
Joint QTL Mapping and Transcriptome Sequencing Analysis Reveal Candidate Seed-Shattering-Related Genes in Common Buckwheat

 ,
, 
Abstract
1. Introduction
2. Results
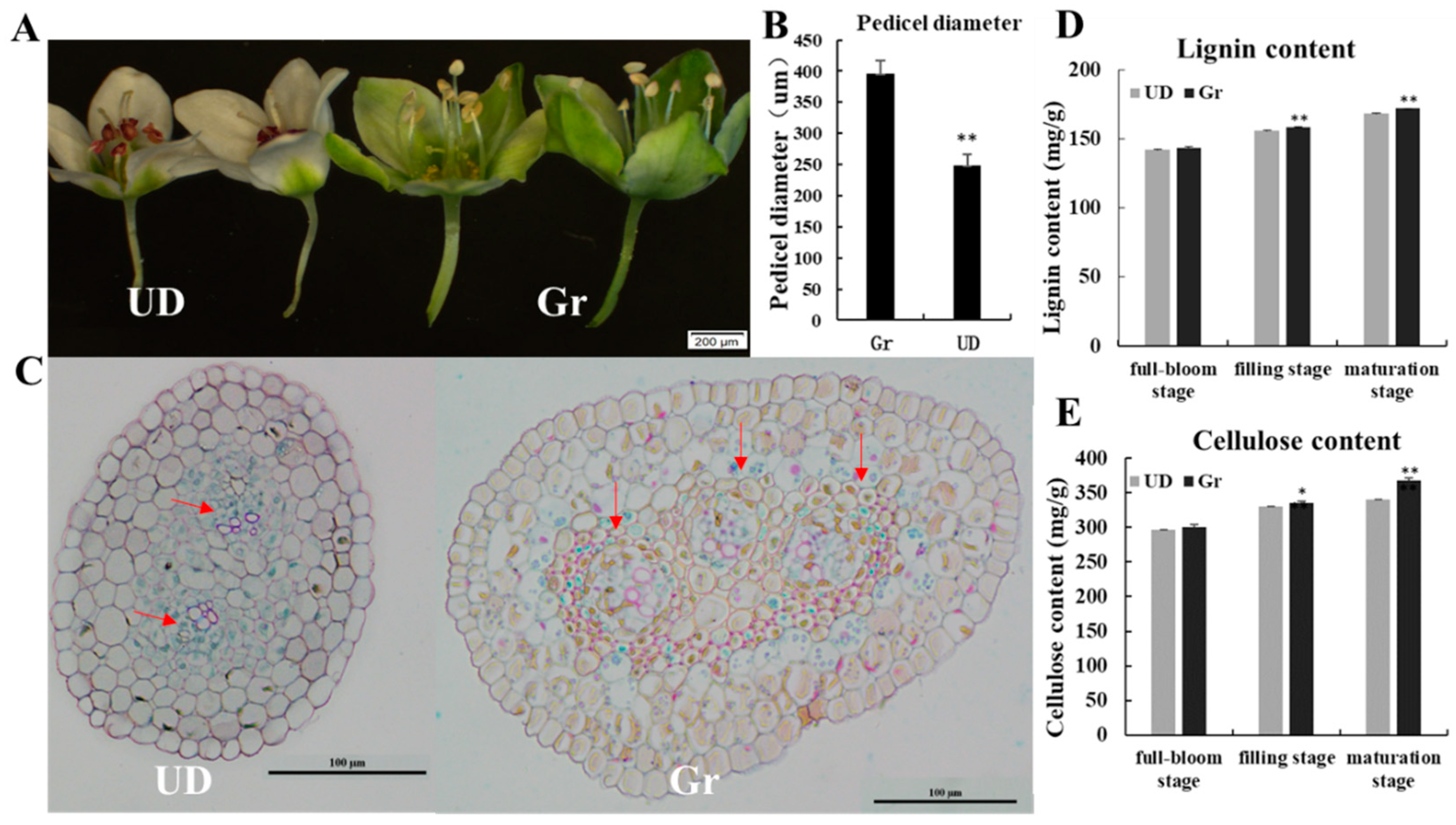
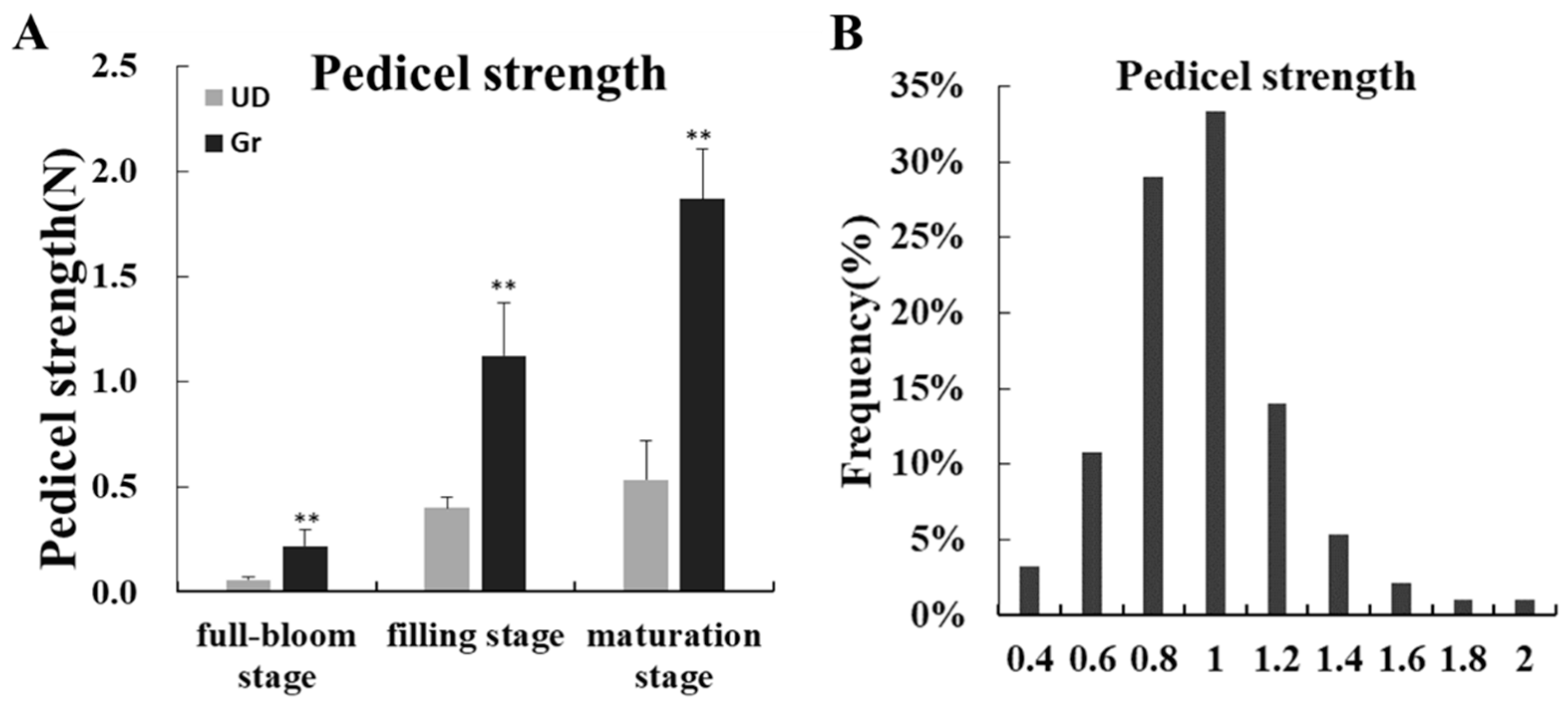
2.1. The Phenotypic Data Analysis of Parents and F2 Population
2.2. Genetic Linkage Map and QTLs Mapping for Pedicel Strength
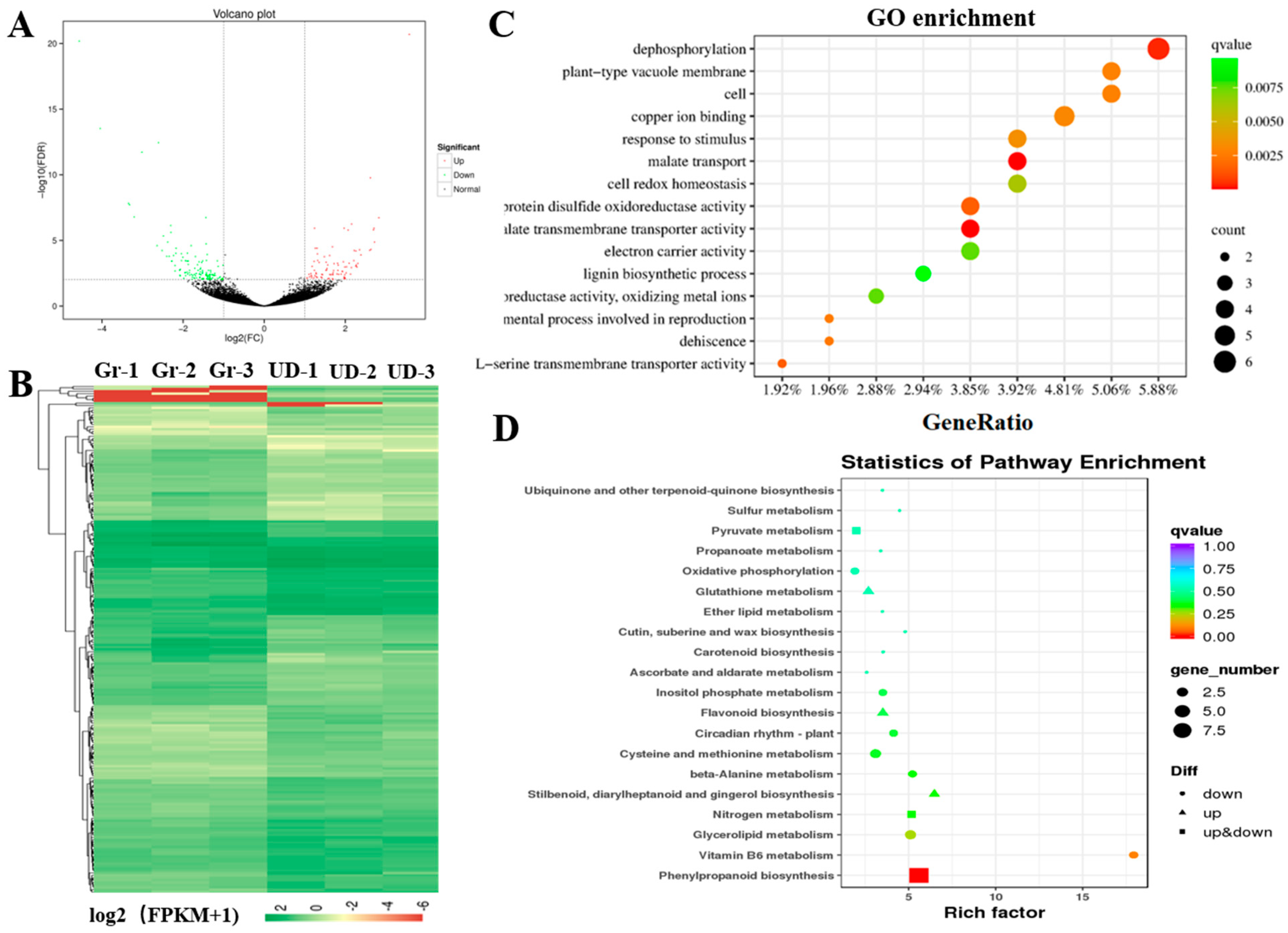
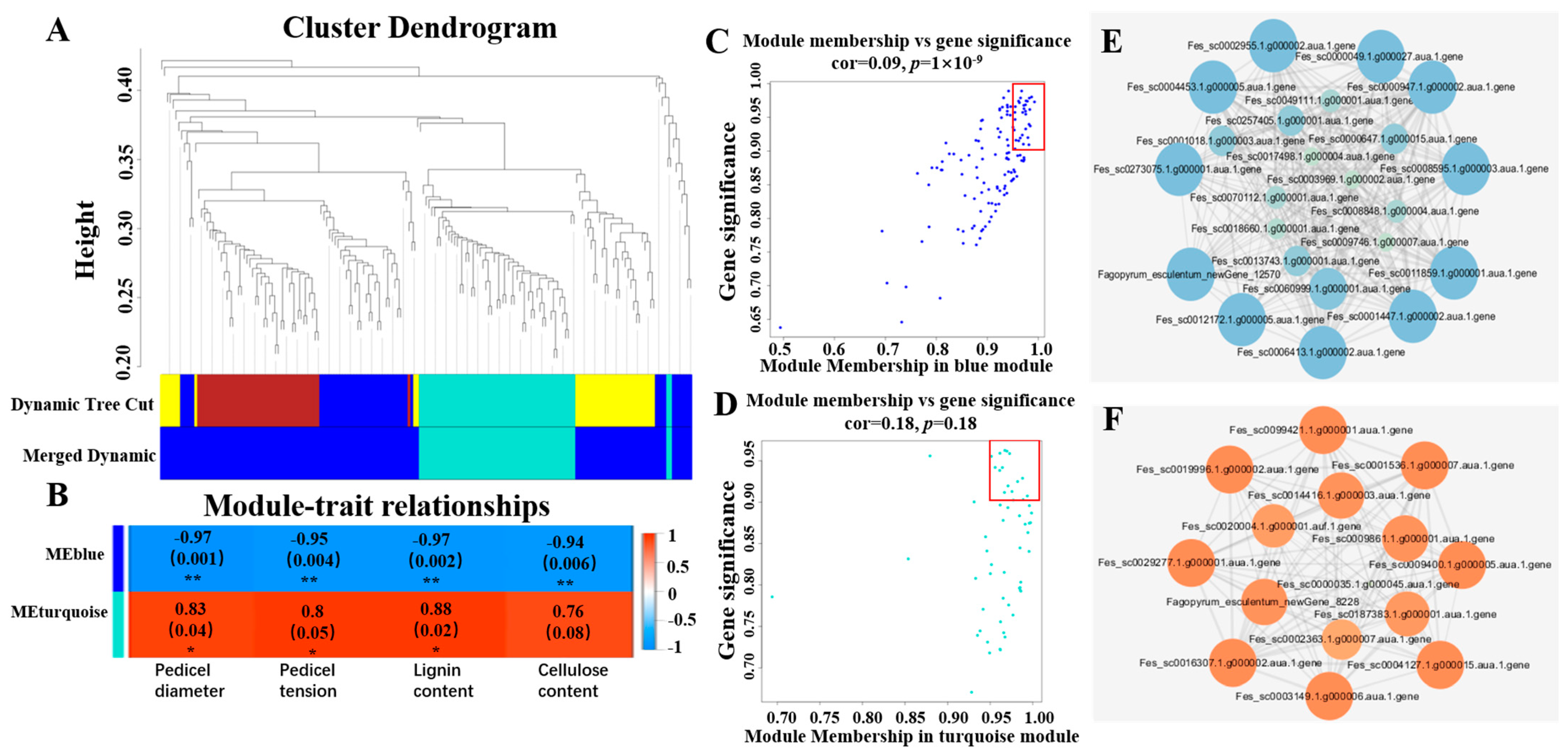
2.3. RNA Sequencing and Weighted Gene Co-Expression Network Analysis
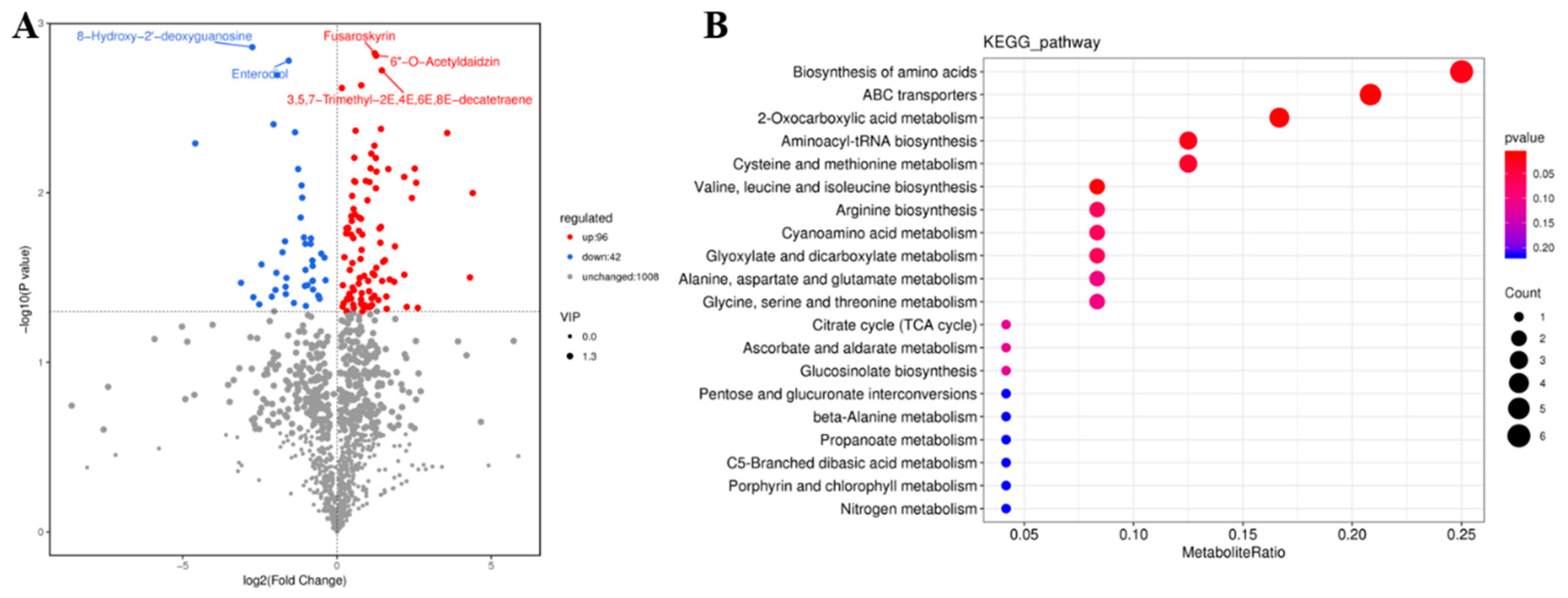
2.4. Metabolome Sequencing and Conjoint Analyses
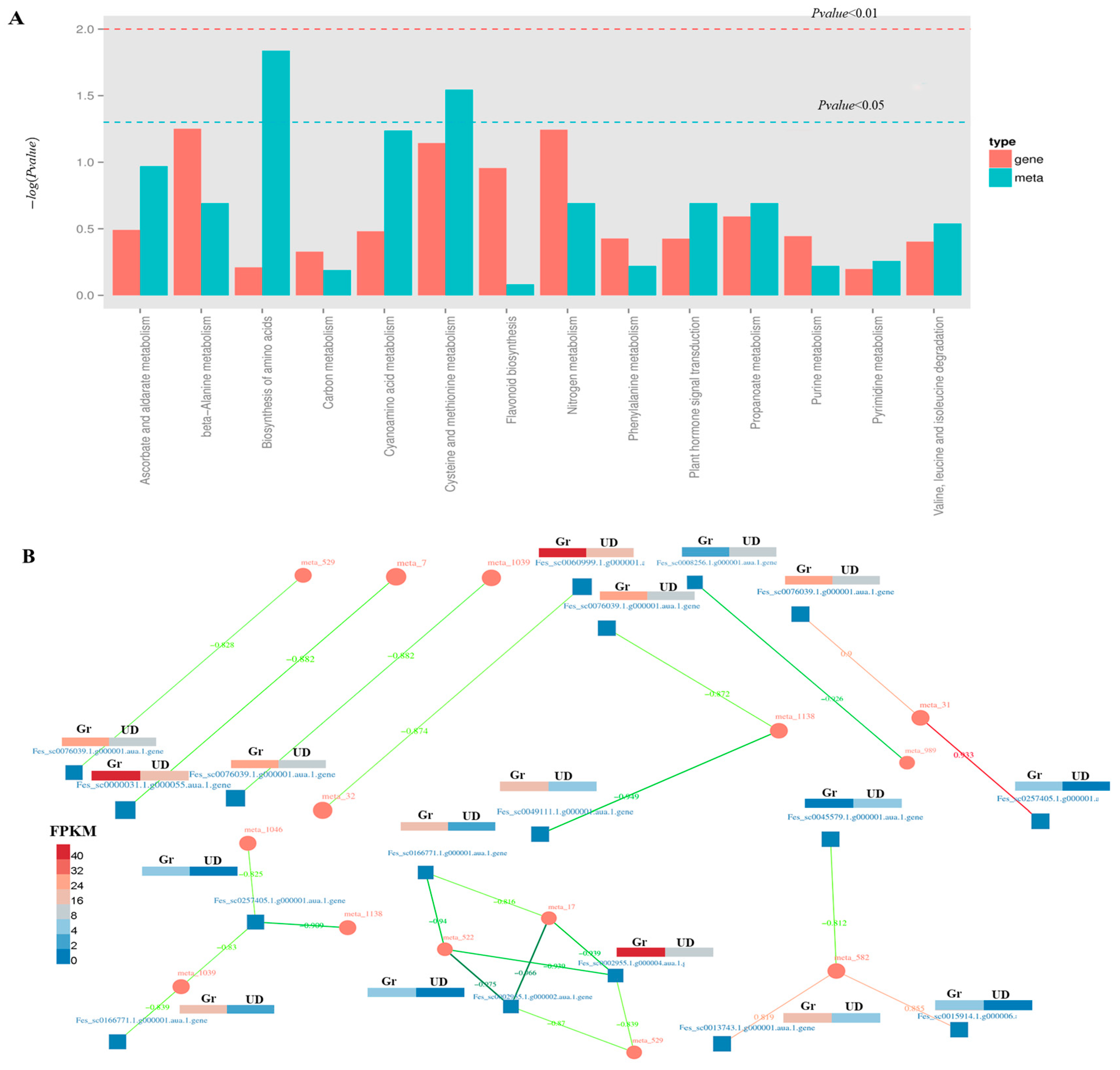
2.5. Integrating QTL Mapping and RNA Sequencing Data
2.6. Screening for Candidate-Shattering Genes in Common Buckwheat
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Shattering-Related Traits Evaluation
4.2. Genetic Map Construction and QTL Mapping
4.3. RNA Sequencing and Differential Expression Analysis
4.4. Metabolome Sequencing Analysis
4.5. Integrating QTL Mapping and RNA Sequencing Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohnishi, O. Search for the wild ancestor of buckwheat III. The wild ancestor of cultivated common buckwheat, and of tatary buckwheat. Econ. Bot. 1998, 52, 123–133. [Google Scholar] [CrossRef]
- Ohnishi, O. Search for the wild ancestor of buckwheat. I. Description of new Fagopyrum (Polygonaceae) species and their distribution in China and Himalayan hills. Fagopyrum 1998, 15, 18–28. [Google Scholar]
- Yasui, Y.; Hirakawa, H.; Ueno, M.; Matsui, K.; Katsube-Tanaka, T.; Yang, S.J.; Aii, J.; Sato, S.; Mori, M. Assembly of the draft genome of buckwheat and its applications in identifying agronomically useful genes. DNA Res. 2016, 23, 215–224. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Zielinski, H. Buckwheat as a functional food and its effects on health. J. Agric. Food Chem. 2015, 63, 7896–7913. [Google Scholar] [CrossRef]
- Lee, J.S.; Bok, S.H.; Jeon, S.M.; Kim, H.J.; Do, K.M.; Park, Y.B.; Choi, M.S. Antihyperlipidemic effects of buckwheat leaf and flower in rats fed a high-fat diet. Food Chem. 2010, 119, 235–240. [Google Scholar] [CrossRef]
- Stringer, D.M.; Taylor, C.G.; Appah, P.; Blewett, H.; Zahradka, P. Consumption of buckwheat modulates the post-prandial response of selected gastrointestinal satiety hormones in individuals with type 2 diabetes mellitus. Metab. Clin. Exp. 2013, 62, 1021–1031. [Google Scholar] [CrossRef]
- Koyama, M.; Hattori, S.; Amano, Y.; Watanabe, M.; Nakamura, K. Blood pressure-lowering peptides from neo-fermented buckwheat sprouts: A new approach to estimating ACE-inhibitory activity. PLoS ONE 2014, 9, 105802. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, E.S.; Malikov, V.G.; Falendysh, L.G. Green-flower form of buckwheat. Fagopyrum 1988, 8, 79–82. [Google Scholar]
- Oba, S.; Suzuki, Y.; Fujimoto, F. Breaking strength of pedicel and grain shattering habit in two species of buckwheat (Fagopyrum spp.). Plant Prod. Sci. 1998, 1, 62–66. [Google Scholar] [CrossRef]
- Suzuki, T.; Mukasa, Y.; Morishita, T.; Takigawa, S.; Noda, T. Traits of shattering resistant buckwheat ‘W/SK86GF’. Breed. Sci. 2012, 62, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C. Inter-specific hybridization in the genus Fagopyrum. In Proceedings of the Sixth International Symposium on Buckwheat, Shinshu, Japan, 24–29 August 1995; pp. 255–264. [Google Scholar]
- Ohnishi, O.; Asano, N. Genetic diversity of Fagopyrum homotropicum, a wild species related to common buckwheat. Genet. Resour. Crop Evol. 1999, 46, 389–398. [Google Scholar] [CrossRef]
- Pan, S.J.; Chen, Q.F. Genetic mapping of common buckwheat using DNA, protein and morphological markers. Hereditas 2010, 147, 27–33. [Google Scholar] [CrossRef]
- Wang, Y.; Scarth, R.; Campbell, G.C. Inheritance of seed shattering in interspecific hybrids between Fagopyrum esculentum and F. homotropicum. Crop Sci. 2005, 45, 693–697. [Google Scholar] [CrossRef]
- Matsui, K.; Tetsuka, T.; Nishio, T.; Hara, T. Heteromorphic incompatibility retained in self-compatible plants produced by a cross between common and wild buckwheat. New Phytol. 2003, 159, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Huang, K.F.; Chen, Q.F. Inheritance of Shattering Habit, Acute Achene, and Red Stem of Common Buckwheat (Fagopyrum esculentum). J. Henan Agric. Sci. 2012, 41, 28–31. [Google Scholar]
- Li, X.; Huang, Z.Q.; Jian, J.; Chen, Q. Screening of Seed-holding in Different Buckwheat Varieities. Guizhou Agric. Sci. 2015, 43, 19–23. [Google Scholar]
- Zhang, L.; Wang, L.; Zeng, X.; Chen, R.; Yang, S.; Pan, S. Comparative transcriptome analysis reveals fruit discoloration mechanisms in postharvest strawberries in response to high ambient temperature. Food Chem. X 2019, 2, 100025. [Google Scholar] [CrossRef]
- Krysan, P.J. An Arabidopsis Mitogen-Activated Protein Kinase Kinase Kinase Gene Family Encodes Essential Positive Regulators of Cytokinesis. Plant Cell Online 2002, 14, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Guo, L.H.; Zhao, Y.J.; Zhao, X.Q.; Yuan, Z.H. Systematic Analysis and Expression Profiles of the 4-Coumarate: CoA Ligase (4CL) Gene Family in Pomegranate (Punica granatum L.). Int. J. Mol. Sci. 2022, 23, 3509. [Google Scholar] [CrossRef] [PubMed]
- Basnet, R.; Zhang, J.; Hussain, N.; Shu, Q. Characterization and Mutational Analysis of a Monogalactosyldiacylglycerol Synthase Gene OsMGD2 in Rice. Front. Plant Sci. 2019, 10, 992. [Google Scholar] [CrossRef]
- Yang, L.; Hu, G.; Li, N.; Habib, S.; Huang, W.; Li, Z. Functional Characterization of SlSAHH2 in Tomato Fruit Ripening. Front. Plant Sci. 2017, 8, 1312. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Cao, G.; Adil, M.F.; Tu, Y.; Wang, W.; Cai, B.; Shamsi, I.H. Changes in water loss and cell wall metabolism during postharvest withering of tobacco (Nicotiana tabacum L.) leaves using tandem mass tag-based quantitative proteomics approach. Plant Physiol. Biochem. 2020, 150, 121–132. [Google Scholar] [CrossRef]
- Tayade, R.; Rana, V.; Shafiqul, M.; Nabi, R.B.S.; Raturi, G.; Dhar, H.; Thakral, V.; Kim, Y. Genome-Wide Identification of Aquaporin Genes in Adzuki Bean (Vigna angularis) and Expression Analysis under Drought Stress. Int. J. Mol. Sci. 2022, 23, 16189. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zeng, L.; Zhu, S.; Wu, L.; Wang, Y.; Tang, S.; Liu, T. Draft genome analysis provides insights into the fiber yield, crude protein biosynthesis, and vegetative growth of domesticated ramie (Boehmeria nivea L. Gaud). DNA Res. 2017, 25, 173–181. [Google Scholar] [CrossRef]
- Takahashi, Y.; Teshima, K.M.; Yokoi, S.; Innan, H.; Shimamoto, K. Variations in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels contribute to diversity of flowering time in cultivated rice. Proc. Natl. Acad. Sci. USA 2009, 106, 4555–4560. [Google Scholar] [CrossRef]
- Liu, H.M.; Xiao, S.Q.; Sui, S.Z.; Huang, R.W.; Wang, X.; Wu, H.F.; Liu, X. A tandem CCCH type zinc finger protein gene CpC3H3 from Chimonanthus praecox promotes flowering and enhances drought tolerance in Arabidopsis. BMC Plant Biol. 2022, 22, 506. [Google Scholar] [CrossRef]
- Liu, Y.B.; Lu, S.M.; Zhang, J.F.; Liu, S.; Lu, Y.T. A xyloglucan endotransglucosylase/hydrolase involves in growth of primary root and alters the deposition of cellulose in Arabidopsis. Planta 2007, 226, 1547–1560. [Google Scholar] [CrossRef]
- Wang, J.H.; Gu, K.D.; Zhang, Q.Y.; Yu, J.Q.; Wang, C.K.; You, C.X.; Cheng, L.; Hu, D.G. Ethylene inhibits malate accumulation in apple by transcriptional repression of aluminum-activated malate transporter 9 via the WRKY31-ERF72 network. New Phytol. 2023. [CrossRef]
- Protsenko, M.A.; Buza, N.L.; Krinitsyna, A.A.; Bulantseva, E.A.; Korableva, N.P. Polygalacturonase-inhibiting protein is a structural component of plant cell wall. Biochemistry 2008, 73, 1053–1062. [Google Scholar] [CrossRef]
- Ding, L.N.; Li, M.; Wang, W.J.; Cao, J.; Wang, Z.; Zhu, K.M.; Tan, X.L. Advances in plant GDSL lipases: From sequences to functional mechanisms. Acta Physiol. Plant. 2019, 41, 151. [Google Scholar] [CrossRef]
- Tan, W.J.; Xiao, S.; Chen, Q.F. Potential role of salicylic acid in modulating diacylglycerol homeostasis in response to freezing temperatures in Arabidopsis. Plant Signal. Behav. 2015, 10, e1082698. [Google Scholar] [CrossRef]
- Serrano, M. Isolation and Gene Expression Analysis of Arabidopsis thaliana Mutants With Constitutive Expression of ATL2, an Early Elicitor-Response RING-H2 Zinc-Finger Gene. Genetics 2004, 167, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Song, K.; Reichardt, I.; Kim, H.; Mayer, U.; Stierhof, Y.-D.; Jurgens, G. Arabidopsis-adaptin subunit AP1M of adaptor protein complex 1 mediates late secretory and vacuolar traffic and is required for growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10318–10323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Xie, J.Q.; Wang, L.T.; Si, L.B.; Zheng, S.; Yang, Y.L.; Yang, H.; Tian, S.G. Wheat TabZIP8, 9, 13 participate in ABA biosynthesis in NaCl-stressed roots regulated by TaCDPK9-1. Plant Physiol. Biochem. 2020, 151, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Aufhammer, W. Development and utilization of the seed yield potential of buckwheat (F. esculentum). Bodenkultur 1994, 45, 37–47. [Google Scholar]
- Inoue, N.; Hagiwara, M. Analysis of the yielding process based on the module concept in common buckwheat. Fagopyrum 1999, 16, 73–77. [Google Scholar]
- Lee, J. Produced. Harvested and utilizable grain yields of the pseudo cereals buckwheat (Fagopyrum esculentum Moench), quinoa (Chenopodium quinoa Willd.) and amaranth (Amaranthus hypochondriacus L. × A. hybridus L.) as affected by production techniques. Bodenkultur 1996, 47, 5–14. [Google Scholar]
- Yasui, Y.; Wang, Y.; Ohnishi, O.; Campbell, C. Amplified fragment length polymorphism linkage analysis of common buckwheat (Fagopyrum esculentum) and its wild self-pollinated relative Fagopyrum homotropicum. Genome 2004, 47, 345–351. [Google Scholar] [CrossRef]
- Konishi, T.; Ohnishi, O. A linkage map for common buckwheat based on microsatellite and AFLP markers. Fagopyrum 2006, 23, 1–6. [Google Scholar]
- Hara, T.; Iwata, H.; Okuno, K.; Matsui, K.; Ohsawa, R. QTL analysis of photoperiod sensitivity in common buckwheat by using markers for expressed sequence tags and photoperiod-sensitivity candidate genes. Breed Sci. 2011, 61, 394–404. [Google Scholar] [CrossRef]
- Yabe, S.; Hara, T.; Ueno, M.; Enoki, H.; Kimura, T.; Nishimura, S.; Yasui, Y.; Ohsawa, R.; Iwata, H. Rapid genotyping with DNA micro-arrays for high-density linkage mapping and QTL mapping in common buckwheat (Fagopyrum esculentum Moench). Breed. Sci. 2014, 64, 291–299. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, Y.; Cui, J.; Yue, L.; Tao, J.; Wang, Y.; Zhang, R.; Liu, J.; Jiang, A.; Zhang, J.; et al. Genetic Map Construction, QTL Mapping, and Candidate Genes Screening of Grain Size Traits in Common Buckwheat (Fagopyrum esculentum M.). Agronomy 2022, 12, 2062. [Google Scholar] [CrossRef]
- He, Q.; Ma, D.; Li, W.; Xing, L.S.; Zhang, H.Y.; Wang, Y.; Du, C.L.; Li, X.Z.; Jia, Z.; Li, X.X.; et al. High-quality Fagopyrum esculentum genome provides insights into the flavonoid accumulation among different tissues and self-incompatibility. J. Integr. Plant Biol. 2023, 1672–9072. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Hou, Z.; Wang, S.; Liu, Z.; Wei, S.; Zhang, Y.; Yin, J. Transcriptome Analysis Reveals the Accumulation Mechanism of Anthocyanins in Buckwheat (Fagopyrum esculentum Moench) Cotyledons and Flowers. Int. J. Mol. Sci. 2019, 20, 1493. [Google Scholar] [CrossRef]
- Lu, Q.H.; Wang, Y.Q.; Song, J.N.; Yang, H.B. Transcriptomic identification of salt-related genes and de novo assembly in common buckwheat (F. esculentum). Plant Physiol. Biochem. 2018, 127, 299–309. [Google Scholar] [CrossRef]
- Xu, J.M.; Fan, W.; Jin, J.F.; Lou, H.Q.; Chen, W.W.; Yang, J.L.; Zheng, S.J. Transcriptome Analysis of Al-Induced Genes in Buckwheat (Fagopyrum esculentum Moench) Root Apex: New Insight into Al Toxicity and Resistance Mechanisms in an Al Accumulating Species. Front. Plant Sci. 2017, 8, 1141. [Google Scholar] [CrossRef]
- Chen, W.; Xu, J.; Jin, J.; Lou, H.; Fan, W.; Yang, J. Genome-Wide Transcriptome Analysis Reveals Conserved and Distinct Molecular Mechanisms of Al Resistance in Buckwheat (Fagopyrum esculentum Moench) Leaves. Int. J. Mol. Sci. 2017, 18, 1859. [Google Scholar] [CrossRef]
- Campbell, R.; Pont SD, A.; Morris, J.A.; McKenzie, G.; Sharma, S.K.; Hedley, P.E.; Taylor, M.A. Genome-wide QTL and bulked transcriptomic analysis reveals new candidate genes for the control of tuber carotenoid content in potato (Solanum tuberosum L.). Theor. Appl. Genet. 2014, 127, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Huang, Z.; Song, S.; Xin, Y.; Mao, D.; Lv, Q.; Zhu, L. Integrated analysis of phenome, genome, and transcriptome of hybrid rice uncovered multiple heterosis-related loci for yield increase. Proc. Natl. Acad. Sci. USA 2016, 113, 6026–6035. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, H.; Chu, S.; Li, H.; Chi, Y.; Triebwasser-Freese, D.; Yu, D. Integrating QTL mapping and transcriptomics identifies candidate genes underlying QTLs associated with soybean tolerance to low-phosphorus stress. Plant Mol. Biol. 2016, 93, 137–150. [Google Scholar] [CrossRef]
- Odilbekov, F.; He, X.; Armoniené, R.; Saripella, G.V.; Henriksson, T.; Singh, P.K.; Chawade, A. QTL Mapping and Transcriptome Analysis to Identify Differentially Expressed Genes Induced by Septoria Tritici Blotch Disease of Wheat. Agronomy 2019, 9, 510. [Google Scholar] [CrossRef]
- Yang, P.; Sun, X.T.; Liu, X.Y.; Wang, W.W.; Hao, Y.S.; Chen, L.; Liu, J.; He, H.L.; Zhang, T.R.; Bao, W.Y.; et al. Identification of Candidate Genes for Lint Percentage and Fiber Quality Through QTL Mapping and Transcriptome Analysis in an Allotetraploid Interspecific Cotton CSSLs Population. Front. Plant Sci. 2022, 13, 882051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q.; Wang, H.; Tao, S.; Cao, H.; Shi, Y.; Bakirov, A.; Xu, A.; Huang, Z. Discovery of common loci and candidate genes for controlling salt-alkali tolerance and yield-related traits in Brassica napus L. Plant Cell Rep. 2023, 42, 1039–1057. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Wang, K.J.; Wang, C.H.; Zhao, Y.B.; Tao, Z.; Li, J.Y.; Wang, L.; Shi, J.; Huang, S.J.; Xie, C.X.; et al. Combining quantitative trait locus mapping with multiomics profiling reveals genetic control of corn leaf aphid (Rhopalosiphum maidis) resistance in maize. J. Exp. Bot. 2023, erad113. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Xiao, Y.H.; Luo, M.; Li, X.B.; Luo, X.Y.; Hou, L.; Li, D.M.; Pei, Y. Construction of a genetic linkage map and QTL analysis of fiber-related traits in upland cotton (Gossypium hirsutum L.). Euphytica 2005, 144, 91–99. [Google Scholar] [CrossRef]
- Van Ooijen, J.W.; Voorrips, R.E. JoinMap 4.0., Software for the Calculation of Genetic Linkage Maps; Plant Research International: Wageningen, The Netherlands, 2006. [Google Scholar]
- Van Ooijen, J.W. MapQTL 6.0., Software for the Mapping of Quantitative Trait Loci in Experimental Populations; Plant Research International: Wageningen, The Netherlands, 2009. [Google Scholar]
- Voorrips, R.E. MapChart 2.2: Software for the Graphical Presentation of Linkage Maps and QTLs; Plant Research International: Wageningen, The Netherlands, 2006. [Google Scholar]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkage Group | Length | Number of Markers | Average Distance (cm) | SD Loci | SD Ratio (%) |
|---|---|---|---|---|---|
| LG 01 | 248.86 | 60 | 4.08 | 48 | 78.68 |
| LG 02 | 161.10 | 13 | 12.39 | 11 | 84.61 |
| LG 03 | 155.79 | 16 | 9.74 | 11 | 68.75 |
| LG 04 | 173.79 | 24 | 7.24 | 16 | 66.67 |
| LG 05 | 159.21 | 13 | 12.25 | 10 | 76.92 |
| LG 06 | 144.20 | 21 | 6.87 | 12 | 57.14 |
| LG 07 | 176.97 | 19 | 9.31 | 17 | 89.47 |
| LG 08 | 139.31 | 8 | 17.41 | 7 | 87.50 |
| Total | 1359.23 | 174 | 7.77 | 132 | 75.42 |
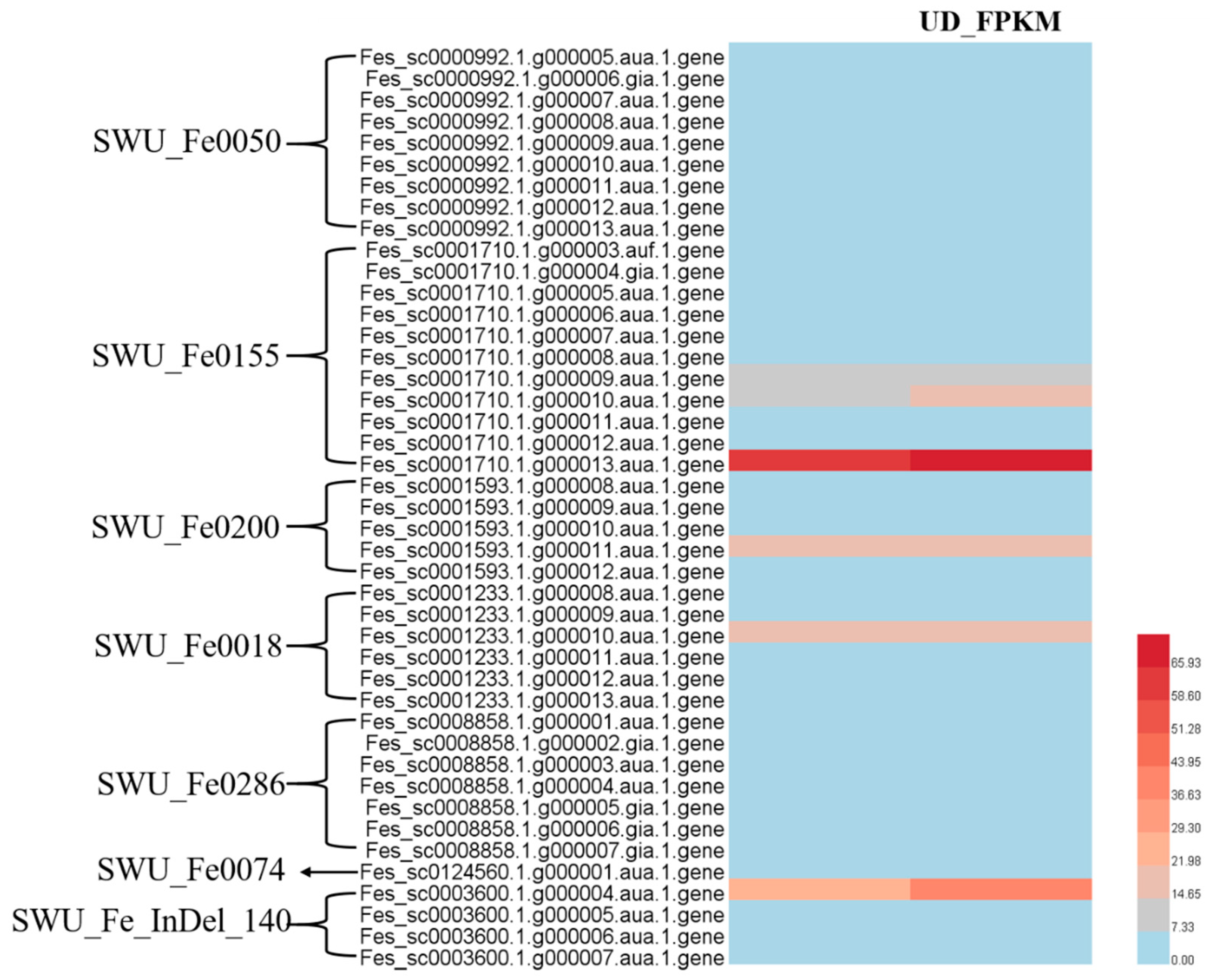
| QTL | Linkage Group | Nearest Marker | LOD | A | Var% |
|---|---|---|---|---|---|
| qPP01.1 | LG 01 | SWU_Fe0050 | 2.01 | −0.01 | 9.9 |
| qPP04.1 | LG 04 | SWU_Fe0155 | 2.06 | 0.58 | 11.8 |
| qPP04.2 | LG 04 | SWU_Fe0200 | 2.19 | 0.14 | 10.4 |
| qPP05.1 | LG 05 | SWU_Fe018 | 3.63 | 1.68 | 16.6 |
| qPP06.1 | LG 06 | SWU_Fe0286 | 2.63 | −0.14 | 7.9 |
| qPP06.2 | LG 06 | SWU_Fe0047 | 2.04 | −0.19 | 9.7 |
| qPP07.1 | LG 07 | SWU_Fe_InDel140 | 2.3 | −0.01 | 10.9 |
| Gene ID | UD_FPKM | Gr_FPKM | log2FC | Regulated | NR_Annotation | Reference |
|---|---|---|---|---|---|---|
| Fes_sc0013567.1.g000001 | 2.05 | 21.99 | 1.97 | Up | Laccase-14 | Zhang et al., [18] |
| Fes_sc0000452.1.g000003 | 1.72 | 6.36 | 1.57 | Up | Mitogen-activated protein kinase kinase kinase ANP1 | Krysan, [19] |
| Fes_sc0060999.1.g000001 | 50.4 | 18.75 | −1.41 | Down | 4-coumarate: CoA ligase | Wang et al., [20] |
| Fes_sc0000049.1.g000027 | 7.21 | 1.08 | −2.18 | Down | Monogalactosyldiacylglycerol synthase 2 | Basnet et al., [21] |
| Fes_sc0002955.1.g000004 | 50.8 | 10.15 | −1.94 | Down | S-adenosylhomocysteine hydrolase | Yang et al., [22] |
| Fes_sc0002955.1.g000002 | 6.51 | 0.9 | −2.61 | Down | S-adenosylhomocysteine hydrolase | Yang et al., [22] |
| Fes_sc0281858.1.g000001 | 34.97 | 8.99 | −1.68 | Down | Xyloglucan endotransglucosylase/hydrolase 27 | Wu et al., [23] |
| Fes_sc0003149.1.g000006 | 22.59 | 61.38 | 1.24 | Up | Aquaporin | Tayade et al., [24] |
| Fes_sc0000947.1.g000002 | 321.51 | 95.22 | −1.57 | Down | WAT1-related protein | Liu et al., [25] |
| Fes_sc0098541.1.g000001 | 0.41 | 9.18 | 2.29 | Up | Laccase-14 | Zhang et al., [18] |
| Fagopyrum_esculentum_newGene_12570 | 11.12 | 1.79 | −2.04 | Down | Protein heading date 3a | Takahashi et al., [26] |
| Fes_sc0010036.1.g000002 | 5.4 | 15.15 | 1.3 | Up | Zinc finger, CCCH-type | Liu et al., [27] |
| Fes_sc0005201.1.g000005 | 1.28 | 6.8 | 1.82 | Up | Xyloglucan endotransglucosylase/hydrolase 8 | Liu et al., [28] |
| Fes_sc0009433.1.g000003 | 40.15 | 14.2 | −1.39 | Down | Aluminum-activated malate transporter 9-like | Wang et al., [29] |
| Fes_sc0005671.1.g000006 | 9.48 | 26.83 | 1.28 | Up | Polygalacturonase-inhibiting protein | Protsenko et al., [30] |
| Fes_sc0003889.1.g000014 | 16.05 | 92.65 | 1.84 | Up | GDSL lipase | Ding et al., [31] |
| Fes_sc0008820.1.g000003 | 6.26 | 1.85 | −1.53 | Down | Monoglyceride lipase-like | Tan et al., [32] |
| Fes_sc0126564.1.g000001 | 4.21 | 12.55 | 1.35 | Up | Polygalacturonase-inhibiting protein | Protsenko et al., [30] |
| Fes_sc0001710.1.g000010 | 21.11 | 12.74 | -- | -- | RING-H2 finger protein ATL2 | Serrano, [33] |
| Fes_sc0001593.1.g000011 | 15.87 | 15.62 | -- | -- | AP-1 complex subunit mu-2 | Park et al., [34] |
| Fes_sc0003600.1.g000004 | 36.83 | 29.21 | -- | -- | bZIP9 | Zhang et al., [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Zhang, Y.; Liu, Y.; Cui, J.; He, X.; Wu, Y.; Yue, L.; Zhang, J.; Ding, M.; Yi, Z.; et al. Joint QTL Mapping and Transcriptome Sequencing Analysis Reveal Candidate Seed-Shattering-Related Genes in Common Buckwheat. Int. J. Mol. Sci. 2023, 24, 10013. https://doi.org/10.3390/ijms241210013
Chen C, Zhang Y, Liu Y, Cui J, He X, Wu Y, Yue L, Zhang J, Ding M, Yi Z, et al. Joint QTL Mapping and Transcriptome Sequencing Analysis Reveal Candidate Seed-Shattering-Related Genes in Common Buckwheat. International Journal of Molecular Sciences. 2023; 24(12):10013. https://doi.org/10.3390/ijms241210013
Chicago/Turabian StyleChen, Chuyi, Yuke Zhang, Yang Liu, Jingbin Cui, Xingxing He, Yichao Wu, Linqing Yue, Jian Zhang, Mengqi Ding, Zelin Yi, and et al. 2023. "Joint QTL Mapping and Transcriptome Sequencing Analysis Reveal Candidate Seed-Shattering-Related Genes in Common Buckwheat" International Journal of Molecular Sciences 24, no. 12: 10013. https://doi.org/10.3390/ijms241210013
APA StyleChen, C., Zhang, Y., Liu, Y., Cui, J., He, X., Wu, Y., Yue, L., Zhang, J., Ding, M., Yi, Z., & Fang, X. (2023). Joint QTL Mapping and Transcriptome Sequencing Analysis Reveal Candidate Seed-Shattering-Related Genes in Common Buckwheat. International Journal of Molecular Sciences, 24(12), 10013. https://doi.org/10.3390/ijms241210013