Crizotinib in a Patient with Lung Cancer Positive for an HLA-DRB1-MET Gene Fusion. JCO Precis. Oncol. 2017, 2017, PO.17.00117. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.C.; Wang, W.X.; Xu, C.W.; Zhang, Q.X.; Du, K.Q.; Chen, G.; Lv, T.F.; Song, Y. Identification of a novel crizotinib-sensitive MET-ATXN7L1 gene fusion variant in lung adenocarcinoma by next generation sequencing. Ann. Oncol. 2018, 29, 2392–2393. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, Y.; Ye, T.; Zhao, Y.; Gao, Z.; Yuan, H.; Zheng, D.; Zheng, S.; Li, H.; Li, Y.; et al. Detection of Novel NRG1, EGFR, and MET Fusions in Lung Adenocarcinomas in the Chinese Population. J. Thorac. Oncol. 2019, 14, 2003–2008. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, M.; Rabushko, E.; Rozenberg, J.M.; Mohammad, T.; Seryakov, A.; Sekacheva, M.; Buzdin, A. Clinically relevant fusion oncogenes: Detection and practical implications. Ther. Adv. Med. Oncol. 2022, 14, 17588359221144108. [Google Scholar] [CrossRef]
- Kazdal, D.; Hofman, V.; Christopoulos, P.; Ilie, M.; Stenzinger, A.; Hofman, P. Fusion-positive non-small cell lung carcinoma: Biological principles, clinical practice, and diagnostic implications. Genes. Chromosomes Cancer 2022, 61, 244–260. [Google Scholar] [CrossRef]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef]
- Benvenuti, S.; Gentile, A.; Lazzari, L.; Arnesano, A.; Trusolino, L.; Comoglio, P.M. An ‘in-cell trial’ to assess the efficacy of a monovalent anti-MET antibody as monotherapy and in association with standard cytotoxics. Mol. Oncol. 2014, 8, 378–388. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Ou, S.H.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Drilon, A.; Fan, P.D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, Y.; Stoopler, M.B.; Shen, Y.; Cheng, H.; Chen, J.; Mansukhani, M.; Koul, S.; Halmos, B.; Borczuk, A.C. Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations. J. Clin. Oncol. 2016, 34, 794–802. [Google Scholar] [CrossRef]
- Waqar, S.N.; Morgensztern, D.; Sehn, J. MET Mutation Associated with Responsiveness to Crizotinib. J. Thorac. Oncol. 2015, 10, e29–e31. [Google Scholar] [CrossRef]
- Mendenhall, M.A.; Goldman, J.W. MET-Mutated NSCLC with Major Response to Crizotinib. J. Thorac. Oncol. 2015, 10, e33–e34. [Google Scholar] [CrossRef] [PubMed]
- Mahjoubi, L.; Gazzah, A.; Besse, B.; Lacroix, L.; Soria, J.C. A never-smoker lung adenocarcinoma patient with a MET exon 14 mutation (D1028N) and a rapid partial response after crizotinib. Investig. New Drugs 2016, 34, 397–398. [Google Scholar] [CrossRef]
- Jorge, S.E.; Schulman, S.; Freed, J.A.; VanderLaan, P.A.; Rangachari, D.; Kobayashi, S.S.; Huberman, M.S.; Costa, D.B. Responses to the multitargeted MET/ALK/ROS1 inhibitor crizotinib and co-occurring mutations in lung adenocarcinomas with MET amplification or MET exon 14 skipping mutation. Lung Cancer 2015, 90, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Oxnard, G.R.; Elkin, S.; Sullivan, E.K.; Carter, J.L.; Barbie, D.A. Response to Crizotinib in a Patient with Lung Adenocarcinoma Harboring a MET Splice Site Mutation. Clin. Lung Cancer 2015, 16, e101–e104. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ignatius Ou, S.H.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029. [Google Scholar] [CrossRef]
- Cortot, A.; Le, X.; Smit, E.; Viteri, S.; Kato, T.; Sakai, H.; Park, K.; Camidge, D.R.; Berghoff, K.; Vlassak, S.; et al. Safety of MET Tyrosine Kinase Inhibitors in Patients With MET Exon 14 Skipping Non-small Cell Lung Cancer: A Clinical Review. Clin. Lung Cancer 2022, 23, 195–207. [Google Scholar] [CrossRef]
- Landi, L.; Chiari, R.; Tiseo, M.; D’Inca, F.; Dazzi, C.; Chella, A.; Delmonte, A.; Bonanno, L.; Giannarelli, D.; Cortinovis, D.L.; et al. Crizotinib in MET-Deregulated or ROS1-Rearranged Pretreated Non-Small Cell Lung Cancer (METROS): A Phase II, Prospective, Multicenter, Two-Arms Trial. Clin. Cancer Res. 2019, 25, 7312–7319. [Google Scholar] [CrossRef]
- Moro-Sibilot, D.; Cozic, N.; Perol, M.; Mazieres, J.; Otto, J.; Souquet, P.J.; Bahleda, R.; Wislez, M.; Zalcman, G.; Guibert, S.D.; et al. Crizotinib in c-MET- or ROS1-positive NSCLC: Results of the AcSe phase II trial. Ann. Oncol. 2019, 30, 1985–1991. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Albiges, L.; Burotto, M.; Szczylik, C.; Zurawski, B.; Yanez Ruiz, E.; Maruzzo, M.; Suarez Zaizar, A.; Fein, L.E.; et al. Cabozantinib plus Nivolumab and Ipilimumab in Renal-Cell Carcinoma. N. Engl. J. Med. 2023, 388, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.W.; Dahlberg, S.E.; Wakelee, H.A.; Aisner, S.C.; Bowden, M.; Huang, Y.; Carbone, D.P.; Gerstner, G.J.; Lerner, R.E.; Rubin, J.L.; et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): A randomised, controlled, open-label, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.B.; Peek, V.L.; Ajamie, R.; Buchanan, S.G.; Graff, J.R.; Heidler, S.A.; Hui, Y.H.; Huss, K.L.; Konicek, B.W.; Manro, J.R.; et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Investig. New Drugs 2013, 31, 833–844. [Google Scholar] [CrossRef] [PubMed]
- He, A.R.; Cohen, R.B.; Denlinger, C.S.; Sama, A.; Birnbaum, A.; Hwang, J.; Sato, T.; Lewis, N.; Mynderse, M.; Niland, M.; et al. First-in-Human Phase I Study of Merestinib, an Oral Multikinase Inhibitor, in Patients with Advanced Cancer. Oncologist 2019, 24, e930–e942. [Google Scholar] [CrossRef]
- Wu, W.; Bi, C.; Credille, K.M.; Manro, J.R.; Peek, V.L.; Donoho, G.P.; Yan, L.; Wijsman, J.A.; Yan, S.B.; Walgren, R.A. Inhibition of tumor growth and metastasis in non-small cell lung cancer by LY2801653, an inhibitor of several oncokinases, including MET. Clin. Cancer Res. 2013, 19, 5699–5710. [Google Scholar] [CrossRef]
- Kawada, I.; Hasina, R.; Arif, Q.; Mueller, J.; Smithberger, E.; Husain, A.N.; Vokes, E.E.; Salgia, R. Dramatic antitumor effects of the dual MET/RON small-molecule inhibitor LY2801653 in non-small cell lung cancer. Cancer Res. 2014, 74, 884–895. [Google Scholar] [CrossRef]
- Engstrom, L.D.; Aranda, R.; Lee, M.; Tovar, E.A.; Essenburg, C.J.; Madaj, Z.; Chiang, H.; Briere, D.; Hallin, J.; Lopez-Casas, P.P.; et al. Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin. Cancer Res. 2017, 23, 6661–6672. [Google Scholar] [CrossRef]
- Kollmannsberger, C.; Hurwitz, H.; Bazhenova, L.; Cho, B.C.; Hong, D.; Park, K.; Reckamp, K.L.; Sharma, S.; Der-Torossian, H.; Christensen, J.G.; et al. Phase I Study Evaluating Glesatinib (MGCD265), An Inhibitor of MET and AXL, in Patients with Non-small Cell Lung Cancer and Other Advanced Solid Tumors. Target Oncol. 2023, 18, 105–118. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Q.; Yang, G.; Marando, C.; Koblish, H.K.; Hall, L.M.; Fridman, J.S.; Behshad, E.; Wynn, R.; Li, Y.; et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin. Cancer Res. 2011, 17, 7127–7138. [Google Scholar] [CrossRef]
- Baltschukat, S.; Engstler, B.S.; Huang, A.; Hao, H.X.; Tam, A.; Wang, H.Q.; Liang, J.; DiMare, M.T.; Bhang, H.C.; Wang, Y.; et al. Capmatinib (INC280) Is Active Against Models of Non-Small Cell Lung Cancer and Other Cancer Types with Defined Mechanisms of MET Activation. Clin. Cancer Res. 2019, 25, 3164–3175. [Google Scholar] [CrossRef]
- Bladt, F.; Faden, B.; Friese-Hamim, M.; Knuehl, C.; Wilm, C.; Fittschen, C.; Gradler, U.; Meyring, M.; Dorsch, D.; Jaehrling, F.; et al. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin. Cancer Res. 2013, 19, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Kurzrock, R.; Amin, H.M.; Xiong, W.; Fu, S.; Piha-Paul, S.A.; Janku, F.; Eskandari, G.; Catenacci, D.V.; Klevesath, M.; et al. First-in-Man Phase I Trial of the Selective MET Inhibitor Tepotinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Sai, Y.; Wang, J.; Yu, M.; Wang, G.; Zhang, L.; Ren, H.; Fan, S.; Ren, Y.; Qing, W.; et al. Preclinical pharmacokinetics, disposition, and translational pharmacokinetic/pharmacodynamic modeling of savolitinib, a novel selective cMet inhibitor. Eur. J. Pharm. Sci. 2019, 136, 104938. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Savolitinib: First Approval. Drugs 2021, 81, 1665–1670. [Google Scholar] [CrossRef]
- Lu, S.; Fang, J.; Li, X.; Cao, L.; Zhou, J.; Guo, Q.; Liang, Z.; Cheng, Y.; Jiang, L.; Yang, N.; et al. Once-daily savolitinib in Chinese patients with pulmonary sarcomatoid carcinomas and other non-small-cell lung cancers harbouring MET exon 14 skipping alterations: A multicentre, single-arm, open-label, phase 2 study. Lancet Respir. Med. 2021, 9, 1154–1164. [Google Scholar] [CrossRef]
- Lu, S.; Fang, J.; Li, X.; Cao, L.; Zhou, J.; Guo, Q.; Liang, Z.; Cheng, Y.; Jiang, L.; Yang, N.; et al. Long-Term Efficacy, Safety, and Subgroup Analysis of Savolitinib in Chinese Patients With NSCLCs Harboring MET Exon 14 Skipping Alterations. JTO Clin. Res. Rep. 2022, 3, 100407. [Google Scholar] [CrossRef]
- Lu, S.; Fang, J.; Li, X.; Cao, L.; Zhou, J.; Guo, Q.; Liang, Z.; Cheng, Y.; Jiang, L.; Yang, L.; et al. Phase II study of SCC244 in NSCLC patients harboring MET Exon 14 skipping (METex14) mutations (GLORY study). In Proceedings of the 2022 AACR Annual Meeting, New Orleans, LA, USA, 8–13 April 2022. [Google Scholar]
- Scagliotti, G.; Moro-Sibilot, D.; Kollmeier, J.; Favaretto, A.; Cho, E.K.; Grosch, H.; Kimmich, M.; Girard, N.; Tsai, C.M.; Hsia, T.C.; et al. A Randomized-Controlled Phase 2 Study of the MET Antibody Emibetuzumab in Combination with Erlotinib as First-Line Treatment for EGFR Mutation-Positive NSCLC Patients. J. Thorac. Oncol. 2020, 15, 80–90. [Google Scholar] [CrossRef]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 50. [Google Scholar] [CrossRef]
- Dersarkissian, M.; Bhak, R.; Lin, H.; Li, S.; Cheng, M.; Lax, A.; Huang, H.; Duh, M.; Ou, S. Real-world treatment patterns and survival in non-small cell lung cancer patients with EGFR exon 20 insertion mutations. J. Thorac. Oncol. 2019, 14, S681. [Google Scholar] [CrossRef]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.Y.; Kim, S.W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- Krebs, M.; Spira, A.I.; Cho, B.C.; Besse, B.; Goldman, J.W.; Janne, P.A.; Ma, Z.; Mansfield, A.S.; Minchom, A.R.; Ou, S.I.; et al. Amivantamab in patients with NSCLC with MET exon 14 skipping mutation: Updated results from the CHRYSALIS study. J. Clin. Oncol. 2022, 40 (Suppl. S16), 9008. [Google Scholar] [CrossRef]
- Cho, B.C.; Ahn, M.J.; Kim, T.M.; Kim, C.; Shim, B.Y.; Han, J.Y.; Drilon, A.E.; Lena, H.; Gomez, J.; Gray, J.E.; et al. 1173P Early safety, tolerability, and efficacy of REGN5093 in patients (pts) with MET-altered advanced non-small cell lung cancer (aNSCLC) from a first in human (FIH) study. Ann. Oncol. 2022, 33, S1085. [Google Scholar] [CrossRef]
- Camidge, R.; Janku, F.; Martinez-Bueno, A.; Catenacci, D.; Lee, J.; Lee, S.; Dowlati, A.; Rohrberg, K.; Navarro, A.; Moon, Y.; et al. Safety and preliminary clinical activity of the MET antibody mixture, Sym015 in advanced non-small cell lung cancer (NSCLC) patients with MET amplification/exon 14 deletion. J. Clin. Oncol. 2020, 38, 9510. [Google Scholar] [CrossRef]
- Desai, A.; Abdayem, P.; Adjei, A.A.; Planchard, D. Antibody-drug conjugates: A promising novel therapeutic approach in lung cancer. Lung Cancer 2022, 163, 96–106. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Marks, S.; Naidoo, J. Antibody drug conjugates in non-small cell lung cancer: An emerging therapeutic approach. Lung Cancer 2022, 163, 59–68. [Google Scholar] [CrossRef]
- Camidge, D.R.; Bar, J.; Horinouchi, H.; Goldman, J.; Moiseenko, F.; Filippova, E.; Cicin, I.; Bradbury, P.; Daaboul, N.; Tomasini, P.; et al. Telisotuzumab vedotin (Teliso-V) monotherapy in patients (pts) with previously treated c-Met–overexpressing (OE) advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2022, 40, 9016. [Google Scholar] [CrossRef]
- Drilon, A.E.; Awad, M.M.; Gadgeel, S.M.; Villaruz, L.; Sabari, J.; Perez, J.; Daly, C.; Patel, S.; Li, S.; Seebach, F.; et al. A phase 1/2 study of REGN5093- M114, a METxMET antibody-drug conjugate, in patients with mesenchymal epithelial transition factor (MET)-overexpressing NSCLC. J. Clin. Oncol. 2022, 40, TPS8593. [Google Scholar] [CrossRef]
- Domenech, M.; Munoz Marmol, A.M.; Mate, J.L.; Estival, A.; Moran, T.; Cucurull, M.; Saigi, M.; Hernandez, A.; Sanz, C.; Hernandez-Gallego, A.; et al. Correlation between PD-L1 expression and MET gene amplification in patients with advanced non-small cell lung cancer and no other actionable oncogenic driver. Oncotarget 2021, 12, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Demuth, C.; Andersen, M.N.; Jakobsen, K.R.; Madsen, A.T.; Sorensen, B.S. Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment. Oncotarget 2017, 8, 68221–68229. [Google Scholar] [CrossRef] [PubMed]
- Albitar, M.; Sudarsanam, S.; Ma, W.; Jiang, S.; Chen, W.; Funari, V.; Blocker, F.; Agersborg, S. Correlation of MET gene amplification and TP53 mutation with PD-L1 expression in non-small cell lung cancer. Oncotarget 2018, 9, 13682–13693. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Sabari, J.K.; Leonardi, G.C.; Shu, C.A.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol. 2018, 29, 2085–2091. [Google Scholar] [CrossRef]
- Mayenga, M.; Assie, J.B.; Monnet, I.; Massiani, M.A.; Tabeze, L.; Friard, S.; Fraboulet, S.; Metivier, A.C.; Chouaid, C.; Zemoura, L.; et al. Durable responses to immunotherapy of non-small cell lung cancers harboring MET exon-14-skipping mutation: A series of 6 cases. Lung Cancer 2020, 150, 21–25. [Google Scholar] [CrossRef]
- Guisier, F.; Dubos-Arvis, C.; Vinas, F.; Doubre, H.; Ricordel, C.; Ropert, S.; Janicot, H.; Bernardi, M.; Fournel, P.; Lamy, R.; et al. Efficacy and Safety of Anti-PD-1 Immunotherapy in Patients With Advanced NSCLC With BRAF, HER2, or MET Mutations or RET Translocation: GFPC 01-2018. J. Thorac. Oncol. 2020, 15, 628–636. [Google Scholar] [CrossRef]
- Dempke, W.C.M.; Fenchel, K. Has programmed cell death ligand-1 MET an accomplice in non-small cell lung cancer?-a narrative review. Transl. Lung Cancer Res. 2021, 10, 2667–2682. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, Q.; Zeng, X.; Wang, M.; Dong, S.; Yang, B.; Tu, X.; Wei, T.; Xie, W.; Zhang, C.; et al. MET Amplification Attenuates Lung Tumor Response to Immunotherapy by Inhibiting STING. Cancer Discov. 2021, 11, 2726–2737. [Google Scholar] [CrossRef]
- Lin, J.J.; Shaw, A.T. Resisting Resistance: Targeted Therapies in Lung Cancer. Trends Cancer 2016, 2, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Waarts, M.R.; Stonestrom, A.J.; Park, Y.C.; Levine, R.L. Targeting mutations in cancer. J. Clin. Investig. 2022, 132, JCI154943. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yang, H.; Zhu, B.; Wang, S.; Pang, J.; Wu, X.; Xu, Y.; Zhang, J.; Zhang, J.; Ou, Q.; et al. Mutations in the MET tyrosine kinase domain and resistance to tyrosine kinase inhibitors in non-small-cell lung cancer. Respir. Res. 2023, 24, 28. [Google Scholar] [CrossRef] [PubMed]
- Rivas, S.; Marin, A.; Samtani, S.; Gonzalez-Feliu, E.; Armisen, R. MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies. Int. J. Mol. Sci. 2022, 23, 13898. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Kobayashi, Y.; Suda, K.; Koga, T.; Nishino, M.; Ohara, S.; Chiba, M.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. Sensitivity and Resistance of MET Exon 14 Mutations in Lung Cancer to Eight MET Tyrosine Kinase Inhibitors In Vitro. J. Thorac. Oncol. 2019, 14, 1753–1765. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Moonsamy, P.; Gainor, J.F.; Lennerz, J.K.; Piotrowska, Z.; Lin, J.J.; Lennes, I.T.; Sequist, L.V.; Shaw, A.T.; Goodwin, K.; et al. A Phase 2 Study of Capmatinib in Patients With MET-Altered Lung Cancer Previously Treated With a MET Inhibitor. J. Thorac. Oncol. 2021, 16, 850–859. [Google Scholar] [CrossRef]
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.; Mattar, M.; Khodos, I.; et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Jamme, P.; Fernandes, M.; Copin, M.C.; Descarpentries, C.; Escande, F.; Morabito, A.; Gregoire, V.; Jamme, M.; Baldacci, S.; Tulasne, D.; et al. Alterations in the PI3K Pathway Drive Resistance to MET Inhibitors in NSCLC Harboring MET Exon 14 Skipping Mutations. J. Thorac. Oncol. 2020, 15, 741–751. [Google Scholar] [CrossRef]
- Coleman, N.; Hong, L.; Zhang, J.; Heymach, J.; Hong, D.; Le, X. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open 2021, 6, 100319. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef]
- Yang, J.J.; Fang, J.; Shu, Y.Q.; Chang, J.H.; Chen, G.Y.; He, J.X.; Li, W.; Liu, X.Q.; Yang, N.; Zhou, C.; et al. A phase Ib study of the highly selective MET-TKI savolitinib plus gefitinib in patients with EGFR-mutated, MET-amplified advanced non-small-cell lung cancer. Investig. New Drugs 2021, 39, 477–487. [Google Scholar] [CrossRef]
- Ahn, M.J.; de Marinis, F.; Bonanno, L.; de Marinis, F.; Bonanno, L.; Cho, B.C.; Kim, T.M.; Cheng, S.; Novello, S.; Proto, C.; et al. MET biomarker-based preliminary efficacy analysis in SAVANNAH: Savolitinib+ osimertinib in EGFRm NSCLC post-osimertinib. J. Clin. Oncol. 2022, 17, S469–S470. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.W.; Soo, R.A.; Kim, S.W.; Pan, H.; et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): An open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 2020, 8, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Kim, T.M.; Lim, B.K.; Wislez, M.; Dooms, C.; Finocchiaro, G.; Hayashi, H.; Liam, C.K.; Raskin, J.; Tho, L.M.; et al. LBA52 Tepotinib + osimertinib for EGFRm NSCLC with MET amplification (METamp) after progression on first-line (1L) osimertinib: Initial results from the INSIGHT 2 study. Ann. Oncol. 2022, 33, S1419–S1420. [Google Scholar] [CrossRef]
- Horinouchi, H.; Goldman, J.W.; Cho, B.C.; Tomasini, P.; Dunbar, M.; Hoffman, D.; Parikh, A.; Blot, V.; Camidge, R. 387P-Telisotuzumab vedotin (Teliso-V) in combination with osimertinib in patients with advanced EGFR-mutated, c-met overexpressing, non-small cell lung cancer (NSCLC): Safety and efficacy results from phase Ib study. Ann. Oncol. 2022, 33, S1560. [Google Scholar] [CrossRef]


{kind=link}
| Drug | Trial | Phase | Treatment | Population | N. of Patients | Results |
|---|---|---|---|---|---|---|
| Crizotinib | PROFILE-1001 (NCT00585195) | I | Crizotinib 250 mg BID | Advanced NSCLC with MET exon 14 skipping or MET amplification: Low (≥1.8–≤2.2 copies) Medium (≥2.2–≤4 copies) High (> 4 copies) | 65 3 14 21 | ORR 32% PFS 7.3 months DOR 9.1 months |
| METROS (NCT02499614) | II | Crizotinib 250 mg BID | Advanced NSCLC with MET exon 14 skipping or MET amplification (MET/CEP7 ratio >2.2) | 26 | ORR 27% PFS 4.4 months DOR 3.7 months | |
| AcSé (NCT02034981) | II | Crizotinib 250 mg BID | Advanced NSCLC with c-MET ≥ 6 copies or all c-MET-mutations | 25 28 | ORR 16%; PFS 3.2 months ORR 10.7%; PFS 2.2 months | |
| Cabozantinib | NCT01866410 | II | Cabozantinib 40 mg daily + erlotinib 150 mg daily | Advanced NSCLC with EGFR mutation and progressive disease on EGFR TKI (no MET mutations) | 37 | ORR: 10.8%; PFS: 3.6 months; OS: 13.1 months |
| NCT01708954 | II | Arm A: erlotinib 150 mg daily Arm B: cabozantinib 60 mg daily Arm C: erlotinib 150 mg + cabozantinib 40 mg | Previously treated advanced NSCLC (MET mutations not evaluated) | 38 38 35 | ORR: 3%; PFS: 1.8 months; OS: 5.1 months ORR: 11%; PFS: 4.3 months; OS: 9.2 months ORR: 3%; PFS: 4.7 months; OS: 13.3 months | |
| Capmatinib | GEOMETRY mono-1 (NCT02414139) | II | Capmatinib 400 mg BID | NSCLC with: MET exon 14 skipping (1st line) | 28 | ORR: 68%; PFS: 12.4 months; DOR: 12.6 months |
| MET exon 14 skipping (subsequent lines) | 69 | ORR: 41%; PFS: 5.4 months; DOR: 9.7 months | ||||
| MET amplification GCN ≥ 10 (1st line) | 69 | ORR: 29%; PFS: 4.1 months; DOR 8.3 months | ||||
| MET amplification GCN ≥ 10 (subsequent lines) | 15 | ORR: 40%; PFS: 4.2 months; DOR 7.5 months | ||||
| Tepotinib | VISION (NCT02864992) | II | Tepotinib 500 mg once daily | NSCLC with MET exon 14 skipping (cohort A) | 99 | ORR: 46%; PFS: 8.5 months; DOR: 11.1 months; |
| Savolitinib | NCT02897479 | II | Savolitinib 600 mg for BW ≥ 50 kg or 400 mg for BW < 50 kg | Advanced NSCLC or pulmonary sarcomatoid carcinoma with MET exon 14 skipping | 61 | ORR 49.2%; PSF 6.9 months; DCR 93.4%; |
| Drug | Trial | Phase | Treatment | Drug Combined | Primary Endpoint (s) | Secondary Endpoint (s) |
|---|---|---|---|---|---|---|
| Crizotinib | Matrix (NCT02664935) | II | Crizotinib 250 mg BD continuous dosing, 21-day cycle | - | OR, PFS, DCB | TTP, OS, Safety |
| MATCH (NCT02465060) | II | Crizotinib 250 mg BD on days 1–28 | - | ORR | OS, PFS | |
| NCT04084717 | II | Crizotinib 250 mg BD every day of each 28-day cycle | - | ORR, PFS, OS | - | |
| Cabozantinib | NCT01639508 | II | Cabozantinib 60 mg every day of each 28-day cycle | - | ORR | PFS, OS, Safety |
| LUNG-IST-127 (NCT05613413) | II | Cabozantinib 40 mg once daily Days 1–21 + pembrolizumab 200 mg iv Q3W as maintenance therapy following 4 cycles of induction therapy with disease control | Pembrolizumab | PFS | OS, ORR, Safety | |
| NCT04310007 | II | Arm A: cabozantinib S-malate QD 21-day cycle Arm B: cabozantinib S-malate QD 21-day cycle and nivolumab Q3W Arm C: ramucirumab IV and docetaxel IV Q3W | Nivolumab | PFS | OS, BOR, safety | |
| CABinMET (NCT03911193) | II | Cabozantinib 60 mg daily (each 28 days) | - | ORR | PFS, OS, DCR, exploratory biomarkers | |
| Merestinib | NCT02920996 | II | Merestinib 120 mg daily (each 28 days) | - | ORR | PFS, OS, DOR, Safety |
| Glesatinib | NCT02544633 | II | Glesatinib 750 mg BD | - | ORR | PFS, OS, DOR |
| NCT02954991 | II | Glesatinib 750 mg BD + Nivolumab 240 mg IV every 2 weeks or 480 mg IV every 4 weeks | Nivolumab | ORR | OS, PFS, Safety | |
| Capmatinb | NCT03693339 | II | Capmatinib 400 mg BD continuously dosing | - | ORR | PFS, OS, DOR |
| NCT04677595 | II | Capmatinib 400 mg BD continuously dosing | - | ORR | DOR, TTR, PFS, OS, IDCR | |
| NCT05567055 | II | Capmatinib 400 mg BD continuously dosing | - | CNS Overall response rate | DOR, PFS, OS, CNS DOR, CNS PFS | |
| Geometry-N (NCT04926831) | II | Capmatinib 400 mg BD (neoadjuvant/adjuvant setting) | - | MPR | PCR, ORR, DFS, Safety | |
| NCT04427072 | II | Capmatinib 400 mg BD | - | PFS | ORR, DOR, DCR, IDCR, OS | |
| Tepotinib | NCT04739358 | I/II | Tepotinib daily in cycles of 21-day duration | Other TKIs | Intracranial ORR, Overall and extracranial ORR | Overall, intracranial and extracranial ORR, PFS, DOR and DCR, safety |
| NCT03940703 | II | Tepotinib 500 mg once daily and osimertinib 80 mg once daily each 21 day | Osimertinib | ORR, Safety | PFS, OS | |
| POTENT NCT05782361 | I | Tepotinib 500 mg or 250 mg once daily and pembrolizumab 200 mg Q3W | Pembrolizumab | Antitumor activity | Safety, tolerability | |
| Savolitinib | NCT04923945 | III | Savolitinib 600 mg once daily continuously in patients with BW ≥ 50 kg and Savolitinib 400 mg once daily in patients with BW < 50 kg | - | ORR | PFS, Safety |
| NCT05777278 | I/II | Savolitinib 300 mg or 200 mg BD and Docetaxel 60 mg/m2 iv Q3W | Docetaxel | ORR | PFS, OS, DCR, DOR | |
| NCT05374603 | II | Savolitinib 600 mg for BW ≥ 50 kg, 400 mg for BW < 50 kg once daily and Durvalumab 1500 mg iv Q4W | Durvalumab | PFS | ORR, DOR, DCR, OS | |
| SAVANNAH NCT03778229 | II | Savolitinib 300 mg once daily or 300 mg BD or 600 mg once daily and osimertinib 80 mg oral once daily | Osimertinib | ORR | PFS, OS, DOR | |
| SAFFRON NCT05261399 | III | Arm A: Pemetrexed (500 mg/m2) with either cisplatin (75 mg/m2) or carboplatin (AUC5) Q3W for 4 cycles, followed by pemetrexed maintenance (500 mg/m2) Q3W Arm B: Savolitinib 300 mg BD + osimertinib 80 mg once daily | Osimertinib | PFS | OS, ORR, DCR, DOR | |
| ORCHARD NCT03944772 | II | Savolitinib 300 mg or 600 mg once daily + Osimertinib 80 mg once daily | Osimertinib | ORR | PFS, DOR, OS | |
| SACHI NCT05015608 | III | Savolitinib once daily + Osimertinib once daily (every 3 weeks) | Osimertinib | PFS | ORR, OS, DOR, DCR, TTR, Safety | |
| FLOWERS NCT05163249 | II | Savolitinib 300 mg BD+, Osimertinib 80 mg once daily | Osimertinib | ORR | PFS, DOR, DCR, OS | |
| SANOVO NCT05009836 | III | Savolitinib 600 mg or 400 mg once daily + Osimertinib 80 once daily (every 3 weeks) | Osimertinib | PFS | ORR, OS, DOR, DCR, TTR, Safety | |
| APL-101 | SPARTA NCT03175224 | I/II | APL-101 28-day cycles at four planned dose levels (100 mg, 200 mg, 300 mg and 400 mg) | - | Safety, ORR | ORR, DOR, PFS, TTP |
| Elzovantinib or TPX-0022 | SHIELD-1 NCT03993873 | I/II | Elzovantinib | - | Safety, tolerability | PFS, OS, ORR, DOR, TTR |
| GluMETinib or SCC244 | GLORY, NCT04270591 | I/II | Glumetinib 300 mg once daily | - | ORR | DOR, OS |
| Drug | Trial | Phase | Treatment | Drug Combined | Primary Endpoint (s) | Secondary Endpoint (s) |
|---|---|---|---|---|---|---|
| Amivantamab | CHRYSALIS NCT02609776 | I | Amivantamab + Lazertinib | Lazertinib | Safety, DLT, ORR, DOR | PFS, TTF, OS |
| CHRYSALIS-2 NCT04077463 | I/Ib | Amivantamab + Lazertinib | Lazertinib | DLT, ORR, Safety, DOR | PFS, TTF, OS, Intracranial PFS | |
| MARIPOSA NCT04487080 | III | Arm A: Amivantamab 1050 mg iv BW less than <80 kg and 1400 mg for BW ≥ 80 kg in 28-day cycles + Lazertinib 240 mg once daily Arm B: osimertinib 80 mg o once daily + lazertinib 240 mg once daily and placebo Arm C: lazertinib 240 mg once daily+ osimertinib 80 mg once daily and placebo | Lazertinib | PFS | ORR, OS, DOR, TTP, PFS2, intracranial PFS, Safety | |
| MARIPOSA-2 NCT04988295 | II | Arm A: Amivantamab + Lazertinib, Pemetrexed and Carboplatin (LACP dosing or ACP-L dosing) Arm B: Pemetrexed + Carboplatin iv for up to 4 cycles Q3W → Pemetrexed as maintenance until progression Arm C: Amivantamab+ Pemetrexed + Carboplatin iv for up to 4 cycles Q3W → Amivantamab + Pemetrexed as maintenance until progression | Lazertinib, pemetrexed, carboplatin | PFS | ORR, OS, DOR, TTST, PFS2 | |
| METalmark NCT05488314 | I/II | Amivantamab 700 mg iv for BW less than 80 kg or 1050 mg for BW greater than or equal to 80 kg + Capmatinib 400 mg twice daily | Capmatinib | DLT, Safety, ORR | PFS, DOR, OS, Safety | |
| REGN5093 | NCT04077099 | I/II | REGN5093 iv Monotherapy in dose escalation cohorts followed by an expansion phase | - | DLT, safety, ORR | PFS, OS, DOR, DCR |
| Sym015 | NCT02648724 | I/II | Sym015 at different dose levels 6, 12, 18, and 24 mg/kg. | - | DLT, ORR | AUC, Tmax |
| Telisotuzumab vedotin | NCT03539536 | II | Telisotuzumab vedotin iv infusion every 14 days | - | ORR, safety | OS, PFS, DOR, DCR |
| REGN5093-M114 | NCT04982224 | I/II | REGN5093-M114 Iv infusion | - | DLT, Safety | ORR, DCR, PFS, OS, DOR, TTR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spagnolo, C.C.; Ciappina, G.; Giovannetti, E.; Squeri, A.; Granata, B.; Lazzari, C.; Pretelli, G.; Pasello, G.; Santarpia, M. Targeting MET in Non-Small Cell Lung Cancer (NSCLC): A New Old Story? Int. J. Mol. Sci. 2023, 24, 10119. https://doi.org/10.3390/ijms241210119
Spagnolo CC, Ciappina G, Giovannetti E, Squeri A, Granata B, Lazzari C, Pretelli G, Pasello G, Santarpia M. Targeting MET in Non-Small Cell Lung Cancer (NSCLC): A New Old Story? International Journal of Molecular Sciences. 2023; 24(12):10119. https://doi.org/10.3390/ijms241210119
Chicago/Turabian StyleSpagnolo, Calogera Claudia, Giuliana Ciappina, Elisa Giovannetti, Andrea Squeri, Barbara Granata, Chiara Lazzari, Giulia Pretelli, Giulia Pasello, and Mariacarmela Santarpia. 2023. "Targeting MET in Non-Small Cell Lung Cancer (NSCLC): A New Old Story?" International Journal of Molecular Sciences 24, no. 12: 10119. https://doi.org/10.3390/ijms241210119
APA StyleSpagnolo, C. C., Ciappina, G., Giovannetti, E., Squeri, A., Granata, B., Lazzari, C., Pretelli, G., Pasello, G., & Santarpia, M. (2023). Targeting MET in Non-Small Cell Lung Cancer (NSCLC): A New Old Story? International Journal of Molecular Sciences, 24(12), 10119. https://doi.org/10.3390/ijms241210119