
New Insights on the Activity and Selectivity of MAO-B Inhibitors through In Silico Methods



Abstract
1. Introduction
2. Results and Discussion
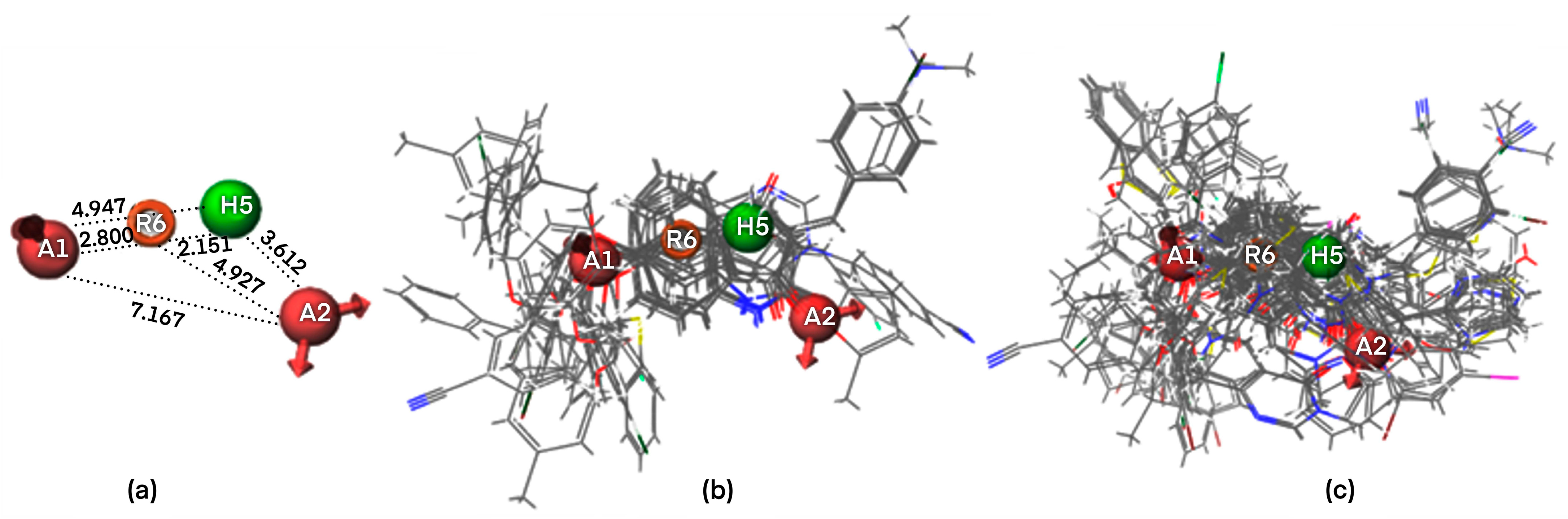
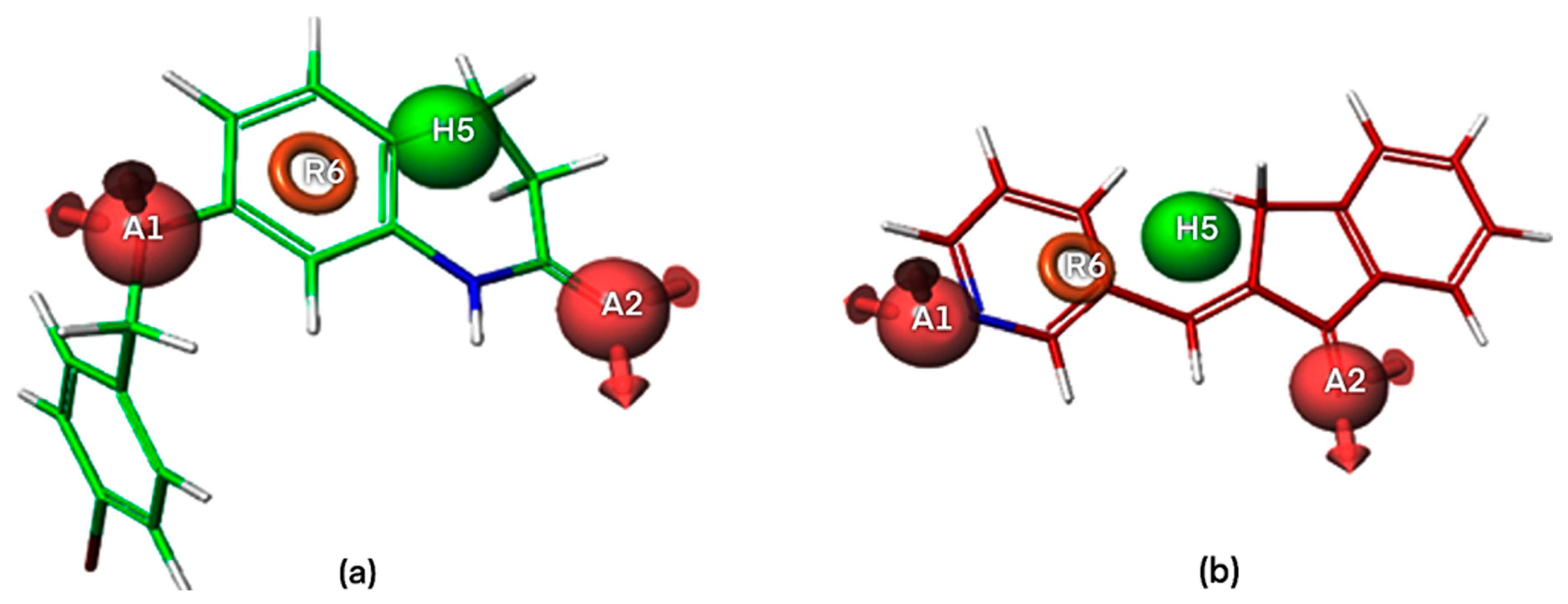
2.1. Pharmacophore Modeling
2.1.1. Pharmacophore Model Validation
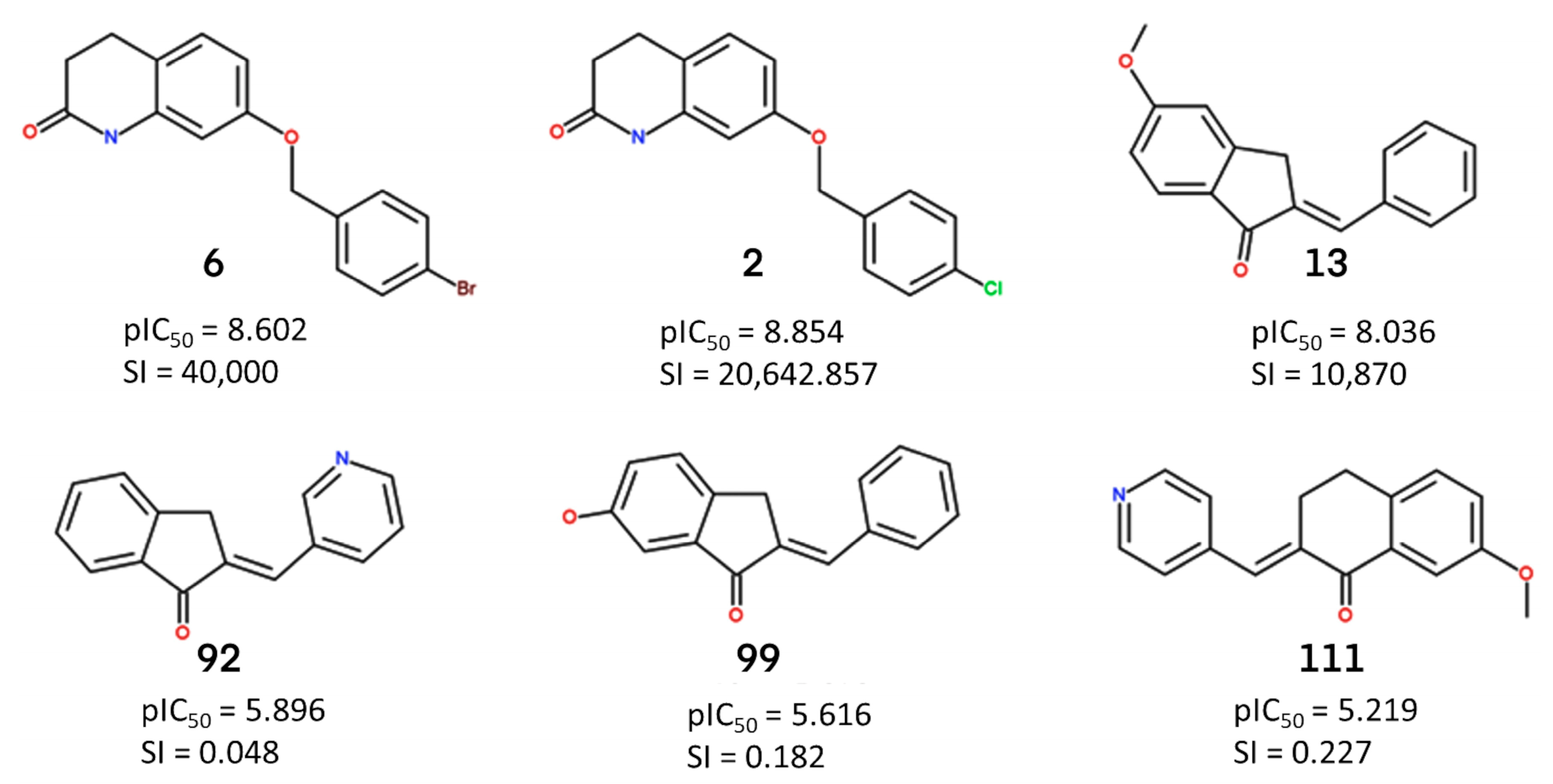
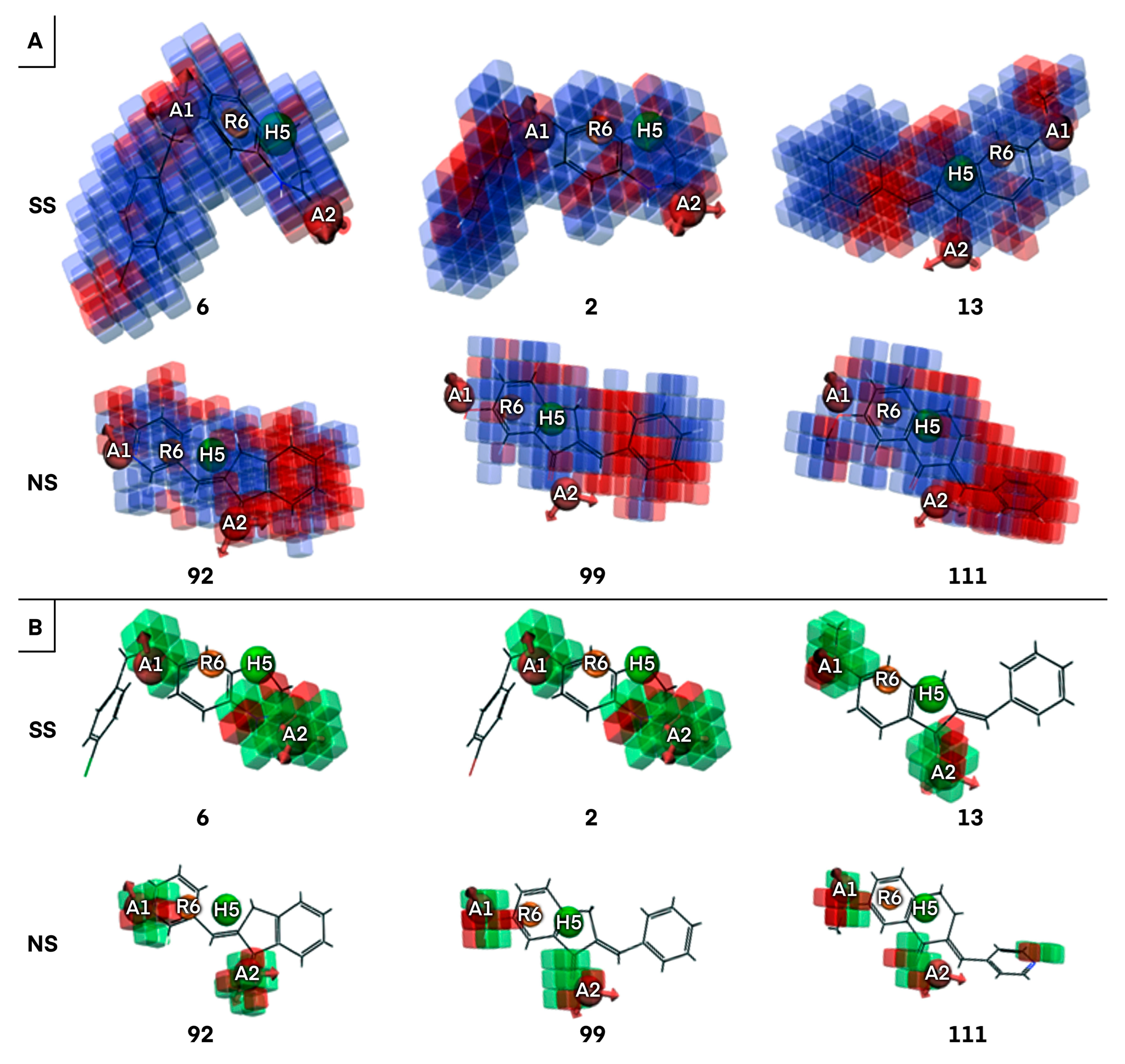
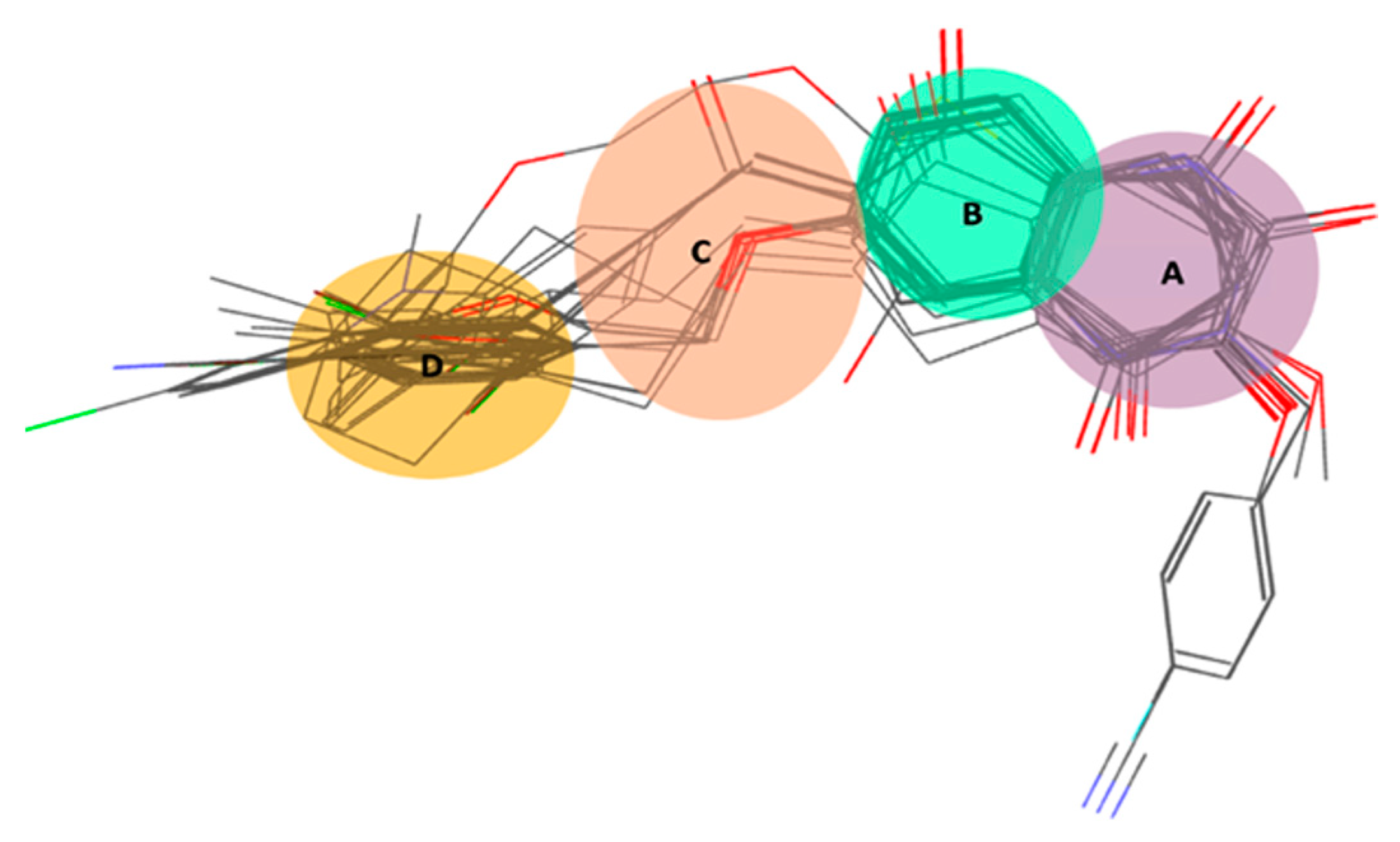
2.1.2. Atom-Based 3D QSAR Approach
2.1.3. Validation of Atom-Based 3D QSAR Model
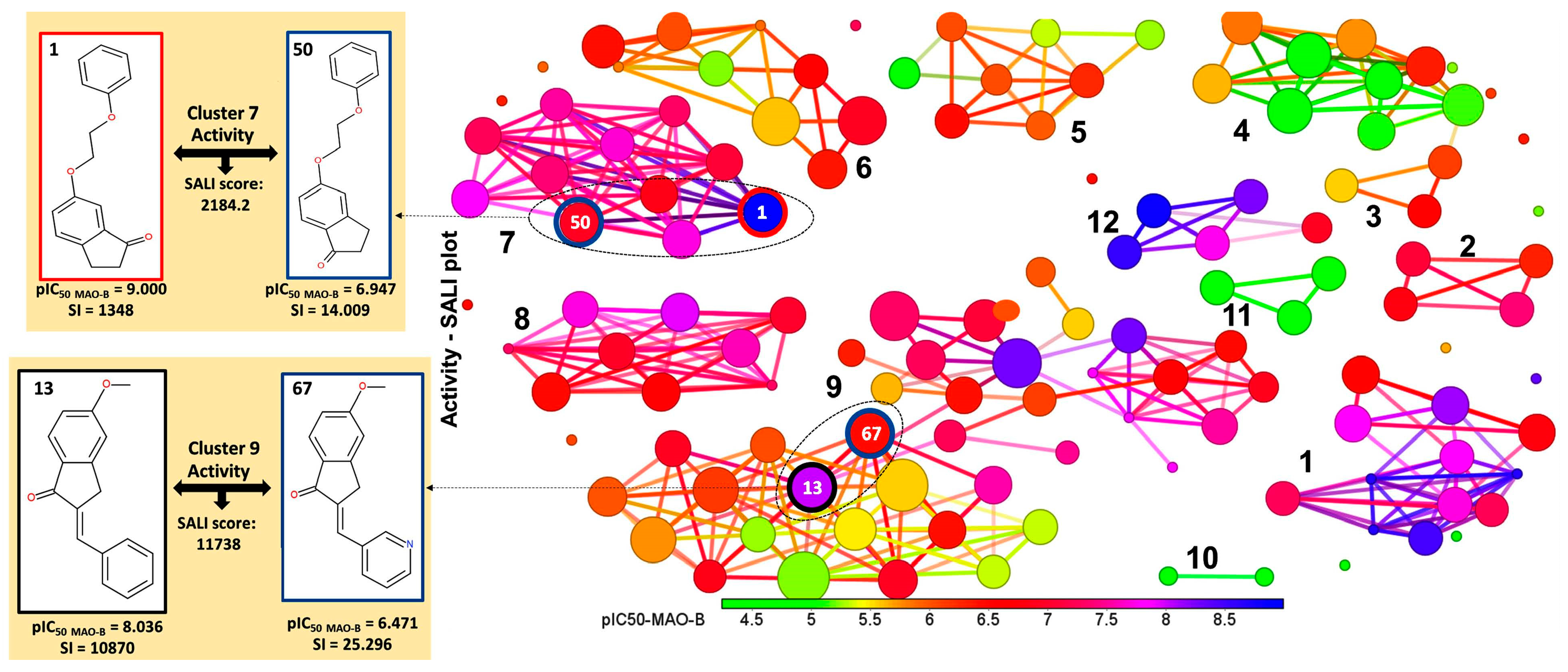
2.2. Identification of Activity-Cliffs
2.3. ECFP (Extended-Connectivity Fingerprints) Analysis
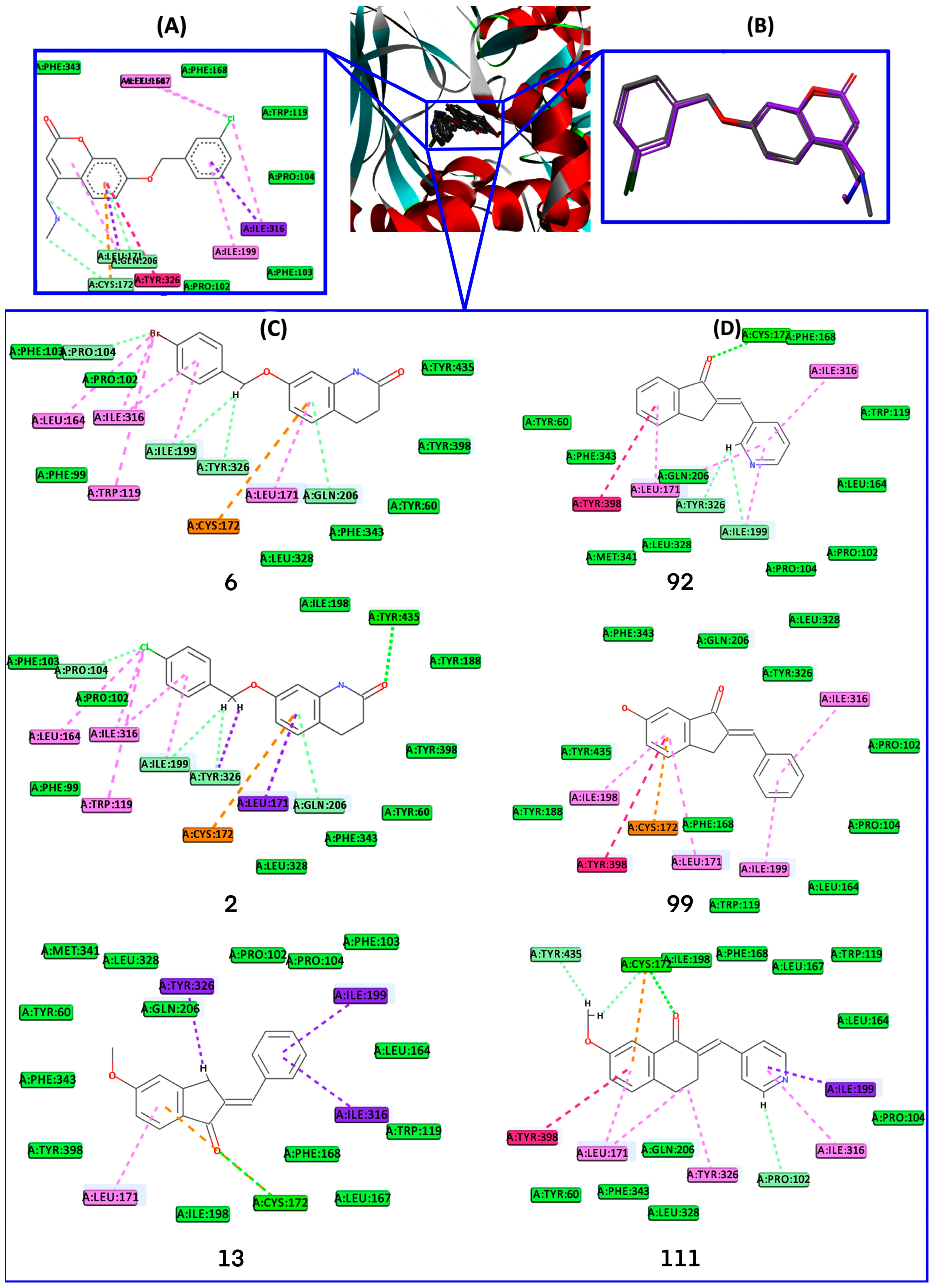
2.4. Docking Analysis
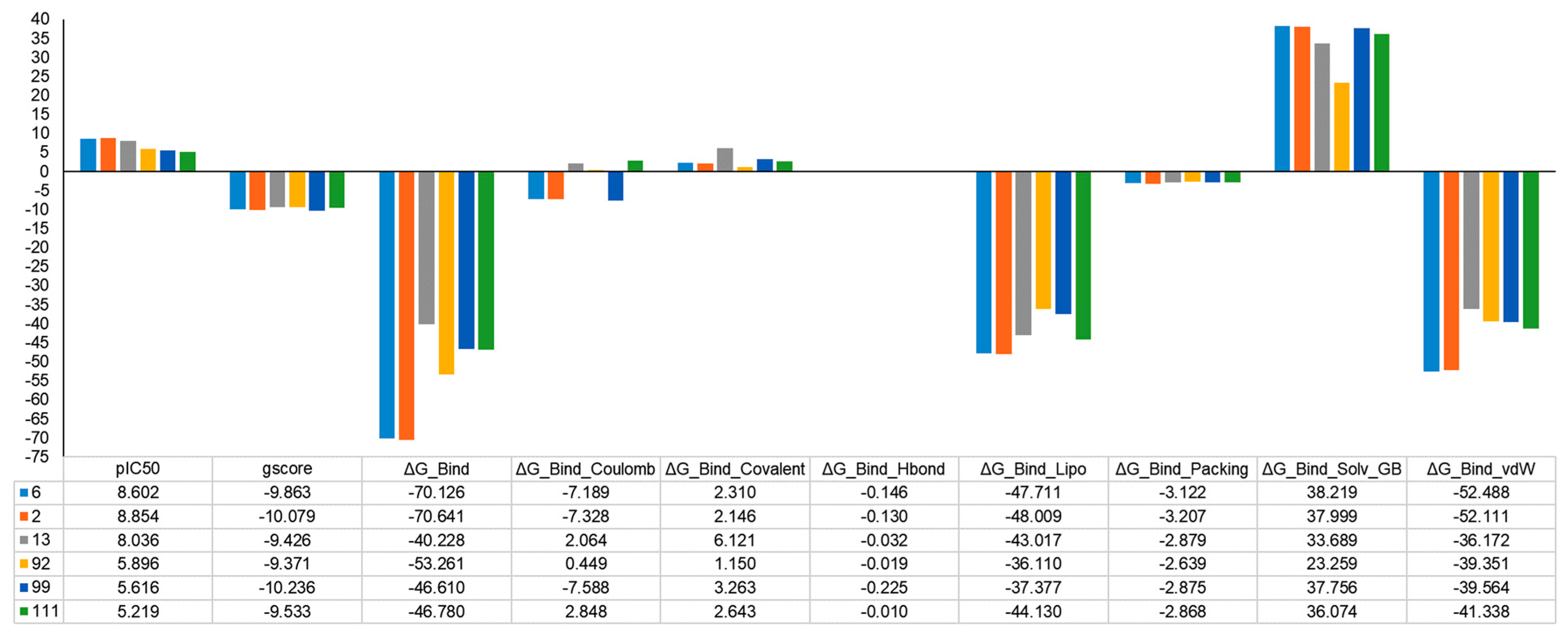
2.5. MM-GBSA Binding Free Energy Analysis
3. Materials and Methods
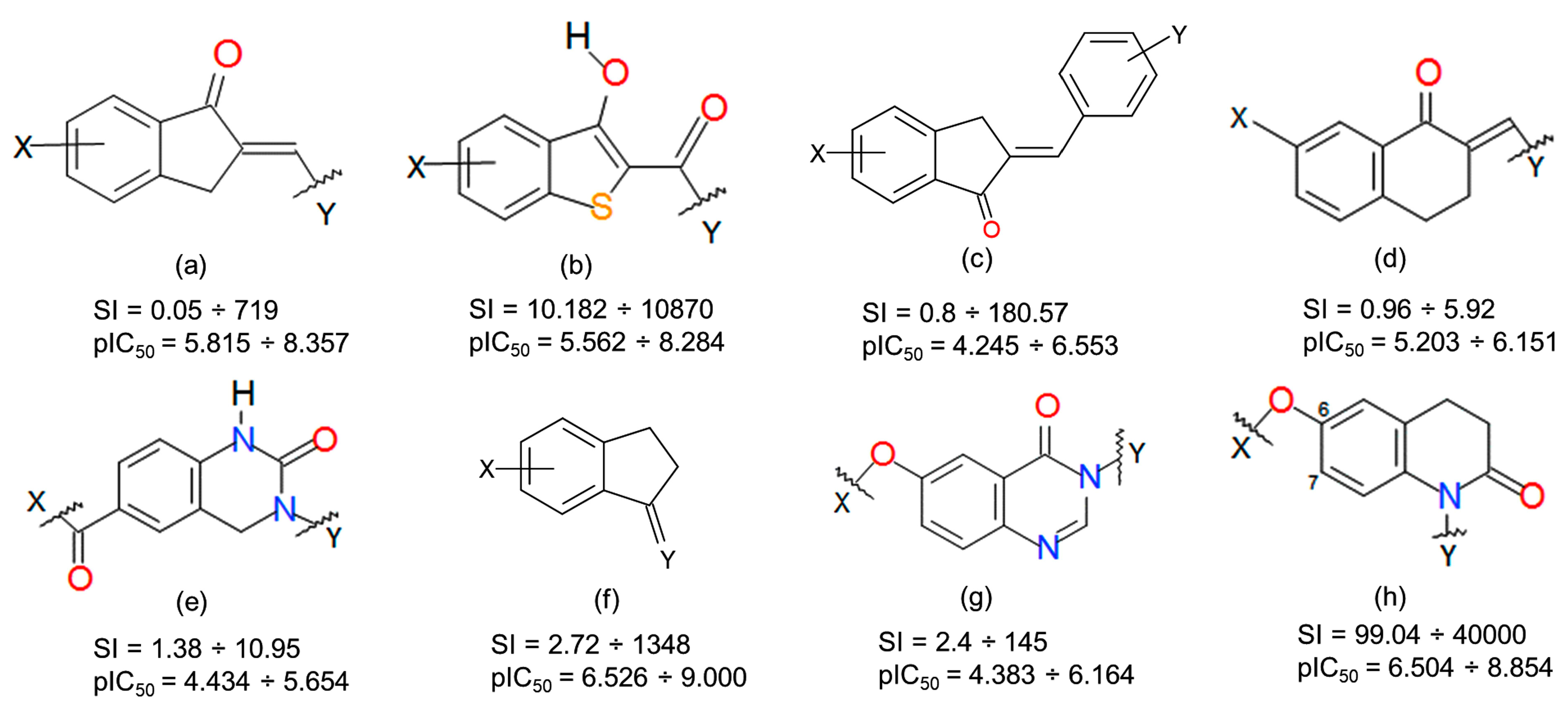
3.1. Dataset
3.2. Pharmacophore Modelling
3.3. Atom-Based 3D QSAR
3.4. Activity Cliffs
3.5. Extended-Connectivity Fingerprints (ECFPs)
3.6. Docking
3.7. MM-GBSA Binding Free Energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary
References
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.W.K.; Georgieva, M.G.; Atanasov, A.G.; Tzvetkov, N.T. Monoamine Oxidases (MAOs) as Privileged Molecular Targets in Neuroscience: Research Literature Analysis. Front. Mol. Neurosci. 2019, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.P.; Laux, G. MAO-inhibitors in Parkinson’s disease. Exp. Neurobiol. 2011, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Weinstock, M. Therapeutic applications of selective and non-selective inhibitors of monoamine oxidase A and B that do not cause significant tyramine potentiation. Neurotoxicology 2004, 25, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Crisan, L.; Istrate, D.; Bora, A.; Pacureanu, L. Virtual screening and drug repurposing experiments to identify potential novel selective MAO-B inhibitors for Parkinson’s disease treatment. Mol. Divers. 2021, 25, 1775–1794. [Google Scholar] [CrossRef]
- Fiedorowicz, J.G.; Swartz, K.L. The Role of Monoamine Oxidase Inhibitors in Current Psychiatric Practice. J. Psychiatr. Pract. 2004, 10, 239–248. [Google Scholar] [CrossRef]
- Finberg, J.P.; Gillman, K. Selective inhibitors of monoamine oxidase type B and the “cheese effect”. Int. Rev. Neurobiol. 2011, 100, 169–190. [Google Scholar]
- Finberg, J.P. Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: Focus on modulation of CNS monoamine neurotransmitter release. Pharmacol. Ther. 2014, 143, 133–152. [Google Scholar] [CrossRef]
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D.E.; Mattevi, A. Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. USA 2005, 102, 12684–12689. [Google Scholar] [CrossRef]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.E.; Blomme, E.A.G.; Van Vleet, T.R. Novel Computational Approach to Predict Off-Target Interactions for Small Molecules. Front. Big Data 2019, 2, 25. [Google Scholar] [CrossRef]
- Baweja, G.S.; Gupta, S.; Kumar, B.; Patel, P.; Asati, V. Recent updates on structural insights of MAO-B inhibitors: A review on target-based approach. Mol. Divers. 2023. [Google Scholar] [CrossRef]
- Crisan, L.; Avram, S.; Pacureanu, L. Pharmacophore-based screening and drug repurposing exemplified on glycogen synthase kinase-3 inhibitors. Mol. Divers. 2017, 21, 385–405. [Google Scholar] [CrossRef]
- Jenkins, J.L.; Kao, R.Y.; Shapiro, R. Virtual screening to enrich hit lists from high-throughput screening: A case study on small-molecule inhibitors of angiogenin. Proteins 2003, 50, 81–93. [Google Scholar] [CrossRef]
- Crisan, L.; Pacureanu, L.; Bora, A.; Avram, S.; Kurunczi, L.; Simon, Z. QSAR study and molecular docking on indirubin inhibitors of Glycogen Synthase Kinase-3. Cent. Eur. J. Chem. 2013, 11, 63–77. [Google Scholar] [CrossRef]
- Ivan, D.; Crisan, L.; Funar-Timofei, S.; Mracec, M. A quantitative structure–activity relationships study for the anti-HIV-1 activities of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine derivatives using the multiple linear regression and partial least squares methodologies. J. Serb. Chem. Soc. 2013, 78, 495–506. [Google Scholar] [CrossRef]
- Avram, S.I.; Crisan, L.; Bora, A.; Pacureanu, L.M.; Avram, S.; Kurunczi, L. Retrospective group fusion similarity search based on eROCE evaluation metric. Bioorg. Med. Chem. 2013, 21, 1268–1278. [Google Scholar] [CrossRef]
- Avram, S.; Bora, A.; Halip, L.; Curpăn, R. Modeling Kinase Inhibition Using Highly Confident Data Sets. J. Chem. Inf. Model. 2018, 58, 957–967. [Google Scholar] [CrossRef]
- Pacureanu, L.; Avram, S.; Bora, A.; Kurunczi, L.; Crisan, L. Portraying the selectivity of GSK-3 inhibitors towards CDK-2 by 3D similarity and docking. Struct. Chem. 2019, 30, 911–923. [Google Scholar] [CrossRef]
- Tanrikulu, Y.; Krüger, B.; Proschak, E. The holistic integration of virtual screening in drug discovery. Drug Discov. Today 2013, 18, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Crisan, L.; Borota, A.; Bora, A.; Pacureanu, P. Diarylthiazole and Diarylimidazole Selective COX-1 Inhibitors Analysis through Pharmacophore Modeling, Virtual Screening, and DFT-Based Approaches. Struct. Chem. 2019, 30, 2311–2326. [Google Scholar] [CrossRef]
- Visa, A.; Maranescu, B.; Lupa, L.; Crisan, L.; Borota, A. New Efficient Adsorbent Materials for the Removal of Cd(II) from Aqueous Solutions. Nanomaterials 2020, 10, 899. [Google Scholar] [CrossRef] [PubMed]
- Visa, A.; Plesu, N.; Maranescu, B.; Ilia, G.; Borota, A.; Crisan, L. Combined Experimental and Theoretical Insights into the Corrosion Inhibition Activity on Carbon Steel Iron of Phosphonic Acids. Molecules 2021, 26, 135. [Google Scholar] [CrossRef] [PubMed]
- Petric, M.; Crisan, L.; Crisan, M.; Micle, A.; Maranescu, B.; Ilia, G. Synthesis and QSRR Study for a Series of Phosphoramidic Acid Derivatives. Heteroat. Chem. 2013, 24, 138–145. [Google Scholar] [CrossRef]
- Visa, A.; Mracec, M.; Maranescu, B.; Maranescu, V.; Ilia, G.; Popa, A.; Mracec, M. Structure simulation into a lamellar supramolecular network and calculation of the metal ions/ligands ratio. Chem. Cent. J. 2012, 6, 91. [Google Scholar] [CrossRef]
- Bora, A.; Suzuki, T.; Funar-Timofei, S. Neonicotinoid insecticide design: Molecular docking, multiple chemometric approaches, and toxicity relationship with Cowpea aphids. Environ. Sci. Pollut. Res. 2019, 26, 14547–14561. [Google Scholar] [CrossRef]
- Maranescu, B.; Visa, A.; Mracec, M.; Ilia, G.; Maranescu, V.; Simon, Z.; Mracec, M. Lamellar Co2+ vinylphosphonate metal organic framework. PM3 semi-empirical analysis of structural properties. Rev. Roum. Chim. 2011, 56, 473–482. [Google Scholar]
- AbdulHameed, M.D.M.; Chaudhury, S.; Singh, N.; Sun, H.; Wallqvist, A.; Tawa, G.J. Exploring Polypharmacology Using a ROCS-Based Target Fishing Approach. J. Chem. Inf. Model. 2012, 52, 492–505. [Google Scholar] [CrossRef]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef]
- Gleeson, M.P.; Hersey, A.; Hannongbua, S. In-silico ADME models: A general assessment of their utility in drug discovery applications. Curr. Top. Med. Chem. 2011, 11, 358–381. [Google Scholar] [CrossRef]
- Crisan, L.; Pacureanu, L.; Avram, S.; Bora, A.; Avram, S.; Kurunczi, L. PLS and shape-based similarity analysis of maleimides–GSK-3 inhibitors. J. Enzym. Inhib. Med. Chem. 2014, 29, 599–610. [Google Scholar] [CrossRef]
- Rodríguez-Enríquez, F.; Viña, D.; Uriarte, E.; Fontenla, J.A.; Matos, M.J. Discovery and optimization of 3-thiophenylcoumarins as novel agents against Parkinson’s disease: Synthesis, in vitro and in vivo studies. Bioorg. Chem. 2020, 101, 103986. [Google Scholar] [CrossRef]
- Rodríguez-Enríquez, F.; Costas-Lago, M.C.; Besada, P.; Alonso-Pena, M.; Torres-Terán, I.; Viña, D.; Fontenla, J.Á.; Sturlese, M.; Moro, S.; Quezada, E.; et al. Novel coumarin-pyri[1]dazine hybrids as selective MAO-B inhibitors for the Parkinson’s disease therapy. Bioorg. Chem. 2020, 104, 104203. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Y.; Zeng, R.F.; Liu, Y.; Xie, S.S.; Lan, J.S.; Ding, Y.; Yang, Y.-T.; Yang, J.; Zhang, T. Design and synthesis of novel 3, 4-dihydrocoumarins as potent and selective monoamine oxidase-B inhibitors with the neuroprotection against Parkinson’s disease. Bioorg. Chem. 2021, 109, 104685. [Google Scholar] [CrossRef]
- Crisan, L.; Bora, A. Small Molecules of Natural Origin as Potential Anti-HIV Agents: A Computational Approach. Life 2021, 11, 722. [Google Scholar] [CrossRef]
- Mellado, M.; Salas, C.O.; Uriarte, E.; Viña, D.; Jara-Gutiérrez, C.; Matos, M.J.; Cuellar, M. Design, Synthesis and Docking Calculations of Prenylated Chalcones as Selective Monoamine Oxidase B Inhibitors with Antioxidant Activity. Chemistryselect 2019, 4, 7698–7703. [Google Scholar] [CrossRef]
- Rehuman, N.A.; Oh, J.M.; Abdelgawad, M.A.; Beshr, E.A.; Abourehab, M.A.; Gambacorta, N.; Nicolotti, O.; Jat, R.K.; Kim, H.; Mathew, B. Development of Halogenated-Chalcones Bearing with Dimethoxy Phenyl Head as Monoamine Oxidase-B Inhibitors. Pharmaceuticals 2022, 15, 1152. [Google Scholar] [CrossRef]
- Osmaniye, D.; Kurban, B.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Novel thiosemicarbazone derivatives: In vitro and in silico evaluation as potential MAO-B inhibitors. Molecules 2021, 26, 6640. [Google Scholar] [CrossRef]
- Li, W.; Yang, X.; Song, Q.; Cao, Z.; Shi, Y.; Deng, Y.; Zhang, L. Pyridoxine-resveratrol hybrids as novel inhibitors of MAO-B with antioxidant and neuroprotective activities for the treatment of Parkinson’s disease. Bioorg. Chem. 2020, 97, 103707. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Ogunrombi, M.O.; Malan, S.F.; Terre’Blanche, G.; Castagnoli Jr, N.; Bergh, J.J.; Petzer, J.P. Structure–activity relationships in the inhibition of monoamine oxidase B by 1-methyl-3-phenylpyrroles. Bioorg. Med. Chem. 2008, 16, 2463–2472. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- ROCS, Version 3.2.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013. Available online: http://www.eyesopen.com (accessed on 18 April 2023).
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of Human Monoamine Oxidase B Complexes with Selective Noncovalent Inhibitors: Safinamide and Coumarin Analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O.; et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef]
- Alagöz, M.A.; Oh, J.M.; Zenni, Y.N.; Özdemir, Z.; Abdelgawad, M.A.; Naguib, I.A.; Ghoneim, M.M.; Gambacorta, N.; Nicolotti, O.; Kim, H.; et al. Development of a Novel Class of Pyridazinone Derivatives as Selective MAO-B Inhibitors. Molecules 2022, 27, 3801. [Google Scholar] [CrossRef]
- BIOVIA. Discovery Studio Visualizer, version 20.1.0; Accelrys Software Inc.: San Diego, CA, USA, 2020.
- Kawasaki, Y.; Freire, E. Finding a better path to drug selectivity. Drug Discov. Today 2011, 16, 985–990. [Google Scholar] [CrossRef]
- Nada, H.; Lee, K.; Gotina, L.; Pae, A.N.; Elkamhawy, A. Identification of novel discoidin domain receptor 1 (DDR1) inhibitors using E-pharmacophore modeling, structure-based virtual screening, molecular dynamics simulation and MM-GBSA approaches. Comput. Biol. Med. 2022, 142, 105217. [Google Scholar] [CrossRef]
- Nel, M.S.; Petzer, A.; Petzer, J.P.; Legoabe, L.J. 2-Heteroarylidene-1-indanone derivatives as inhibitors of monoamine oxidase. Bioorg. Chem. 2016, 69, 20–28. [Google Scholar] [CrossRef]
- Nel, M.S.; Petzer, A.; Petzer, J.P.; Legoabe, L.J. 2-Benzylidene-1-indanone derivatives as inhibitors of monoamine oxidase. Bioorg. Med. Chem. Lett. 2016, 26, 4599–4605. [Google Scholar] [CrossRef]
- Guglielmi, P.; Secci, D.; Petzer, A.; Bagetta, D.; Chimenti, P.; Rotondi, G.; Ferrante, C.; Recinella, L.; Leone, S.; Alcaro, S.; et al. Benzo[b]tiophen-3-ol derivatives as effective inhibitors of human monoamine oxidase: Design, synthesis, and biological activity. J. Enzym. Inhib. Med. Chem. 2019, 31, 1511–1525. [Google Scholar] [CrossRef]
- Amakali, K.T.; Legoabe, L.J.; Petzer, A.; Petzer, J.P. Synthesis and in vitro Evaluation of 2-heteroarylidene-1-tetralone Derivatives as Monoamine Oxidase Inhibitors. Drug Res. 2018, 68, 687–695. [Google Scholar] [CrossRef]
- Marais, L.; Petzer, A.; Petzer, J.P.; Legoabe, L.J. The monoamine oxidase inhibition properties of C6- and N1-substituted 3-methyl-3,4-dihydroquinazolin-2(1H)-one derivatives. Mol. Divers. 2020, 24, 391–406. [Google Scholar] [CrossRef]
- Mostert, S.; Petzer, A.; Petzer, J.P. Indanones As High-Potency Reversible Inhibitors of Monoamine Oxidase. Chemmedchem 2015, 10, 862–873. [Google Scholar] [CrossRef]
- Qhobosheane, M.A.; Legoabe, L.J.; Petzer, A.; Petzer, J.P. The monoamine oxidase inhibition properties of C6-mono- and N3/C6-disubstituted derivatives of 4(3H)-quinazolinone. Bioorg. Chem. 2019, 85, 60–65. [Google Scholar] [CrossRef]
- Meiring, L.; Petzer, J.P.; Petzer, A. C6- and C7-Substituted 3,4-dihydro-2(1H)-quinolinones as Inhibitors of Monoamine Oxidase. Drug Res. 2017, 67, 170–178. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Instant JChem, version 6.03; ChemAxon Kft: Budapest, Hungary, 2013.
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2014, 1, 337–341. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A Conformational Search Method for Efficient Generation of Bioactive Conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Mathea, M.; Klingspohn, W.; Baumann, K. Chemoinformatic Classification Methods and their Applicability Domain. Mol. Inform. 2016, 35, 160–180. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Papa, E.; Gramatica, P. QSAR Prediction of Estrogen Activity for a Large Set of Diverse Chemicals under the Guidance of OECD Principles. Chem. Res. Toxicol. 2006, 19, 1540–1548. [Google Scholar] [CrossRef]
- Alexander, D.L.J.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef]
- Lindgren, F.; Hansen, B.; Karcher, W.; Sjostrom, M.; Eriksson, L. Model Validation by Permutation Tests: Applications to Variable Selection. J. Chemom. 1996, 10, 521–532. [Google Scholar] [CrossRef]
- Shi, L.M.; Fang, H.; Tong, W.; Wu, J.; Perkin, R.; Blair, R.M.; Branham, W.S.; Dial, S.L.; Moland, C.L.; Sheehan, D.M. QSAR Models Using a Large Diverse Set of Estrogens. J. Chem. Inf. Comput. Sci. 2001, 41, 186–195. [Google Scholar] [CrossRef]
- Schüürmann, G.; Ebert, R.-U.; Chen, J.; Wang, B.; Kühne, R. External Validation and Prediction Employing the Predictive Squared Correlation Coefficient—Test Set Activity Mean vs Training Set Activity Mean. J. Chem. Inf. Model. 2008, 48, 2140–2145. [Google Scholar] [CrossRef]
- Consonni, V.; Ballabio, D.; Todeschini, R. Comments on the definition of the Q2 parameter for QSAR validation. J. Chem. Inf. Model. 2009, 49, 1669–1678. [Google Scholar] [CrossRef]
- Gramatica, P. On the development and validation of QSAR models. Methods Mol. Biol. Vol. II 2013, 930, 499–526. [Google Scholar]
- Goodarzi, M.; Deshpande, S.; Murugesan, V.; Katti, S.B.; Prabhakar, Y.S. Is Feature Selection Essential for ANN Modeling? QSAR Comb. Sci. 2009, 28, 1487–1499. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models: How To Evaluate It? Comparison of Different Validation Criteria and Proposal of Using the Concordance Correlation Coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef]
- Lajiness, M. Evaluation of the Performance of Dissimilarity Selection Methodology. In QSAR: Rational Approaches to the Design of Bioactive Compounds; Silipo, C., Vittoria, A., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; pp. 201–204. [Google Scholar]
- Stumpfe, D.; Hu, H.; Bajorath, J. Evolving Concept of Activity Cliffs. ACS Omega 2019, 4, 14360–14368. [Google Scholar] [CrossRef]
- Maggiora, G.M. On Outliers and Activity CliffsWhy QSAR Often Disappoints. J. Chem. Inf. Model. 2006, 46, 1535. [Google Scholar] [CrossRef]
- Bajorath, J. Duality of activity cliffs in drug discovery. Expert Opin. Drug Discov. 2019, 14, 517–520. [Google Scholar] [CrossRef]
- Guha, R.; Van Drie, J.H. Assessing How Well a Modeling Protocol Captures a Structure−Activity Landscape. J. Chem. Inf. Model. 2008, 48, 1716–1728. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Von Korff, M.; Freyss, J.; Sander, T. Flexophore, a New Versatile 3D Pharmacophore Descriptor That Considers Molecular Flexibility. J. Chem. Inf. Model. 2008, 48, 797–810. [Google Scholar] [CrossRef]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–112. [Google Scholar] [CrossRef]
- Hu, Y.; Lounkine, E.; Bajorath, J. Improving the Search Performance of Extended Connectivity Fingerprints through Activity-Oriented Feature Filtering and Application of a Bit-Density-Dependent Similarity Function. Chem. Med. Chem. 2009, 4, 540–548. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Glide. Schrödinger Release 2018-4; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Maestro. Schrödinger Release 2018-4; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Prime. Schrödinger Release 2018-4; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Kumar, S.; Nair, A.S.; Bhashkar, V.; Sudevan, S.T.; Koyiparambath, V.P.; Khames, A.; Abdelgawad, M.A.; Mathew, B. Navigating into the Chemical Space of Monoamine Oxidase Inhibitors by Artificial Intelligence and Cheminformatics Approach. ACS Omega 2021, 6, 23399–23411. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Model for Pharm-1 | ||||
|---|---|---|---|---|
| Statistical parameters | #1 | #2 | #3 | #4 |
| % of molecules in the training set | 70 | 70 | 70 | 70 |
| % of molecules in the test set | 30 | 30 | 30 | 30 |
| Training set, r2 | 0.592 | 0.794 | 0.851 | 0.900 |
| Test set, q2 | 0.624 | 0.665 | 0.777 | 0.774 |
| Pearson correlation coefficient (Pearson-R) | 0.807 | 0.835 | 0.902 | 0.884 |
| Stability | 0.898 | 0.811 | 0.772 | 0.736 |
| Standard deviation (SD) | 0.752 | 0.537 | 0.461 | 0.381 |
| Variance ratio (F-value) | 113.200 | 148.700 | 144.500 | 167.200 |
| Significance level of variance ratio (p-value) | 7.57 × 10−17 | 3.61 × 10−27 | 2.55 × 10−31 | 1.49 × 10−36 |
| Pharm-1 | CCCtr | CCCtest | Q2F1 | Q2F2 | Q2F3 | R2pred | RMSEtr | MAEtr | RMSEtest | MAEtest |
|---|---|---|---|---|---|---|---|---|---|---|
| M1 | 0.947 | 0.869 | 0.774 | 0.774 | 0.794 | 0.861 | 0.369 | 0.275 | 0.527 | 0.456 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacureanu, L.; Bora, A.; Crisan, L. New Insights on the Activity and Selectivity of MAO-B Inhibitors through In Silico Methods. Int. J. Mol. Sci. 2023, 24, 9583. https://doi.org/10.3390/ijms24119583
Pacureanu L, Bora A, Crisan L. New Insights on the Activity and Selectivity of MAO-B Inhibitors through In Silico Methods. International Journal of Molecular Sciences. 2023; 24(11):9583. https://doi.org/10.3390/ijms24119583
Chicago/Turabian StylePacureanu, Liliana, Alina Bora, and Luminita Crisan. 2023. "New Insights on the Activity and Selectivity of MAO-B Inhibitors through In Silico Methods" International Journal of Molecular Sciences 24, no. 11: 9583. https://doi.org/10.3390/ijms24119583
APA StylePacureanu, L., Bora, A., & Crisan, L. (2023). New Insights on the Activity and Selectivity of MAO-B Inhibitors through In Silico Methods. International Journal of Molecular Sciences, 24(11), 9583. https://doi.org/10.3390/ijms24119583