
Integrated IBD Analysis, GWAS Analysis and Transcriptome Analysis to Identify the Candidate Genes for White Spot Disease in Maize
 ,
,
Abstract
1. Introduction
2. Results
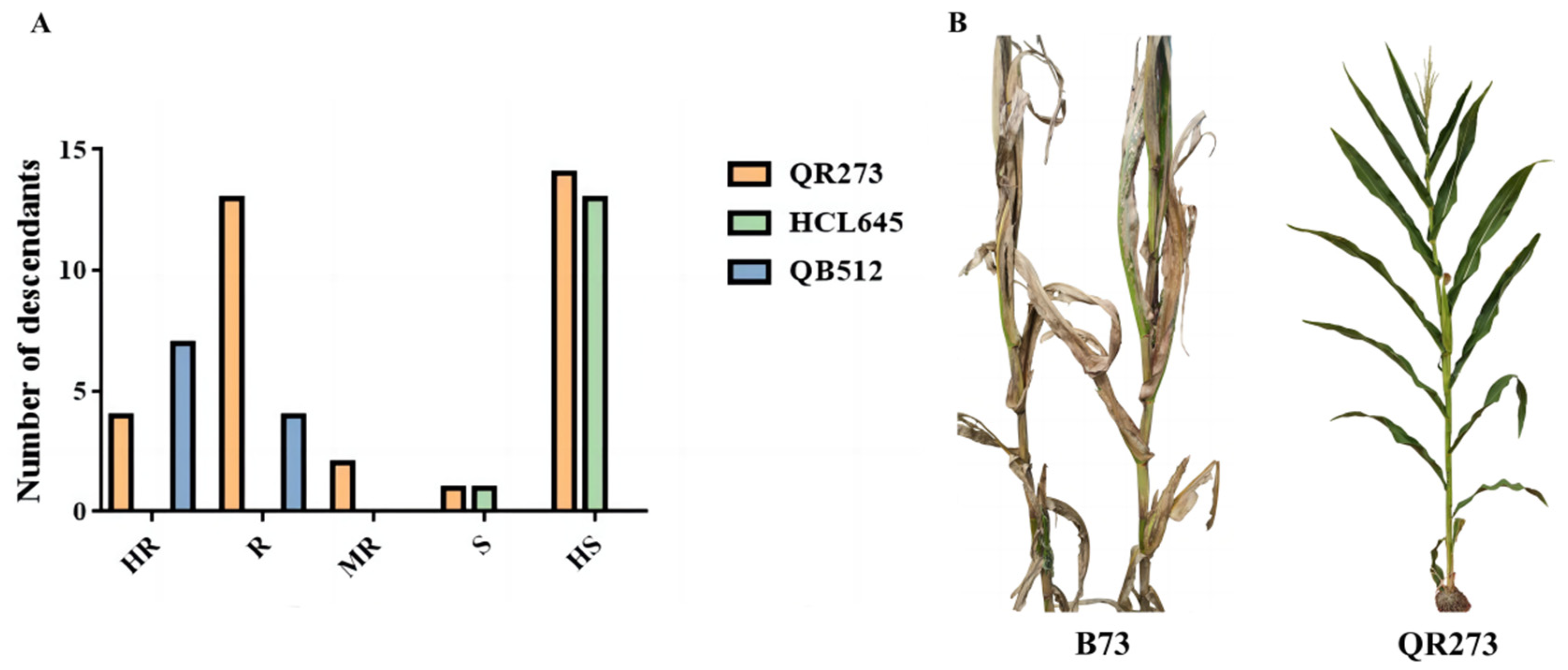
2.1. Analysis of Resistance to MWS
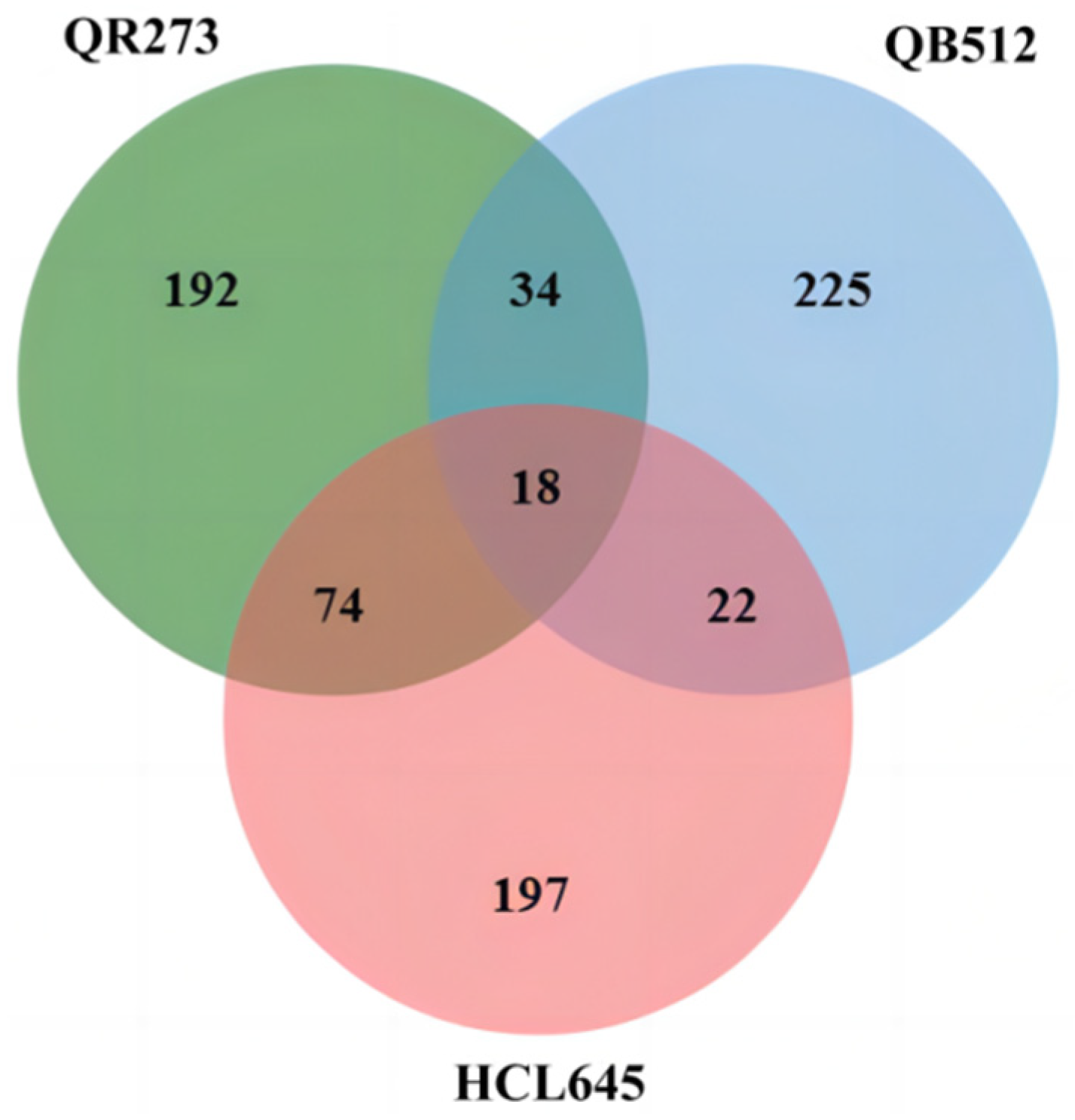
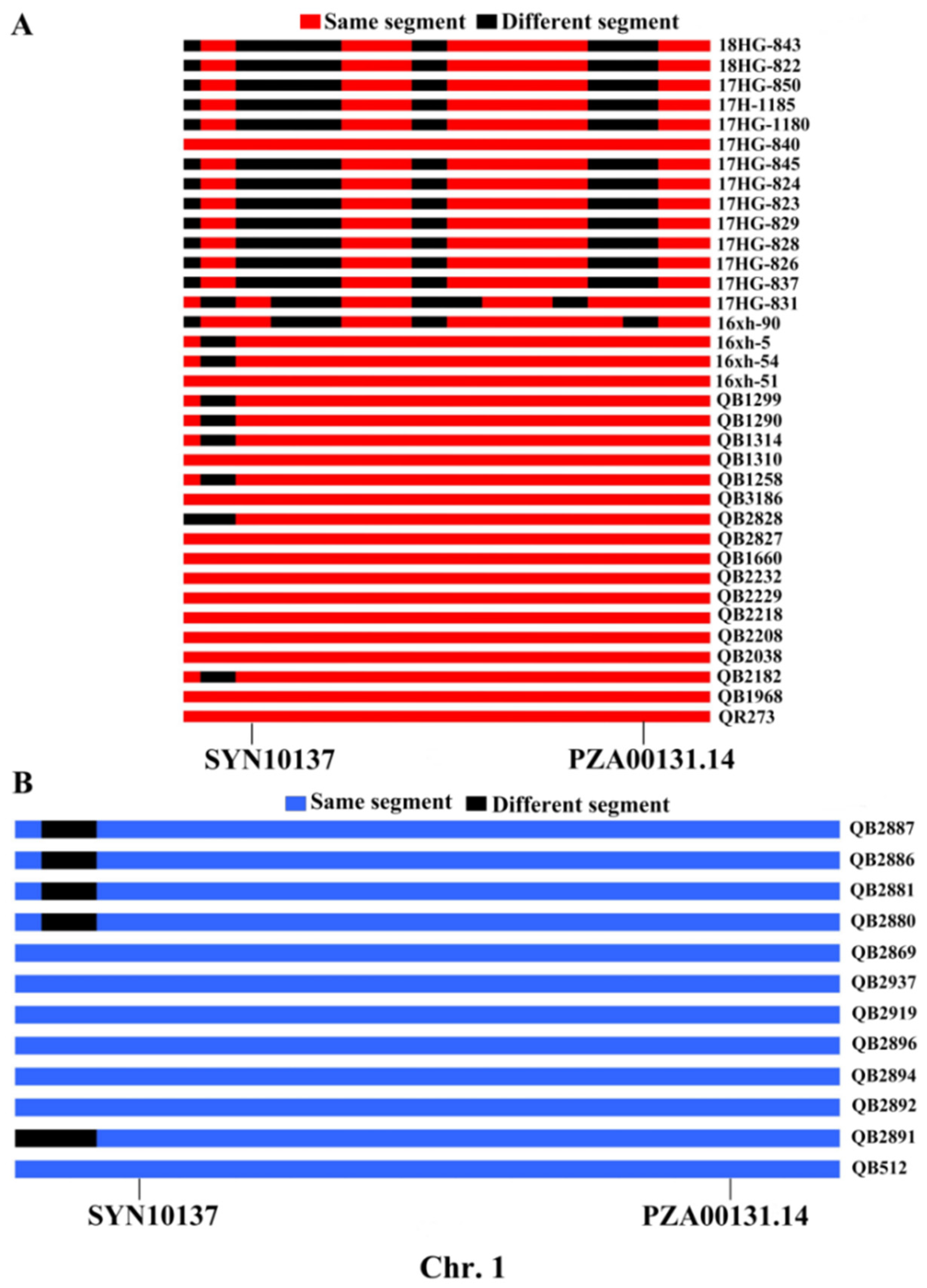
2.2. Excavation of the IBD Segments
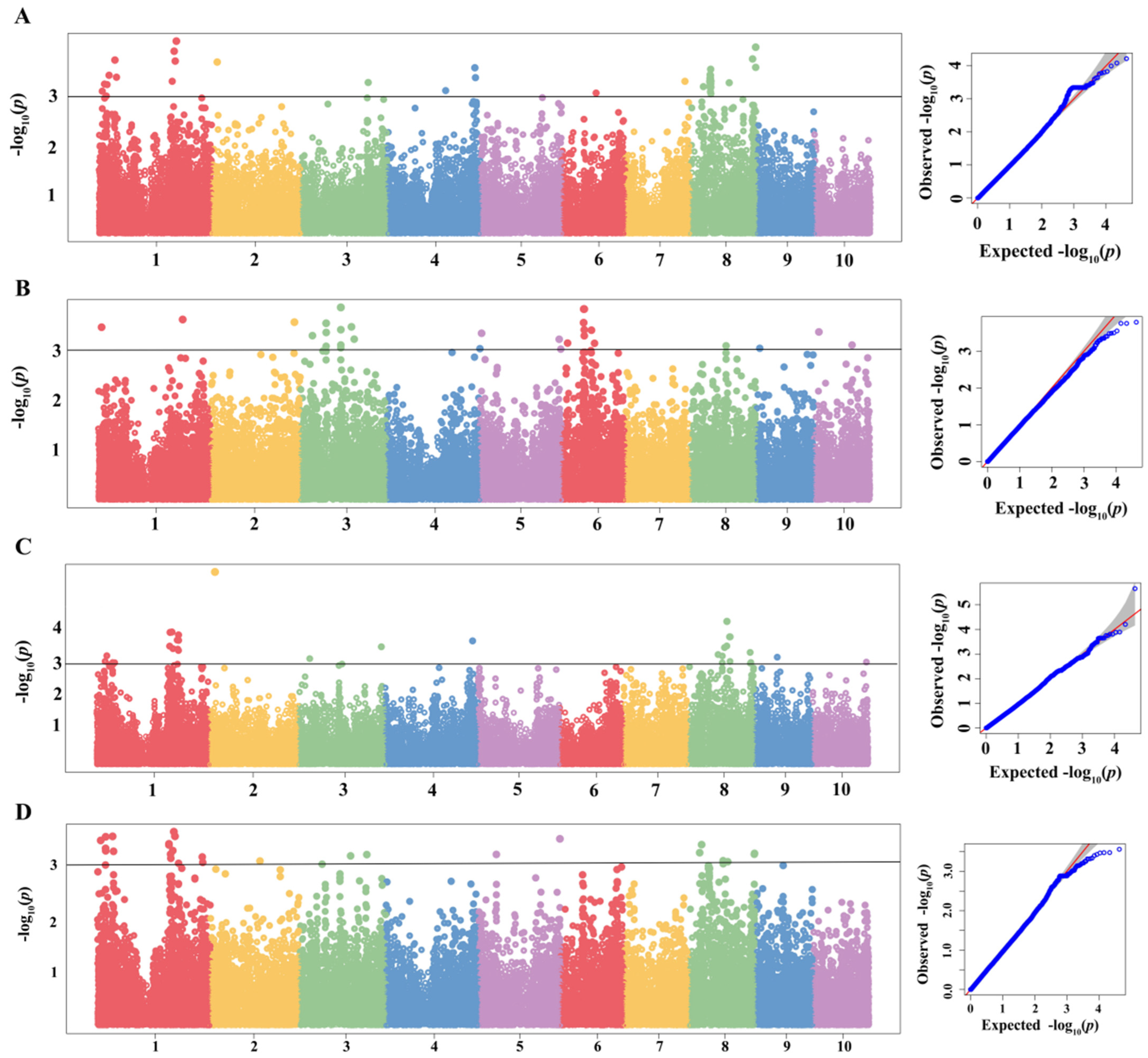
2.3. GWAS of MWS
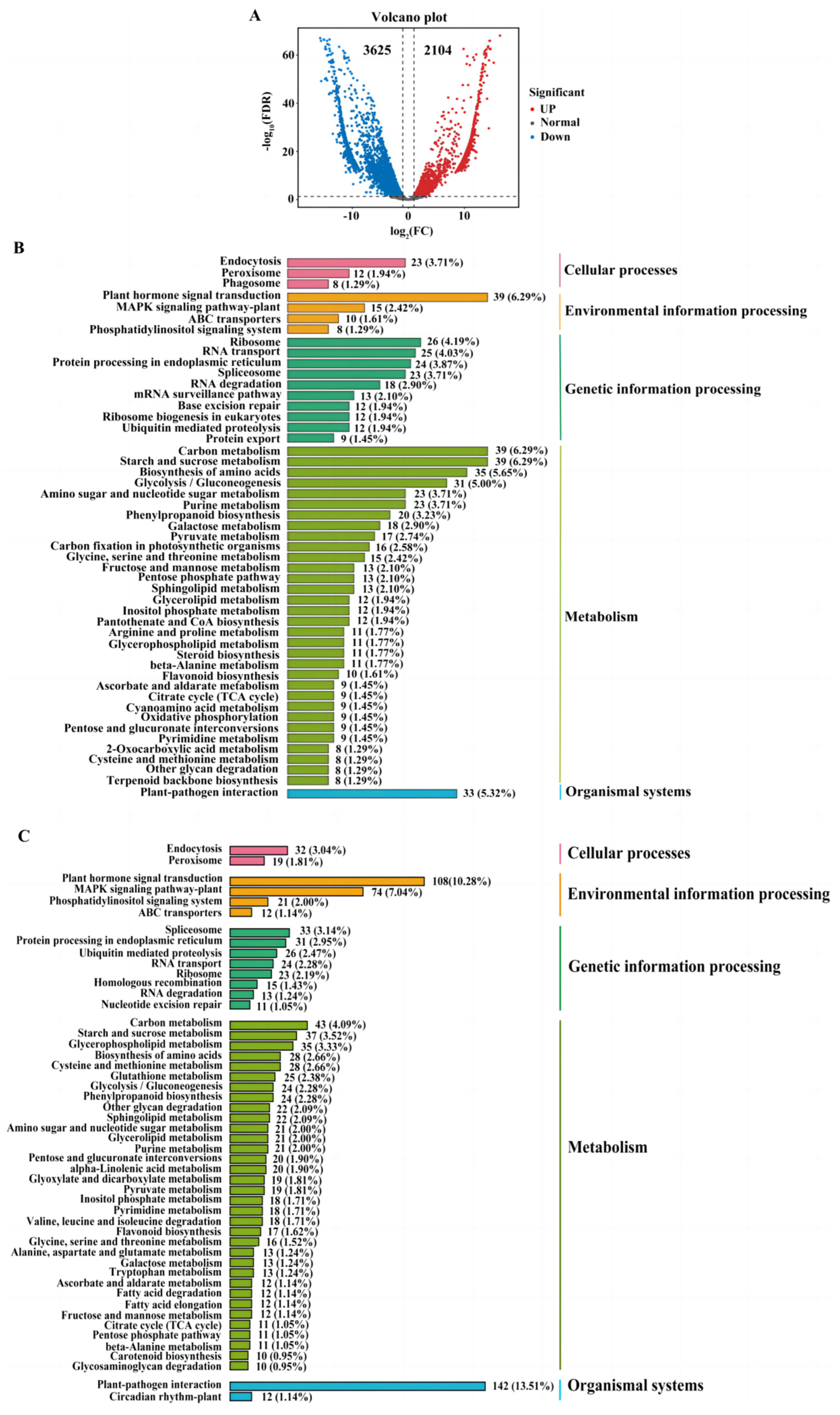
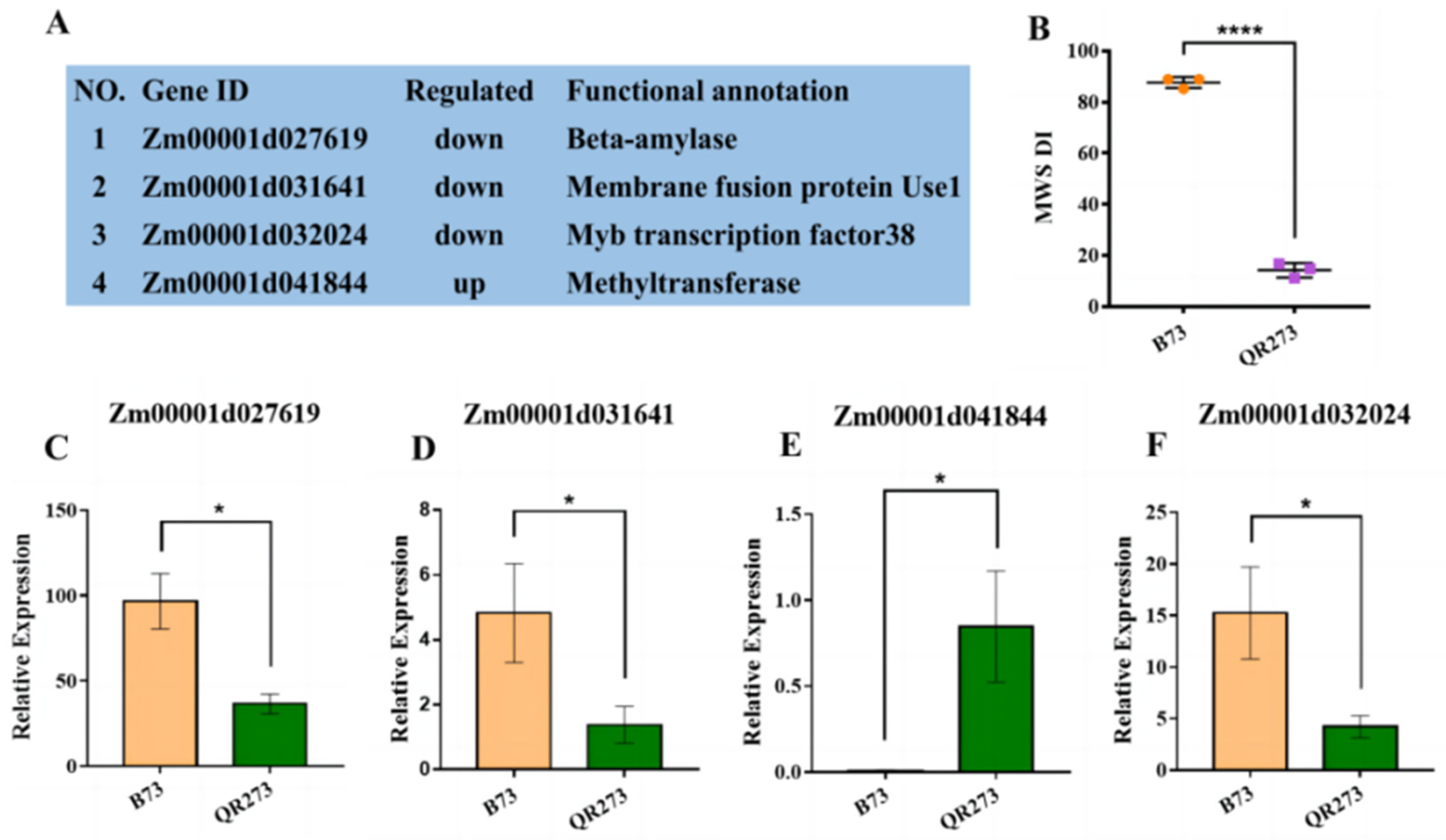
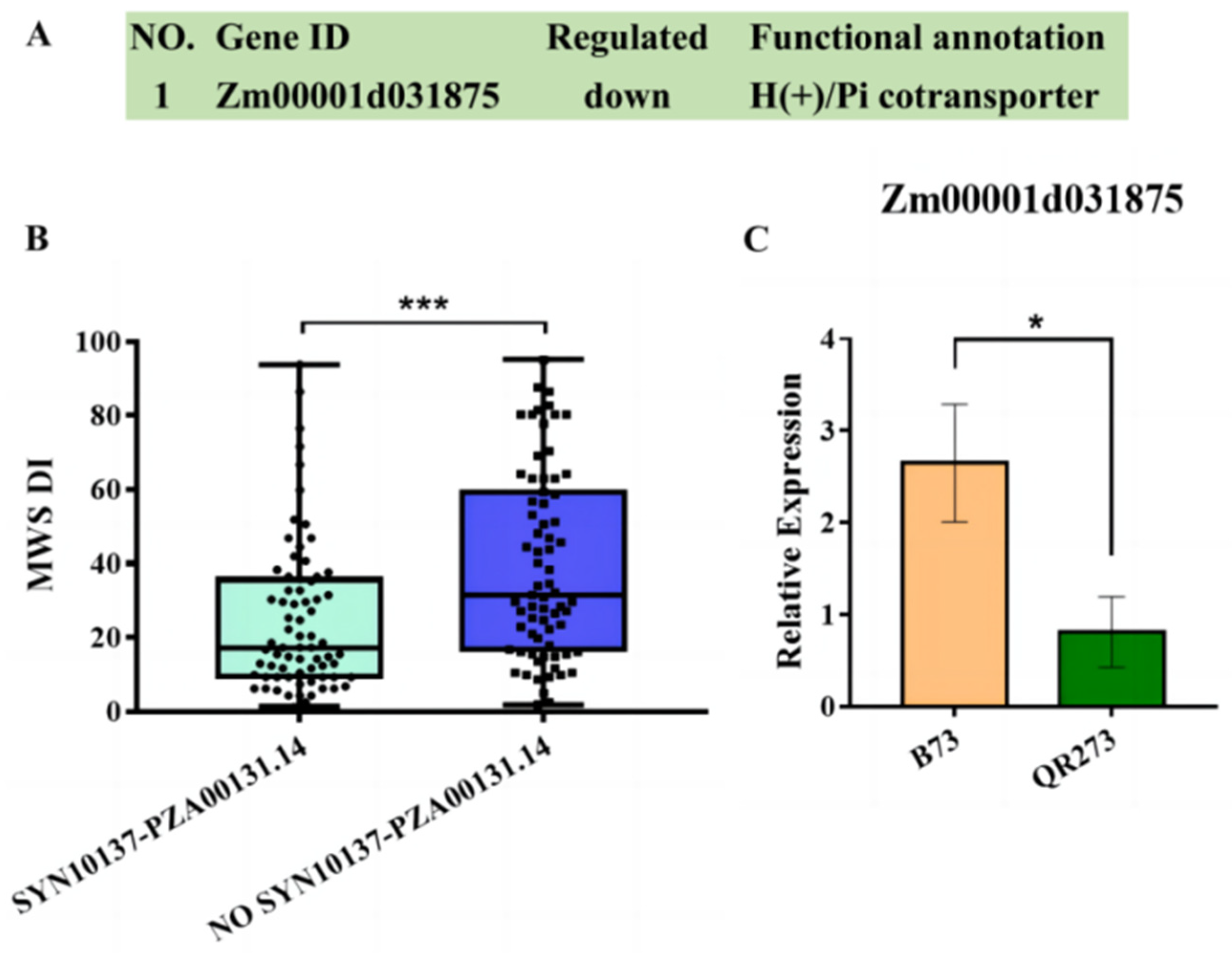
2.4. The Comparision of Quantitative Real-Time PCR (qRT-PCR) and Transcriptome Analysis
2.5. Combining the IBD Analysis, GWAS and Transcriptome Analysis to Identify Candidate Genes
3. Discussion
3.1. Inheritance of MWS Resistance
3.2. Transmission of the IBD Segments
3.3. Specific Pathways Response to Diseases in Maize
3.4. Integrating GWAS and Transcriptome Analysis to Reveal the Function of IBD Segments in MWS Resistance
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Evaluation and Analysis
4.3. Genotyping
4.4. Identification of the IBD Segments
4.5. Analysis of the Functions of the IBD Segments in MWS
4.6. Candidate Gene Prediction and Transcriptome Data Analysis
4.7. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiao, Y.; Jiang, S.; Cheng, Q.; Wang, X.; Yan, J.; Zhang, R.; Qiao, F.; Ma, C.; Luo, J.; Li, W.; et al. The genetic mechanism of heterosis utilization in maize improvement. Genome Biol. 2021, 22, 148. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hou, X.; Xiao, Q.; Yi, Q.; Bian, S.; Hu, Y.; Liu, H.; Zhang, J.; Hao, X.; Cheng, W.; et al. Genetic Analysis in Maize Foundation Parents with Mapping Population and Testcross Population: Ye478 Carried More Favorable Alleles and Using QTL Information Could Improve Foundation Parents. Front. Plant Sci. 2016, 7, 1417. [Google Scholar] [CrossRef] [PubMed]
- Smith, S. Pedigree background changes in US hybrid maize between 1980 and 2004. Crop Sci. 2007, 47, 1914–1926. [Google Scholar] [CrossRef]
- Troyer, A.F.; Mikel, M.A. Minnesota Corn Breeding History: Department of Agronomy Plant Genetics Centennial. Crop Sci. 2010, 50, 1141–1150. [Google Scholar] [CrossRef]
- Hirsch, C.N.; Hirsch, C.D.; Brohammer, A.B.; Bowman, M.J.; Soifer, I.; Barad, O.; Shem-Tov, D.; Baruch, K.; Lu, F.; Hernandez, A.G.; et al. Draft Assembly of Elite Inbred Line PH207 Provides Insights into Genomic and Transcriptome Diversity in Maize. Plant Cell 2016, 28, 2700–2714. [Google Scholar] [CrossRef]
- Mikel, M.A. Genetic Composition of Contemporary U.S. Commercial Dent Corn Germplasm. Crop Sci. 2011, 51, 592–599. [Google Scholar] [CrossRef]
- Smith, J.S.C.; Chin, E.C.L.; Shu, H.; Smith, O.S.; Wall, S.J.; Senior, M.L.; Mitchell, S.E.; Kresovich, S.; Ziegle, J. An evaluation of the utility of SSR loci as molecular markers in maize (Zea mays L.): Comparisons with data from RFLPS and pedigree. Theor. Appl. Genet. 1997, 95, 163–173. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Fu, J.; Li, X.; Li, C.; Zhang, D.; Shi, Y.; Song, Y.; Li, Y.; Wang, T. Exploring Identity-By-Descent Segments and Putative Functions Using Different Foundation Parents in Maize. PLoS ONE 2016, 11, e0168374. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. High-resolution detection of identity by descent in unrelated individuals. Am. J. Hum. Genet. 2010, 86, 526–539. [Google Scholar] [CrossRef]
- Thompson, E.A. Identity by Descent: Variation in Meiosis, Across Genomes, and in Populations. Genetics 2013, 194, 301–326. [Google Scholar] [CrossRef]
- Ge, J.; Xu, Y.; Zhao, M.; Zhan, M.; Cao, C.; Chen, C.; Zhou, B. Effect of Climatic Conditions Caused by Seasons on Maize Yield, Kernel Filling and Weight in Central China. Agronomy 2022, 12, 1816. [Google Scholar] [CrossRef]
- Pechanova, O.; Pechan, T. Maize-Pathogen Interactions: An Ongoing Combat from a Proteomics Perspective. Int. J. Mol. Sci. 2015, 16, 28429–28448. [Google Scholar] [CrossRef] [PubMed]
- Rane, M.S.; Payak, M.M.; Renfro, B.L. A Phaeosphaeria leaf spot of maize. Indian Phytopathol. Soc. Bull. 1966, 3, 8–10. [Google Scholar]
- Carson, M.L.; Goodman, M.M.; Glawe, D.A. Phaeosphaeria leaf spot of maize in Florida. Plant Dis. 1991, 75, 968. [Google Scholar] [CrossRef]
- Paccola-Meirelles, L.D.; Ferreira, A.S.; Meirelles, W.F.; Marriel, I.E.; Casela, C.R. Detection of a bacterium associated with a leaf spot disease of maize in Brazil. J. Phytophathology 2001, 149, 275–279. [Google Scholar] [CrossRef]
- Miller, A.M.; Figueiredo, J.E.F.; Linde, G.A.; Colauto, N.B.; Paccola-Meirelles, L.D. Characterization of the inaA gene and expression of ice nucleation phenotype in Pantoea ananatis isolates from Maize White Spot disease. Genet. Mol. Res. 2016, 15, gmr15017863. [Google Scholar] [CrossRef]
- Adam, E.; Deng, H.; Odindi, J.; Abdel-Rahman, E.M.; Mutanga, O. Detecting the early stage of phaeosphaeria leaf spot infestations in maize crop using in situ hyperspectral data and guided regularized random forest algorithm. J. Spectrosc. 2017, 2017, 6961387. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, Z.P.; He, P.B.; Li, Y.M.; He, P.F.; Chen, Q.; Chang, W.; Wu, Y.X.; Liu, Y.L.; Tan, W.Z.; et al. Identification of the pathogen causing maize white spot and field evaluation of maize cultivars for resistance to the disease. J. Plant Prot. 2022, 49, 840–847. (In Chinese) [Google Scholar]
- Carson, M.L. Vulnerability of U.S. Maize Germ Plasm to Phaeosphaeria Leaf Spot. Plant Dis. 1999, 83, 462–464. [Google Scholar] [CrossRef][Green Version]
- Maag, D.; Erb, M.; Bernal, J.S.; Wolfender, J.L.; Turlings, T.C.; Glauser, G. Maize Domestication and Anti-Herbivore Defences: Leaf-Specific Dynamics during Early Ontogeny of Maize and Its Wild Ancestors. PLoS ONE 2015, 10, e0135722. [Google Scholar] [CrossRef]
- Li, C.; Song, W.; Luo, Y.; Gao, S.; Zhang, R.; Shi, Z.; Wang, X.; Wang, R.; Wang, F.; Wang, J.; et al. The HuangZaoSi Maize Genome Provides Insights into Genomic Variation and Improvement History of Maize. Mol. Plant 2019, 12, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.Y.; Chen, Z.H.; Hu, X.; Lan, Q.Y.; Zhu, Y.F.; Wang, A.G.; Chen, J.J.; Ren, H. Improvement Effects of Donors on Inbred Line QR273 in Maize. J. Maize Sci. 2016, 24, 15–19. (In Chinese) [Google Scholar]
- Wu, X.; Wang, A.G.; Guo, X.Y.; Liu, P.F.; Zhu, Y.F.; Li, X.S.; Chen, Z.H. Genetic characterization of maize germplasm derived from Suwan population and temperate resources. Hereditas 2019, 156, 2. [Google Scholar] [CrossRef]
- Wang, A.G.; Chen, Z.H.; Zhu, Y.F.; Guo, X.Y.; Wu, X.; Liu, P.F. Breeding and Application of Excellent Corn Inbred Line ‘QB506′. Seed 2020, 39, 142–146. (In Chinese) [Google Scholar]
- Wu, X.; Li, Y.; Shi, Y.; Song, Y.; Wang, T.; Huang, Y.; Li, Y. Fine genetic characterization of elite maize germplasm using high-throughput SNP genotyping. Theor. Appl. Genet. 2014, 127, 621–631. [Google Scholar] [CrossRef]
- Liu, C.; Hao, Z.; Zhang, D.; Xie, C.; Li, M.; Zhang, X.; Yong, H.; Zhang, S.; Weng, J.; Li, X. Genetic properties of 240 maize inbred lines and identity-by-descent segments revealed by high-density SNP markers. Mol. Breed. 2015, 35, 146. [Google Scholar] [CrossRef]
- Wang, H.; Liang, Q.J.; Hu, X.J.; Li, K.; Huang, C.L.; Wang, Q.; He, W.Z.; Wang, H.W.; Liu, Z.F. QTL mapping for ear architectural traits under three plant densities in maize. Acta Agron. Sin. 2016, 42, 1592–1600. (In Chinese) [Google Scholar] [CrossRef]
- Shao, X.Y.; Song, X.Y.; Pan, S.X.; Zhao, M.A. Genome-wide association study and Meta-QTL analysis of ear diameter trait in maize. Plant Physiol. J. 2017, 53, 2091–2102. (In Chinese) [Google Scholar]
- Wu, L.; Dai, L.Q.; Dong, Q.S.; Shi, T.T.; Wang, P.W. Genome-wide association analysis of kernel number per row in maize. Acta Agron. Sin. 2017, 43, 1559–1564. (In Chinese) [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.X.; Li, C.; Wu, X.; Qin, W.; Li, X.; Jiao, F.; Zhang, X.; Zhang, D.; Shi, Y.; et al. Fine-mapping of qGW4.05, a major QTL for kernel weight and size in maize. BMC Plant Biol. 2016, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Wu, L.; Dong, Q.; Zhang, Z.; Wu, N.; Song, Y.; Lu, S.; Wang, P. Genome-wide association study of field grain drying rate after physiological maturity based on a resequencing approach in elite maize germplasm. Euphytica 2017, 213, 182. [Google Scholar] [CrossRef]
- Zila, C.T.; Ogut, F.; Romay, M.C.; Gardner, C.A.; Buckler, E.S.; Holland, J.B. Genome-wide association study of Fusarium ear rot disease in the U.S.A. maize inbred line collection. BMC Plant Biol. 2014, 14, 372. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Pu, H.J.; Liu, P.F.; Chen, Q.C.; Zhang, Z.L.; Jiang, F. QTL mapping for Southern rust resistance in fresh corn. J. China Agric. Univ. 2020, 25, 82–88. (In Chinese) [Google Scholar]
- Lana, U.G.P.; Prazeres de Souza, I.R.; Noda, R.W.; Pastina, M.M.; Magalhaes, J.V.; Guimaraes, C.T. Quantitative Trait Loci and Resistance Gene Analogs Associated with Maize White Spot Resistance. Plant Dis. 2017, 101, 200–208. [Google Scholar] [CrossRef]
- Zhang, F.; Wan, X.Q.; Pan, G.T. Molecular Mapping of QTL for Resistance to Maize Ear Rot Caused by Fusarium moniliforme. Acta Agron. Sin. 2007, 33, 491–496. (In Chinese) [Google Scholar]
- Rossi, E.S.; Kuki, M.C.; Pinto, R.J.B.; Scapim, C.A.; Faria, M.V.; Leon, N.D. Genomic-wide association study for white spot resistance in a tropical maize germplasm. Euphytica 2020, 216, 15. [Google Scholar] [CrossRef]
- Fei, J.; Jiang, Q.; Guo, M.; Lu, J.; Wang, P.; Liu, S.; Qu, J.; Ma, Y.; Guan, S. Fine Mapping and Functional Research of Key Genes for Photoperiod Sensitivity in Maize. Front. Plant Sci. 2022, 13, 890780. [Google Scholar] [CrossRef]
- Lv, C.X. Genome Wide Association Study of Grain Water Content and Related Traits in Maize. Master’s Thesis, Sichuan Agricultural University, Ya’an, China, 2019; pp. 1–62. (In Chinese). [Google Scholar]
- Stein, O.; Granot, D. An Overview of Sucrose Synthases in Plants. Front. Plant Sci. 2019, 10, 95. [Google Scholar] [CrossRef]
- González-Cruz, J.; Pastenes, C. Water-stress-induced thermotolerance of photosynthesis in bean (Phaseolus vulgaris L.) plants: The possible involvement of lipid composition and xanthophyll cycle pigments. Environ. Exp. Bot. 2012, 77, 127–140. [Google Scholar] [CrossRef]
- Pinto, V.B.; Almeida, V.C.; Pereira-Lima, Í.A.; Vale, E.M.; Araújo, W.L.; Silveira, V.; Viana, J.M.S. Deciphering the major metabolic pathways associated with aluminum tolerance in popcorn roots using label-free quantitative proteomics. Planta 2021, 254, 132. [Google Scholar] [CrossRef] [PubMed]
- Gkizi, D.; Santos-Rufo, A.; Rodríguez-Jurado, D.; Paplomatas, E.J.; Tjamos, S.E. The β-amylase genes: Negative regulators of disease resistance for Verticillium dahliae. Plant Pathol. 2015, 64, 1484–1490. [Google Scholar] [CrossRef]
- Chen, T.; Xie, Y.; Sun, Q.; Shi, X.; Wang, S.; Laborda, P. First Report of Epicoccum sorghinum Causing Leaf Sheath and Leaf Spot on Maize in China. Plant Dis. 2021, 105, 3741. [Google Scholar] [CrossRef]
- Carson, M.L.; Stuber, C.W.; Senior, M.L. Quantitative Trait Loci Conditioning Resistance to Phaeosphaeria Leaf Spot of Maize Caused by Phaeosphaeria maydis. Plant Dis. 2005, 89, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.U.; Bento, D.A.; de Souza, A.P.; de Souza, C.L.J. QTL mapping for reaction to Phaeosphaeria leaf spot in a tropical maize population. Theor. Appl. Genet. 2009, 119, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Kistner, M.B.; Galiano-Carneiro, A.L.; Kessel, B.; Presterl, T.; Miedaner, T. Multi-parental QTL mapping of resistance to white spot of maize (Zea mays) in southern Brazil and relationship to QTLs of other foliar diseases. Plant Breed. 2021, 140, 801–811. [Google Scholar] [CrossRef]
- Guo, X.Y.; Wu, X.; Wu, W.Q.; Wang, A.G.; Liu, P.F.; Zhu, Y.F.; Ren, H.; Chen, Z.H.; Lou, H.B. Dissection of identical-by-descent segments during the formation of foundation parents derived from Suwan germplasm of maize. Plant Breed. 2020, 139, 862–869. [Google Scholar] [CrossRef]
- Li, J.; Han, G.; Sun, C.; Sui, N. Research advances of MYB transcription factors in plant stress resistance and breeding. Plant Signal. Behav. 2019, 14, 1613131. [Google Scholar] [CrossRef]
- Lv, P.; Wan, J.; Zhang, C.; Hina, A.; Al Amin, G.M.; Begum, N.; Zhao, T. Unraveling the Diverse Roles of Neglected Genes Containing Domains of Unknown Function (DUFs): Progress and Perspective. Int. J. Mol. Sci. 2023, 24, 4187. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, J.J.; Yu, H.; Bryksa, B.C.; Dupuis, J.H.; Ou, X.; Qian, Z.; Song, C.; Wang, S.; Yada, R.Y. Insights into the mechanism of membrane fusion induced by the plant defense element, plant-specific insert. J. Biol. Chem. 2020, 295, 14548–14562. [Google Scholar] [CrossRef]
- Chung, C.L.; Poland, J.; Kump, K.; Benson, J.; Longfellow, J.; Walsh, E.; Balint-Kurti, P.J.; Nelson, R. Targeted discovery of quantitative trait loci for resistance to northern leaf blight and other diseases of maize. Theor. Appl. Genet. 2011, 123, 307–326. [Google Scholar] [CrossRef]
- Lopez-Zuniga, L.O.; Wolters, P.; Davis, S.; Weldekidan, T.; Kolkman, J.M.; Nelson, R.; Hooda, K.S.; Rucker, E.; Thomason, W.; Wisser, R.; et al. Using Maize Chromosome Segment Substitution Line Populations for the Identification of Loci Associated with Multiple Disease Resistance. G3-Genes Genomes Genet. 2019, 9, 189–201. [Google Scholar] [CrossRef]
- Chen, L.; Liu, L.; Li, Z.; Zhang, Y.D.; Kang, M.S.; Wang, Y.Y.; Fan, X.M. High-density mapping for gray leaf spot resistance using two related tropical maize recombinant inbred line populations. Mol. Biol. Rep. 2021, 48, 3379–3392. [Google Scholar] [CrossRef]
- Zheng, H.J.; Chen, J.F.; Mu, C.H.; Makumbi, D.; Xu, Y.B.; Mahuku, G. Combined linkage and association mapping reveal QTL for host plant resistance to common rust (Puccinia sorghi) in tropical maize. BMC Plant Biol. 2018, 18, 310. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, Y.; Kim, Y.S.; Yoo, S.C.; Hörtensteiner, S.; Paek, N.C. 7-Hydroxymethyl chlorophyll a reductase functions in metabolic channeling of chlorophyll breakdown intermediates during leaf senescence. Biochem. Biophys. Res. Commun. 2013, 430, 32–37. [Google Scholar] [CrossRef]
- Casati, P.; Drincovich, M.F.; Edwards, G.E.; Andreo, C.S. Malate metabolism by NADP-malic enzyme in plant defense. Photosynth. Res. 1999, 61, 99–105. [Google Scholar] [CrossRef]
- Versaw, W.K.; Harrison, M.J. A Chloroplast Phosphate Transporter, PHT2;1, Influences Allocation of Phosphate within the Plant and Phosphate-Starvation Responses. Plant Cell 2002, 14, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Schön, C.C.; Melchinger, A.E.; Boppenmaier, J.; Brunklaus-Jung, E.; Herrmann, R.G.; Seitzer, J.F. RFLP Mapping in Maize: Quantitative Trait Loci Affecting Testcross Performance of Elite European Flint Lines. Crop Sci. 1994, 34, 378–389. [Google Scholar] [CrossRef]
- Veldboom, L.R.; Lee, M. Genetic Mapping of Quantitative Trait Loci in Maize in Stress and Nonstress Environments: I. Grain Yield and Yield Components. Crop Sci. 1996, 36, 1310–1319. [Google Scholar] [CrossRef]
- Emaus, M.N.; Cagliero, C.; Gostel, M.R.; Johnson, G.; Anderson, J.L. Simple and efficient isolation of plant genomic DNA using magnetic ionic liquids. Plant Methods 2022, 18, 37. [Google Scholar] [CrossRef] [PubMed]
- Lehne, B.; Lewis, C.M.; Schlitt, T. From SNPs to genes: Disease association at the gene level. PLoS ONE 2011, 6, e20133. [Google Scholar] [CrossRef]
- Žiga, A.; Vikram, A.; Daniel, V.; Joseph, R.L.; Agnieszka, G.; Kyle, R.T.; Yannis, A.; John, J.; Pushmeet, K.; David, R.K. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 2021, 18, 1196–1203. [Google Scholar]
- David, S. A current guide to candidate gene association studies. Trends Genet. 2021, 37, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Shahjaman, M.; Manir Hossain Mollah, M.; Rezanur Rahman, M.; Islam, S.M.S.; Nurul Haque Mollah, M. Robust identification of differentially expressed genes from RNA-seq data. Genomics 2020, 112, 2000–2010. [Google Scholar] [CrossRef]
- Wu, X.; Liu, Y.; Lu, X.; Tu, L.; Gao, Y.; Wang, D.; Guo, S.; Xiao, Y.; Xiao, P.; Guo, X.; et al. Integration of GWAS, linkage analysis and transcriptome analysis to reveal the genetic basis of flowering time-related traits in maize. Front. Plant Sci. 2023, 14, 1145327. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect | Mean Square | F-Value |
|---|---|---|
| Genotype | 3444.291 | 78.533 *** |
| Genotype × environment | 777.392 | 17.725 *** |
| Error | 43.858 |
| QTN | Chr. | Position | Environment | Candidate Gene |
|---|---|---|---|---|
| SYNGENTA14387 | 1 | 9031311 | Mojiang and joint environments | Zm00001d027619 |
| PHM13619.5 | 1 | 22281738 | Shilin and joint environments | Zm00001d028088 |
| PZE-101040783 | 1 | 27552892 | Shilin and Wenshan | |
| SYN11249 | 1 | 43111965 | Shilin and joint environments | Zm00001d000328 |
| PZE-101151153 | 1 | 194604587 | Wenshan and joint environments | Zm00001d031641 |
| SYN10891 | 1 | 195359337 | Shilin and joint environments | Zm00001d031660 |
| SYN10137 | 1 | 200556614 | Shilin, Wenshan and joint environments | |
| PZA00131.14 | 1 | 203385274 | Shilin, Wenshan and joint environments | |
| SYN37674 | 1 | 206370340 | Shilin and joint environments | Zm00001d032024 |
| PZE-102031753 | 2 | 14800008 | Shilin and Wenshan | Zm00001d002549 |
| PZE-103084298 | 3 | 135901771 | Mojiang and joint environments | Zm00001d041844 |
| ZM012464-0529 | 3 | 179012477 | Shilin and joint environments | |
| SYN30108 | 4 | 231898350 | Shilin and Wenshan | Zm00001d053432 |
| PZE-108057528 | 8 | 101489430 | Shilin, Wenshan and joint environments | Zm00001d010230 |
| SYN4935 | 8 | 171846715 | Shilin and joint environments | Zm00001d012665 |
| Scale | Area Occupied by Disease Spots |
|---|---|
| 0 | 0 |
| 1 | ≤5% |
| 3 | 6–10% |
| 5 | 11–30% |
| 7 | 31–50% |
| 9 | ≥50% |
| Scale | Disease Index (DI) | Resistance |
|---|---|---|
| 0 | DI = 0 | Immunity (IM) |
| 1 | DI ≤ 10 | High resistance (HR) |
| 3 | 10 < DI ≤ 30 | Resistance (R) |
| 5 | 30 < DI ≤ 50 | Moderate resistance (MR) |
| 7 | 50 < DI ≤ 70 | Susceptibility (S) |
| 9 | DI > 70 | High susceptibility (HS) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; He, Y.; Nie, L.; Guo, S.; Tu, L.; Guo, X.; Wang, A.; Liu, P.; Zhu, Y.; Wu, X.; et al. Integrated IBD Analysis, GWAS Analysis and Transcriptome Analysis to Identify the Candidate Genes for White Spot Disease in Maize. Int. J. Mol. Sci. 2023, 24, 10005. https://doi.org/10.3390/ijms241210005
Wang D, He Y, Nie L, Guo S, Tu L, Guo X, Wang A, Liu P, Zhu Y, Wu X, et al. Integrated IBD Analysis, GWAS Analysis and Transcriptome Analysis to Identify the Candidate Genes for White Spot Disease in Maize. International Journal of Molecular Sciences. 2023; 24(12):10005. https://doi.org/10.3390/ijms241210005
Chicago/Turabian StyleWang, Dong, Yue He, Lei Nie, Shuang Guo, Liang Tu, Xiangyang Guo, Angui Wang, Pengfei Liu, Yunfang Zhu, Xun Wu, and et al. 2023. "Integrated IBD Analysis, GWAS Analysis and Transcriptome Analysis to Identify the Candidate Genes for White Spot Disease in Maize" International Journal of Molecular Sciences 24, no. 12: 10005. https://doi.org/10.3390/ijms241210005
APA StyleWang, D., He, Y., Nie, L., Guo, S., Tu, L., Guo, X., Wang, A., Liu, P., Zhu, Y., Wu, X., & Chen, Z. (2023). Integrated IBD Analysis, GWAS Analysis and Transcriptome Analysis to Identify the Candidate Genes for White Spot Disease in Maize. International Journal of Molecular Sciences, 24(12), 10005. https://doi.org/10.3390/ijms241210005