
Functional Characterisation of the Rare SCN5A p.E1225K Variant, Segregating in a Brugada Syndrome Familial Case, in Human Cardiomyocytes from Pluripotent Stem Cells
 ,
,  , ,
, ,  , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Case Description
2.1. Clinical Evaluation
2.2. Genetic Investigation
2.3. Generation of pLenti-EF1alpha-SCN5A-GFP p.E1225D Lentiviral Vector
2.4. Lentiviral Particles Production and Cardiomyocytes Transduction
2.5. Human Pluripotent Stem Cells Maintenance and Differentiation
2.6. Functional Characterization: Assessment of INa Currents by Patch-Clamp
2.7. Clinical Case
2.7.1. Clinical Features
2.7.2. Genetic Testing Result
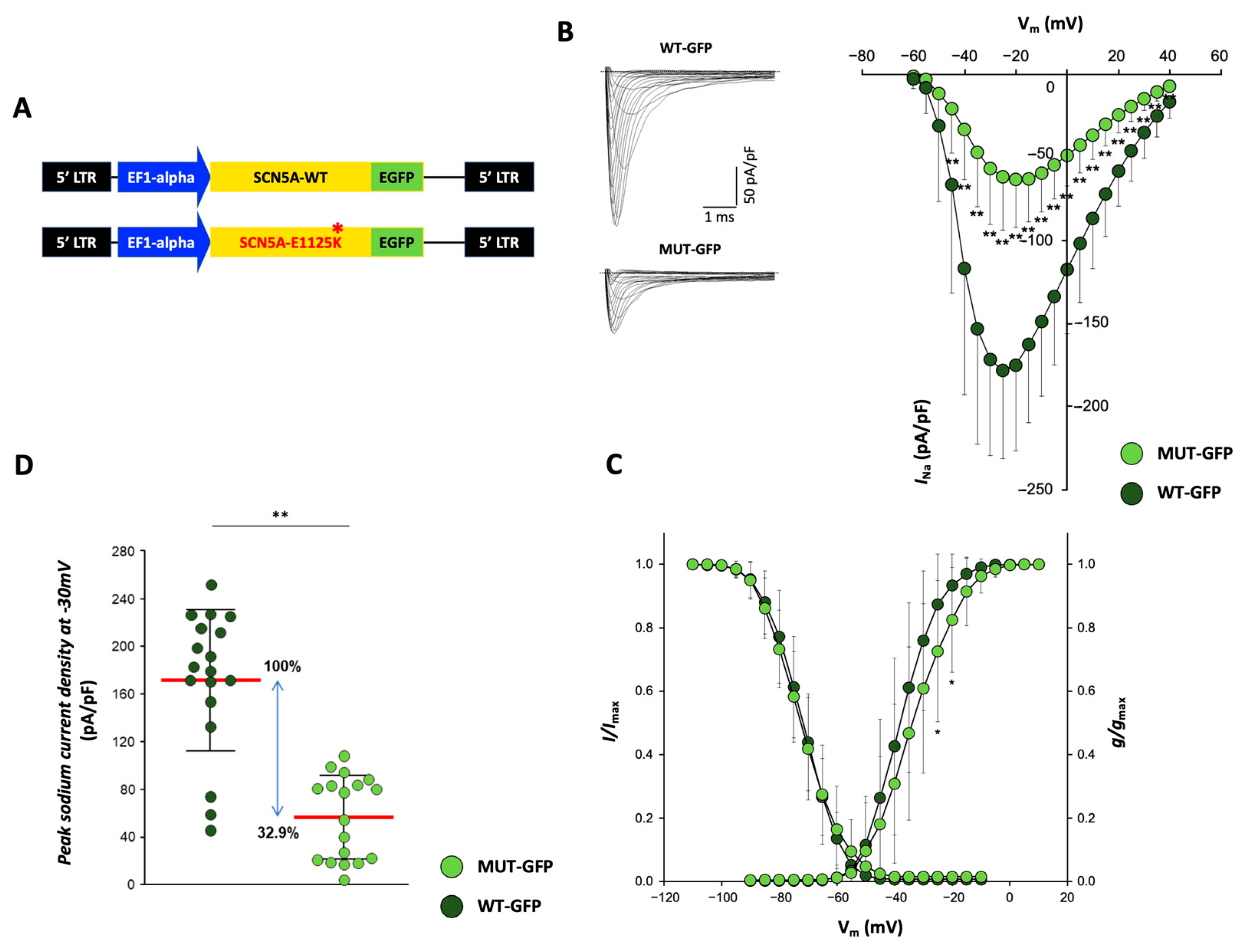
2.7.3. Functional Testing Indicates Reduced Peak Sodium Currents in CMs Overexpressing p.E1225K Nav1.5 Channel
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Di Resta, C.; Berg, J.; Villatore, A.; Maia, M.; Pili, G.; Fioravanti, F.; Tomaiuolo, R.; Sala, S.; Benedetti, S.; Peretto, G. Concealed Substrates in Brugada Syndrome: Isolated Channelopathy or Associated Cardiomyopathy? Genes 2022, 13, 1755. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick Eduardo, B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. J. Arrhythmia 2022, 38, 491–553. [Google Scholar] [CrossRef] [PubMed]
- Wijeyeratne, Y.D.; Tanck, M.W.; Mizusawa, Y.; Batchvarov, V.; Barc, J.; Crotti, L.; Bos, J.M.; Tester, D.J.; Muir, A.; Veltmann, C.; et al. SCN5A Mutation Type and a Genetic Risk Score Associate Variably With Brugada Syndrome Phenotype in SCN5A Families. Circ. Genom. Precis. Med. 2020, 13, E002911. [Google Scholar] [CrossRef]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Sacilotto, L.; Scanavacca, M.I.; Olivetti, N.; Lemes, C.; Pessente, G.D.; Wulkan, F.; Hachul, D.T.; Krieger, J.E.; Pereira, A.C.; Darrieux, F.C.C. Low rate of life-threatening events and limitations in predicting invasive and noninvasive markers of symptoms in a cohort of type 1 Brugada syndrome patients: Data and insights from the GenBra registry. J. Cardiovasc. Electrophysiol. 2020, 31, 2920–2928. [Google Scholar] [CrossRef] [PubMed]
- Probst, V.; Allouis, M.; Sacher, F.; Pattier, S.; Babuty, D.; Mabo, P.; Mansourati, J.; Victor, J.; Nguyen, J.M.; Schott, J.J.; et al. Progressive cardiac conduction defect is the prevailing phenotype in carriers of a Brugada syndrome SCN5A mutation. J. Cardiovasc. Electrophysiol. 2006, 17, 270–275. [Google Scholar] [CrossRef]
- Sommariva, E.; Pappone, C.; Martinelli Boneschi, F.; Di Resta, C.; Rosaria Carbone, M.; Salvi, E.; Vergara, P.; Sala, S.; Cusi, D.; Ferrari, M.; et al. Genetics can contribute to the prognosis of Brugada syndrome: A pilot model for risk stratification. Eur. J. Hum. Genet. EJHG 2013, 21, 911–917. [Google Scholar] [CrossRef]
- Kalarus, Z.; Mairesse, G.H.; Sokal, A.; Boriani, G.; Średniawa, B.; Arroyo, R.C.; Wachter, R.; Frommeyer, G.; Traykov, V.; Dagres, N.; et al. Searching for atrial fibrillation: Looking harder, looking longer, and in increasingly sophisticated ways. An EHRA position paper. Eurospace 2022, 25, 185–198. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, S.; Di Resta, C.; Rocchetti, M.; Barile, L.; Rizzetto, R.; Summa, A.; Severi, S.; Sommariva, E.; Pappone, C.; Ferrari, M.; et al. A Brugada syndrome mutation (p.S216L) and its modulation by p.H558R polymorphism: Standard and dynamic characterization. Cardiovasc. Res. 2011, 91, 606–616. [Google Scholar] [CrossRef]
- My, I.; Di Pasquale, E. Genetic Cardiomyopathies: The Lesson Learned from hiPSCs. J. Clin. Med. 2021, 10, 1149. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Morone, D.; Denegri, M.; Bongianino, R.; Nakahama, H.; Rutigliano, L.; Gosetti, R.; Rizzo, G.; Vollero, A.; Buonocore, M.; et al. Adeno-associated virus-mediated CASQ2 delivery rescues phenotypic alterations in a patient-specific model of recessive catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2016, 7, e2393. [Google Scholar] [CrossRef]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 2267. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, E000043. [Google Scholar] [CrossRef]
- Davis, R.P.; Casini, S.; Van Den Berg, C.W.; Hoekstra, M.; Remme, C.A.; Dambrot, C.; Salvatori, D.; Van Oostwaard, D.W.; Wilde, A.A.M.; Bezzina, C.R.; et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 2012, 125, 3079–3091. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Latronico, M.V.G.; Jotti, G.S.; Condorelli, G. Lentiviral vectors and cardiovascular diseases: A genetic tool for manipulating cardiomyocyte differentiation and function. Gene Ther. 2012, 19, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Glazer, A.M.; Wada, Y.; Li, B.; Muhammad, A.; Kalash, O.R.; O’Neill, M.J.; Shields, T.; Hall, L.; Short, L.; Blair, M.A.; et al. High-Throughput Reclassification of SCN5A Variants. Am. J. Hum. Genet. 2020, 107, 111–123. [Google Scholar] [CrossRef]
- Teekakirikul, P.; Kelly, M.A.; Rehm, H.L.; Lakdawala, N.K.; Funke, B.H. Inherited cardiomyopathies: Molecular genetics and clinical genetic testing in the postgenomic era. J. Mol. Diagn. 2013, 15, 158–170. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Peretto, G.; Sala, S.; Basso, C.; Della Bella, P. Programmed ventricular stimulation in patients with active vs previous arrhythmic myocarditis. J. Cardiovasc. Electrophysiol. 2020, 31, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Pappone, C.; Brugada, J.; Vicedomini, G.; Ciconte, G.; Manguso, F.; Saviano, M.; Vitale, R.; Cuko, A.; Giannelli, L.; Calovic, Z.; et al. Electrical Substrate Elimination in 135 Consecutive Patients With Brugada Syndrome. Circulation 2017, 10, e005053. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Sala, S.; Basso, C.; Rizzo, S.; Radinovic, A.; Frontera, A.; Limite, L.R.; Paglino, G.; Bisceglia, C.; De Luca, G.; et al. Inflammation as a Predictor of Recurrent Ventricular Tachycardia After Ablation in Patients With Myocarditis. J. Am. Coll. Cardiol. 2020, 76, 1644–1656. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Kim, Y.N.; Lee, Y.S.; Jung, B.C.; Lee, S.H.; Shin, D.G.; Cho, Y.; Bae, M.H.; Han, S.M.; Lee, M.H. Genetic Analysis of SCN5A in Korean Patients Associated with Atrioventricular Conduction Block. Genom. Inform. 2012, 10, 110. [Google Scholar] [CrossRef]
- GnomAD. Available online: https://gnomad.broadinstitute.org/about (accessed on 1 March 2023).
- NCBI. Available online: https://www.ncbi.nlm.nih.gov (accessed on 1 March 2023).
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef]
- Rosales Gerpe, M.C.; Van Lieshout, L.P.; Domm, J.M.; Van Vloten, J.P.; Datu, J.; Ingrao, J.C.; Yu, D.L.; De Jong, J.; Moraes, T.J.; Krell, P.J.; et al. Optimized Pre-Clinical Grade Production of Two Novel Lentiviral Vector Pseudotypes for Lung Gene Delivery. Hum. Gene Ther. 2020, 31, 459–471. [Google Scholar] [CrossRef]
- Nakahama, H.; Di Pasquale, E. Generation of Cardiomyocytes from Pluripotent Stem Cells. Methods Mol. Biol. 2016, 1353, 181–190. [Google Scholar] [CrossRef]
- Schulze-Bahr, E.; Eckardt, L.; Breithardt, G.; Seidl, K.; Wichter, T.; Wolpert, C.; Borggrefe, M.; Haverkamp, W. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: Different incidences in familial and sporadic disease. Hum. Mutat. 2003, 21, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflug. Arch. Eur. J. Physiol. 1981, 391, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Tensegrity-based mechanosensing from macro to micro. Prog. Biophys. Mol. Biol. 2008, 97, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2006–H2017. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Yan, G.X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Eurospace 2017, 19, 665–694. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kimoto, H.; Mishima, H.; Yamagata, K.; Ogata, S.; Aizawa, Y.; Hayashi, K.; Morita, H.; Nakajima, T.; Nakano, Y.; et al. Functionally validated SCN5A variants allow interpretation of pathogenicity and prediction of lethal events in Brugada syndrome. Eur. Heart J. 2021, 42, 2854–2863. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Muhammad, A.; Li, B.; Wada, Y.; Hall, L.; Solus, J.F.; Short, L.; Roden, D.M.; Glazer, A.M. Dominant negative effects of SCN5A missense variants. Genet. Med. 2022, 24, 1238–1248. [Google Scholar] [CrossRef]
- Kroncke, B.M.; Smith, D.K.; Zuo, Y.; Glazer, A.M.; Roden, D.M.; Blume, J.D. A Bayesian method to estimate variant-induced disease penetrance. PLoS Genet. 2020, 16, e1008862. [Google Scholar] [CrossRef]
- Kroncke, B.M.; Glazer, A.M.; Smith, D.K.; Blume, J.D.; Roden, D.M. SCN5A (NaV1.5) Variant Functional Perturbation and Clinical Presentation: Variants of a Certain Significance. Circ. Genom. Precis. Med. 2018, 11, e002095. [Google Scholar] [CrossRef]
- Pearman, C.M.; Denham, N.C.; Mills, R.W.; Ding, W.Y.; Modi, S.S.; Hall, M.C.S.; Todd, D.M.; Mahida, S. Relationship between sodium channel function and clinical phenotype in SCN5A variants associated with Brugada syndrome. Hum. Mutat. 2020, 41, 2195–2204. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Rook, M.B.; Groenewegen, W.A.; Herfst, L.J.; Van der Wal, A.C.; Lam, J.; Jongsma, H.J.; Wilde, A.A.M.; Mannens, M.M.A.M. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ. Res. 2003, 92, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Veltmann, C.; Barajas-Martinez, H.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Pfeiffer, R.; Cáceres, G.; Burashnikov, E.; Antzelevitch, C.; Hu, D. Further Insights in the Most Common SCN5A Mutation Causing Overlapping Phenotype of Long QT Syndrome, Brugada Syndrome, and Conduction Defect. J. Am. Heart Assoc. 2016, 5, e003379. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lang, S.; Akin, I.; Zhou, X.; El-Battrawy, I. Brugada Syndrome: Different Experimental Models and the Role of Human Cardiomyocytes From Induced Pluripotent Stem Cells. J. Am. Heart Assoc. 2022, 11, e024410. [Google Scholar] [CrossRef]
- de la Roche, J.; Angsutararux, P.; Kempf, H.; Janan, M.; Bolesani, E.; Thiemann, S.; Wojciechowski, D.; Coffee, M.; Franke, A.; Schwanke, K.; et al. Comparing human iPSC-cardiomyocytes versus HEK293T cells unveils disease-causing effects of Brugada mutation A735V of NaV1.5 sodium channels. Sci. Rep. 2019, 9, 11173. [Google Scholar] [CrossRef] [PubMed]
- Stazi, F.; Battisti, P. When Brugada syndrome is at risk of sudden death: Clinical and anatomical aspects. Eur. Heart J. Suppl. 2022, 24, I165–I169. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Vicedomini, G.; Locati, E.T.; Ciconte, G.; Giannelli, L.; Giordano, F.; Crisà, S.; Vecchi, M.; Borrelli, V.; et al. Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants. Europace 2019, 21, 1550–1558. [Google Scholar] [CrossRef]
- Milman, A.; Gourraud, J.B.; Andorin, A.; Postema, P.G.; Sacher, F.; Mabo, P.; Conte, G.; Giustetto, C.; Sarquella-Brugada, G.; Hochstadt, A.; et al. Gender differences in patients with Brugada syndrome and arrhythmic events: Data from a survey on arrhythmic events in 678 patients. Heart Rhythm 2018, 15, 1457–1465. [Google Scholar] [CrossRef]
- Di Resta, C.; Becchetti, A. Introduction to ion channels. Adv. Exp. Med. Biol. 2010, 674, 9–21. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Barc, J.; Tadros, R.; Glinge, C.; Chiang, D.Y.; Jouni, M.; Simonet, F.; Jurgens, S.J.; Baudic, M.; Nicastro, M.; Potet, F.; et al. Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility. Nat. Genet. 2022, 54, 232–239. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvarani, N.; Peretto, G.; Silvia, C.; Villatore, A.; Thairi, C.; Santoni, A.; Galli, C.; Carrera, P.; Sala, S.; Benedetti, S.; et al. Functional Characterisation of the Rare SCN5A p.E1225K Variant, Segregating in a Brugada Syndrome Familial Case, in Human Cardiomyocytes from Pluripotent Stem Cells. Int. J. Mol. Sci. 2023, 24, 9548. https://doi.org/10.3390/ijms24119548
Salvarani N, Peretto G, Silvia C, Villatore A, Thairi C, Santoni A, Galli C, Carrera P, Sala S, Benedetti S, et al. Functional Characterisation of the Rare SCN5A p.E1225K Variant, Segregating in a Brugada Syndrome Familial Case, in Human Cardiomyocytes from Pluripotent Stem Cells. International Journal of Molecular Sciences. 2023; 24(11):9548. https://doi.org/10.3390/ijms24119548
Chicago/Turabian StyleSalvarani, Nicolò, Giovanni Peretto, Crasto Silvia, Andrea Villatore, Cecilia Thairi, Anna Santoni, Camilla Galli, Paola Carrera, Simone Sala, Sara Benedetti, and et al. 2023. "Functional Characterisation of the Rare SCN5A p.E1225K Variant, Segregating in a Brugada Syndrome Familial Case, in Human Cardiomyocytes from Pluripotent Stem Cells" International Journal of Molecular Sciences 24, no. 11: 9548. https://doi.org/10.3390/ijms24119548
APA StyleSalvarani, N., Peretto, G., Silvia, C., Villatore, A., Thairi, C., Santoni, A., Galli, C., Carrera, P., Sala, S., Benedetti, S., Di Pasquale, E., & Di Resta, C. (2023). Functional Characterisation of the Rare SCN5A p.E1225K Variant, Segregating in a Brugada Syndrome Familial Case, in Human Cardiomyocytes from Pluripotent Stem Cells. International Journal of Molecular Sciences, 24(11), 9548. https://doi.org/10.3390/ijms24119548