
Reversal of Tau-Dependent Cognitive Decay by Blocking Adenosine A1 Receptors: Comparison of Transgenic Mouse Models with Different Levels of Tauopathy


 ,
,
Abstract
1. Introduction
2. Results
2.1. Adenosine A1 Receptor Binding of [18F]CPFPX Is Reduced by Rolofylline Treatment
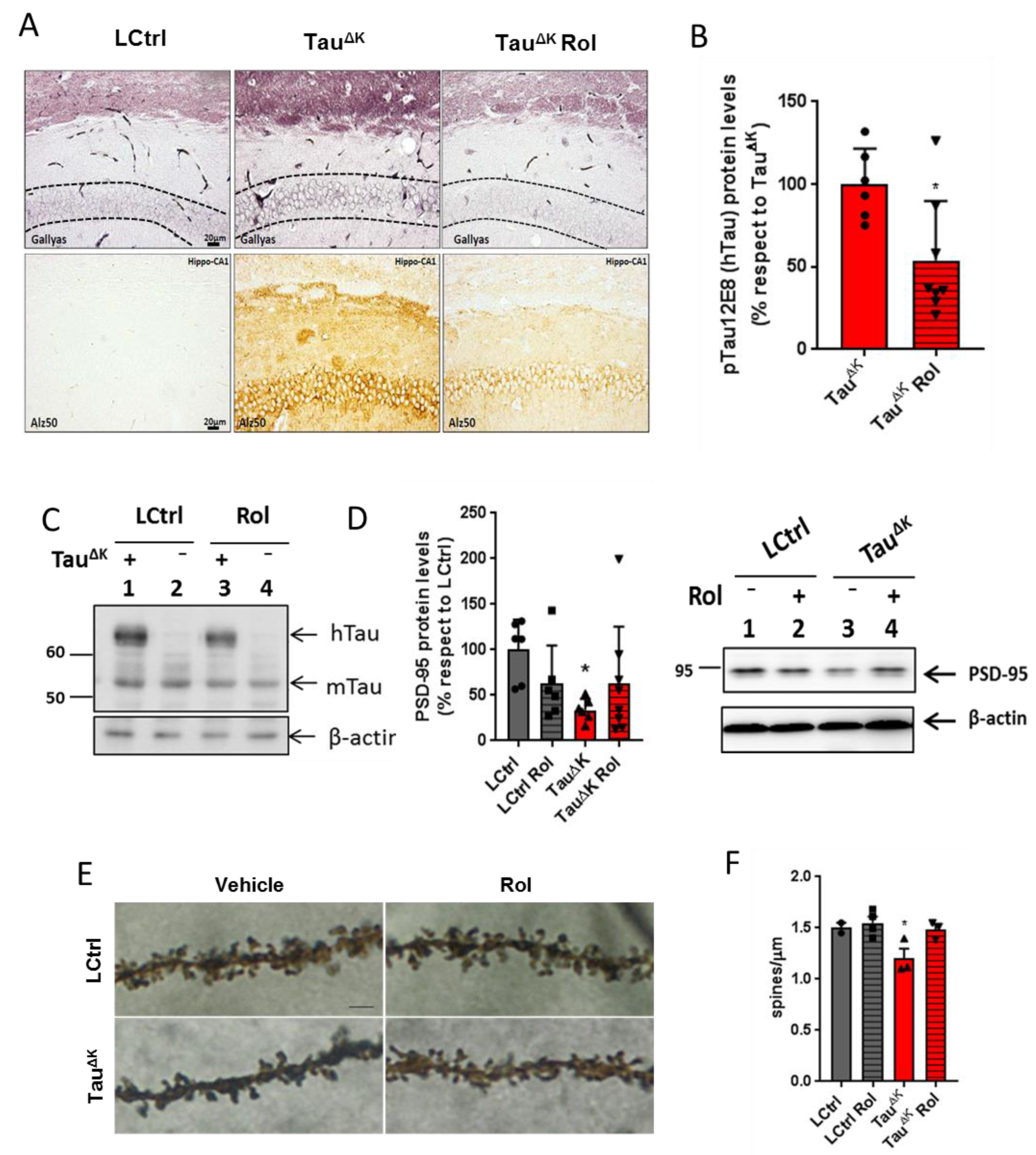
2.2. Decreased Tau Accumulation and Synaptopathology after Blockade of Adenosine A1 Receptors
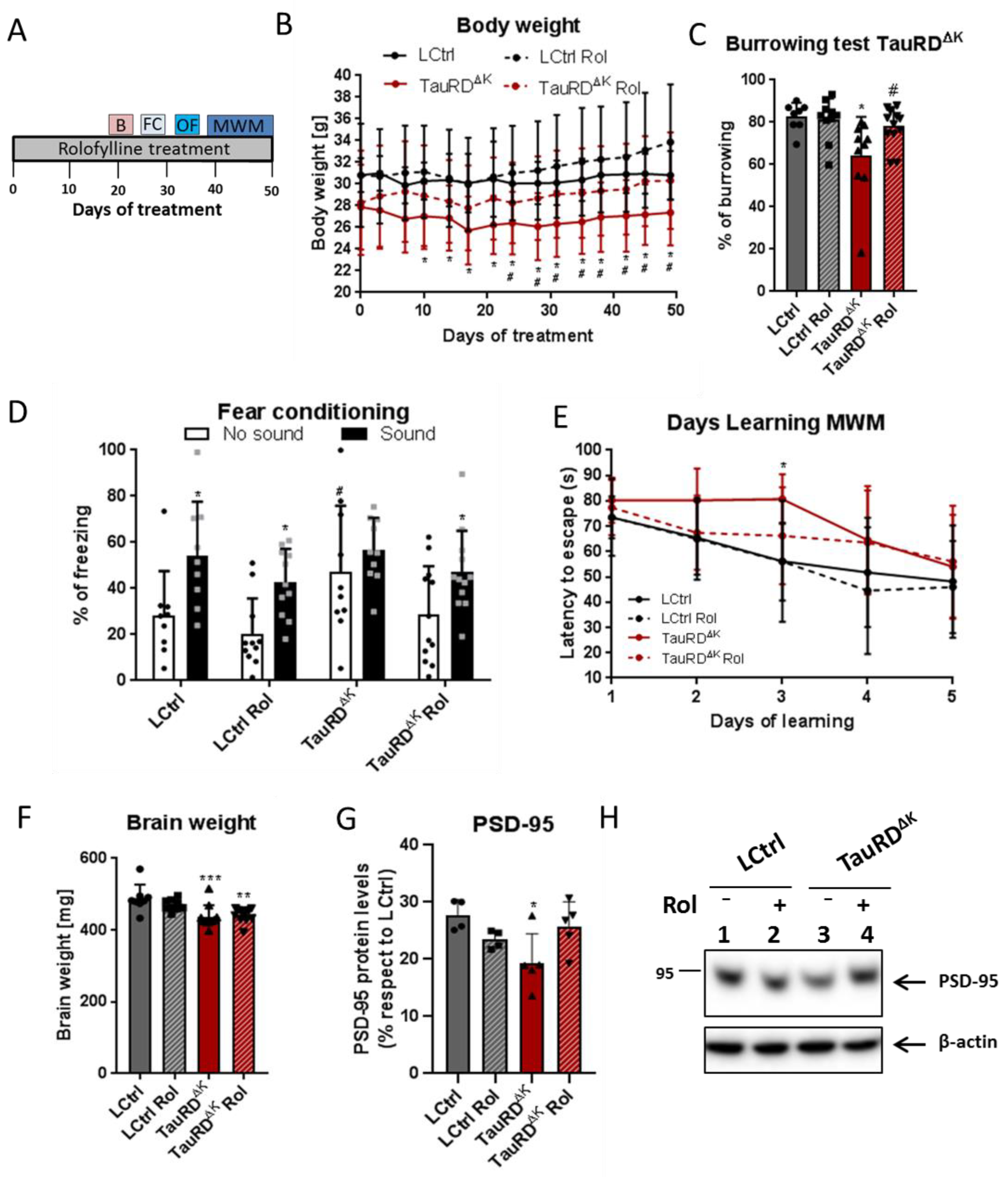
2.3. Rolofylline Decreases Cognitive Deficits and Tauopathy in Mice Expressing Tau-Repeat Domain (TauRDΔK)
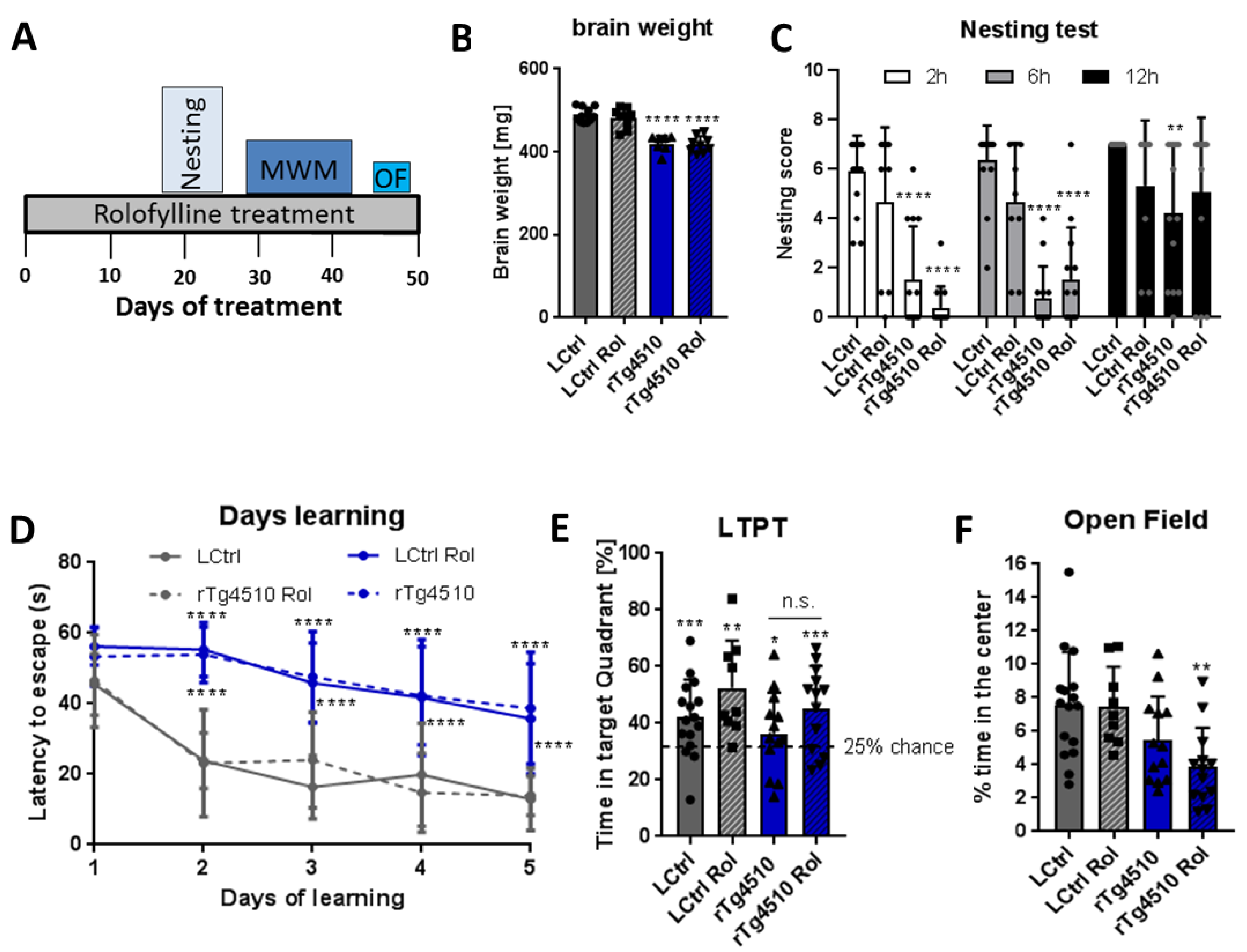
2.4. Rolofylline Does Not Improve Behavior in rTg4510 Mice Strongly Overexpressing Mutant TauP301L
3. Discussion
3.1. Reversal of Tau Pathology in Mouse Models with Near-Physiological Expression of Tau Mutants
3.2. No Reversal of Tau Pathology in Mouse Models with High Non-Physiological Expression of Tau Mutants
3.3. Synaptic Effects vs. Rolofylline Treatment
4. Materials and Methods
4.1. Animals and Rolofylline Treatment
4.2. Histology and Golgi Staining
4.3. Biochemical Analysis of Brain Tissue
4.4. Behavioral Tests
4.5. Positron Emission Tomography (PET)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Treatment | Preventive Treatment | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LCtrl | LCtrl Rol | Tau∆K | Tau∆K Rol | LCtrl | LCtrl Rol | TauRD∆K | TauRD∆K Rol | LCtrl | LCtrl Rol | rTg4510 | rTg4510 Rol | LCtrl | LCtrl Rol | rTg4510 | rTg4510 Rol | ||
| Behavior experiments | Y-maze | - | - | ↓* | ↑* | N/A | N/A | N/A | N/A | (n = 14) | (n = 14) | ↓ (n = 9) | ↓ (n = 9) | N/A | N/A | N/A | N/A |
| NORT | - | - | ↓* | ↑* | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| FC | - | - | ↓* | ↑* | (n = 9) | (n = 11) | ↓ (n = 10) | ↑ (n = 12) | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| Body weight | (n = 7) | (n = 9) | ´=(n = 9) | ´=(n = 8) | (n = 9) | (n = 11) | ↓ (n = 10) | ↑ (n = 13) | (n = 12) | (n = 14) | ↓ (n = 10) | ↓ (n = 10) | N/A | N/A | N/A | N/A | |
| Burrowing test | N/A | N/A | N/A | N/A | (n = 9) | (n = 10) | ↓ (n = 10) | ↑ (n = 13) | (n = 14) | (n = 14) | ↓ (n = 9) | ↓ (n = 10) | N/A | N/A | N/A | N/A | |
| Learning MWM | N/A | N/A | N/A | N/A | (n = 9) | (n = 11) | ↓ (n = 10) | ↑ (n = 13) | N/A | N/A | N/A | N/A | (n = 16) | (n = 9) | ↓ (n = 13) | ↓ (n = 13) | |
| LTPT MWM | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | (n = 16) | (n = 9) | ´=(n = 13) | ´=(n = 13) | |
| Nesting test | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | (n = 14) | (n = 14) | ↓ (n = 9) | ↓↓ (n = 9) | (n = 16) | (n = 9) | ↓ (n = 13) | ↓ (n = 13) (12 h not different than LCtrl) | |
| OF | N/A | N/A | N/A | N/A | (n = 9) | (n = 11) | ´=(n = 10) | ´=(n = 13) | (n = 14) | (n = 14) | ´=(n = 9) | ↑ (n = 9) (hyperactivity) | (n = 16) | (n = 9) | ´=(n = 13) | ´=(n = 13) | |
| OF time in center | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | (n = 16) | (n = 9) | ´=(n = 13) | ↓ (n = 13) (anxiety) | |
| Taupathology | Gallyas silver staining | (n =4) | (n = 4) | ++ (n = 4) | + (n = 4) | (n = 4) | (n = 4) | ++ (n = 4) | + (n = 4) | (n = 4) | (n = 4) | ++ (n = 4) | ++ (n = 4) | N/A | N/A | N/A | N/A |
| pTau12E8 | N/A | N/A | ↑ (n = 6) | ↓ (n = 8) | (n = 4) | (n = 4) | ++ (n = 4) | + (n = 4) | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| Alz50 | (n = 4) | (n = 4) | ↑ (n = 4) | ↓ (n = 4) | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| PHF1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | (n = 4) | (n = 4) | ++ (n = 4) | ++ (n = 4) | N/A | N/A | N/A | N/A | |
| MC1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | (n = 4) | (n = 4) | ++ (n = 4) | ++ (n = 4) | N/A | N/A | N/A | N/A | |
| Synaptic markers | PSD-95 | (n = 6) | (n = 6) | ↓ (n = 6) | ↑ (n = 8) | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Spines | (n = 2) | (n = 4) | ↓ (n = 3) | ↑ (n = 3) | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | |
| Brain weight/ HIP Volume | (n = 5) | (n = 5) | ´=(n = 6) | ´=(n = 6) | (n = 9) | (n = 10) | ↓ (n = 10) | ↓ (n = 12) | (n = 13) | (n = 13) | ↓ (n = 8) | ↓ (n = 9) | (n = 12) | (n = 9) | ↓ (n = 7) | ↓ (n = 10) | |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Tau 2N4R | Largest tau isoform in human CNS, 441 residues |
| [18F]CPFPX | (18)F-8-cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine |
| rolofylline | KW-3902, 1,3-dipropyl-8-(3-noradamantyl)xanthine, 8-(Hexahydro-2,5-methanopentalen-3a(1H)-yl)-3,7-dihydro-1,3-dipropyl-1H-purine-2,6-dione |
| VOI | Volume of interest |
| TauΔK | Full-length tau (2N4R) with mutation ΔK280 |
| TauRDΔK | Tau repeat domain (4R) with mutation ΔK280 |
| AD | Alzheimer’s disease |
| FTLD | Frontotemporal lobar degeneration |
| CMRglu | Cerebral metabolic rate of glucose |
| CaMKIIα | Ca2+/Calmodulin-dependent protein kinase type II |
| PHF | Paired helical filament |
| GFAP | Glial fibrillary acidic protein |
| Iba 1 | Ionized calcium-binding adapter molecule 1 |
| DAB | 3,3′-diaminobenzidine |
| NeuN | Neuronal nuclear protein |
| WT | Wild type |
| MB | Methylene blue |
| MWM | Morris water maze |
| NORT | Novel object recognition test |
| PET | Positron emission tomography |
| PSD-95 | Post-synaptic density protein-95 |
| CA1 | Hippocampal area Cornu Ammonis 1 |
| LCtrl | Littermate controls |
References
- Selkoe, D.; Mandelkow, E.; Holtzman, D. Deciphering Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011460. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, S.G.; Prinelli, F.; Adorni, F.; Caltagirone, C.; Musicco, M. A meta-analysis of the efficacy of donepezil, rivastigmine, galantamine, and memantine in relation to severity of Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 35, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef]
- Takashima, A. Tauopathies and tau oligomers. J. Alzheimer’s Dis. 2013, 37, 565–568. [Google Scholar] [CrossRef]
- Gotz, J.J.; Gotz, J. Experimental models of tauopathy—From mechanisms to therapies. Adv. Exp. Med. Biol. 2019, 1184, 381–391. [Google Scholar]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef]
- SantaCruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef]
- Sydow, A.; Van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Drexler, D.; Zhou, L.; Rune, G.; Mandelkow, E.; et al. Reversibility of tau-related cognitive defects in a regulatable FTD mouse model. J. Mol. Neurosci. 2011, 45, 432–437. [Google Scholar] [CrossRef]
- Van der Jeugd, A.; Hochgräfe, K.; Ahmed, T.; Decker, J.M.; Sydow, A.; Hofmann, A.; Wu, D.; Messing, L.; Balschun, D.; D’hooge, R.; et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012, 123, 787–805. [Google Scholar] [CrossRef] [PubMed]
- de Leon, M.J.; George, A.E.; Ferris, S.H.; Rosenbloom, S.; Christman, D.R.; Gentes, C.I.; Reisberg, B.; Kricheff, I.I.; Wolf, A.P. Regional correlation of PET and CT in senile dementia of the Alzheimer type. AJNR Am. J. Neuroradiol. 1983, 4, 553–556. [Google Scholar] [PubMed]
- Mosconi, L.; Brys, M.; Glodzik-Sobanska, L.; De Santi, S.; Rusinek, H.; de Leon, M. Early detection of Alzheimer’s disease using neuroimaging. Exp. Gerontol. 2007, 42, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Nacmias, B.; Sorbi, S.; De Cristofaro, M.T.; Fayazz, M.; Tedde, A.; Bracco, L.; Herholz, K.; Pupi, A. Brain metabolic decreases related to the dose of the ApoE e4 allele in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.; Huang, S.C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L.; et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Caselli, R.J.; Chen, K.; Alexander, G.E.; Bandy, D.; Frost, J. Declining brain activity in cognitively normal apolipoprotein E ε4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 3334–3339. [Google Scholar] [CrossRef]
- Costantini, L.C.; Barr, L.J.; Vogel, J.L.; Henderson, S.T. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9 (Suppl. S2), S16. [Google Scholar] [CrossRef]
- Bouter, C.; Bouter, Y. 18F-FDG-PET in Mouse Models of Alzheimer’s Disease. Front Med. 2019, 6, 71. [Google Scholar] [CrossRef]
- Decker, J.M.; Krüger, L.; Sydow, A.; Zhao, S.; Frotscher, M.; Mandelkow, E.; Mandelkow, E.-M. Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca++ dysregulation. Acta Neuropathol. Commun. 2015, 3, 23. [Google Scholar] [CrossRef]
- Dunwiddie, T.V.; Masino, S.A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef]
- Trinh, P.N.H.; Baltos, J.-A.; Hellyer, S.D.; May, L.T.; Gregory, K.J. Adenosine receptor signalling in Alzheimer’s disease. Purinergic Signal. 2022, 18, 359–381. [Google Scholar] [CrossRef] [PubMed]
- Dennissen, F.J.; Anglada-Huguet, M.; Sydow, A.; Mandelkow, E.; Mandelkow, E.M. Adenosine A1 receptor antagonist rolofylline alleviates axonopathy caused by human Tau ΔK280. Proc. Natl. Acad. Sci. USA 2016, 113, 11597–11602. [Google Scholar] [CrossRef]
- Bauer, A.; Holschbach, M.H.; Cremer, M.; Weber, S.; Boy, C.; Shah, N.J.; Olsson, R.A.; Halling, H.; Coenen, H.H.; Zilles, K. Evaluation of 18F-CPFPX, a novel adenosine A1 receptor ligand: In vitro autoradiography and high-resolution small animal PET. J. Nucl. Med. 2003, 44, 1682–1689. [Google Scholar]
- Bauer, A.; Holschbach, M.H.; Meyer, P.T.; Boy, C.; Herzog, H.; Olsson, R.A.; Coenen, H.H.; Zilles, K. In vivo imaging of adenosine A1 receptors in the human brain with [18F]CPFPX and positron emission tomography. Neuroimage 2003, 19, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.M.; Nissen, A.; Eckermann, K.; Khlistunova, I.; Biernat, J.; Drexler, D.; Petrova, O.; Schonig, K.; Bujard, H.; Mandelkow, E.; et al. The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J. Neurosci. 2008, 28, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Hochgrafe, K.; Sydow, A.; Mandelkow, E.M. Regulatable transgenic mouse models of Alzheimer disease: Onset, reversibility and spreading of Tau pathology. FEBS J. 2013, 280, 4371–4381. [Google Scholar] [CrossRef]
- Joseph, M.; Anglada-Huguet, M.; Paesler, K.; Mandelkow, E.; Mandelkow, E.-M. Anti-aggregant tau mutant promotes neurogenesis. Mol. Neurodegener. 2017, 12, 88. [Google Scholar] [CrossRef]
- Sydow, A.; Van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Mandelkow, E.M. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 2011, 31, 2511–2525. [Google Scholar] [CrossRef]
- Deacon, R. Assessing burrowing, nest construction, and hoarding in mice. J. Vis. Exp. 2012, e2607. [Google Scholar] [CrossRef]
- Pickhardt, M.; Tassoni, M.; Denner, P.; Kurkowsky, B.; Kitanovic, A.; Möhl, C.; Fava, E.; Mandelkow, E. Screening of a neuronal cell model of tau pathology for therapeutic compounds. Neurobiol. Aging 2019, 76, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Schafer, K.N.; Cisek, K.; Huseby, C.J.; Chang, E.; Kuret, J. Structural determinants of tau aggregation inhibitor potency. J. Biol. Chem. 2013, 288, 32599–32611. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Gamache, J.; Benzow, K.; Forster, C.; Kemper, L.; Hlynialuk, C.; Furrow, E.; Ashe, K.H.; Koob, M.D. Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat. Commun. 2019, 10, 2479. [Google Scholar] [CrossRef]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Mandelkow, E.-M.; Mandelkow, E. Biochemistry and cell biology of Tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Eckermann, K.; Mocanu, M.M.; Khlistunova, I.; Biernat, J.; Nissen, A.; Hofmann, A.; Schonig, K.; Bujard, H.; Haemisch, A.; Mandelkow, E.; et al. The β-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J. Biol. Chem. 2007, 282, 31755–31765. [Google Scholar] [CrossRef] [PubMed]
- Messing, L.; Decker, J.M.; Joseph, M.; Mandelkow, E.; Mandelkow, E.-M. Cascade of tau toxicity in inducible hippocampal brain slices and prevention by aggregation inhibitors. Neurobiol. Aging 2013, 34, 1343–1354. [Google Scholar] [CrossRef]
- Thompson, S.M.; Haas, H.L.; Gahwiler, B.H. Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro. J. Physiol. 1992, 451, 347–363. [Google Scholar] [CrossRef]
- Ochiishi, T.; Chen, L.; Yukawa, A.; Saitoh, Y.; Sekino, Y.; Arai, T.; Nakata, H.; Miyamoto, H. Cellular localization of adenosine A1 receptors in rat forebrain: Immunohistochemical analysis using adenosine A1 receptor-specific monoclonal antibody. J. Comp. Neurol. 1999, 411, 301–316. [Google Scholar] [CrossRef]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A receptors in the brain: Current research and their role in neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef]
- Massie, B.M.; O’Connor, C.M.; Metra, M.; Ponikowski, P.; Teerlink, J.R.; Cotter, G.; Weatherley, B.D.; Cleland, J.G.; Givertz, M.M.; Voors, A.; et al. Dittrich, protect investigators, and committees. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N. Engl. J. Med. 2010, 363, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The relationship between Amyloid and Tau. J. Mol. Neurosci. 2003, 20, 203–206. [Google Scholar] [CrossRef]
- Dudai, Y. The neurobiology of consolidations, or, how stable is the engram? Annu. Rev. Psychol. 2004, 55, 51–86. [Google Scholar] [CrossRef] [PubMed]
- Curzon, P.; Rustay, N.R.; Browman, K.E. Cued and contextual fear conditioning for rodents. In Methods of Behavior Analysis in Neuroscience; Buccafusco, J.J., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009. [Google Scholar]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef] [PubMed]
- McInnes, J.; Wierda, K.; Snellinx, A.; Bounti, L.; Wang, Y.-C.; Stancu, I.-C.; Apóstolo, N.; Gevaert, K.; Dewachter, I.; Spires-Jones, T.L.; et al. Synaptogyrin-3 mediates presynaptic dysfunction induced by Tau. Neuron 2018, 97, 823–835.e8. [Google Scholar] [CrossRef]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Attwell, D. Brain’s immune cells put the brakes on neurons. Nature 2020, 586, 366–367. [Google Scholar] [CrossRef]
- Carnemolla, S.E.; Kumfor, F.; Liang, C.T.; Foxe, D.; Ahmed, R.M.; Piguet, O. Olfactory bulb integrity in frontotemporal dementia and Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 89, 51–66. [Google Scholar] [CrossRef]
- Murphy, C. Olfactory and other sensory impairments in Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 11–24. [Google Scholar] [CrossRef]
- Attems, J.; Walker, L.; Jellinger, K.A. Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathol. 2014, 127, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, T.; Meftah, S.; Murray, T.K.; Craig, P.J.; Blockeel, A.; Phillips, K.; Eastwood, B.; O’neill, M.J.; Marston, H.; Ahmed, Z.; et al. Tracking progressive pathological and functional decline in the rTg4510 mouse model of tauopathy. Alzheimer’s Res. Ther. 2017, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- Tracy, T.E.; Madero-Pérez, J.; Swaney, D.L.; Chang, T.S.; Moritz, M.; Konrad, C.; Ward, M.E.; Stevenson, E.; Hüttenhain, R.; Kauwe, G.; et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell 2022, 185, 712–728.e14. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Tampellini, D.; Rahman, N.; Gallo, E.F.; Huang, Z.; Dumont, M.; Capetillo-Zarate, E.; Ma, T.; Zheng, R.; Lu, B.; Nanus, D.M.; et al. Synaptic activity reduces intraneuronal Aβ, promotes APP transport to synapses, and protects against Aβ-related synaptic alterations. J. Neurosci. 2009, 29, 9704–9713. [Google Scholar] [CrossRef]
- Kremer, A.; Maurin, H.; Demedts, D.; Devijver, H.; Borghgraef, P.; Van Leuven, F.; Takata, N.; Mishima, T.; Hisatsune, C.; Nagai, T.; et al. Early improved and late defective cognition is reflected by dendritic spines in Tau.P301L Mice. J. Neurosci. 2011, 31, 18036–18047. [Google Scholar] [CrossRef]
- Moolman, D.L.; Vitolo, O.V.; Vonsattel, J.-P.G.; Shelanski, M.L. Dendrite and dendritic spine alterations in Alzheimer models. J. Neurocytol. 2004, 33, 377–387. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Marco, S.; Pérez-Otaño, I.; García-Osta, A. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 2012, 22, 1040–1050. [Google Scholar] [CrossRef]
- Xu, W. PSD-95-like membrane associated guanylate kinases (PSD-MAGUKs) and synaptic plasticity. Curr. Opin. Neurobiol. 2011, 21, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Hochgräfe, K.; Sydow, A.; Matenia, D.; Cadinu, D.; Könen, S.; Petrova, O.; Pickhardt, M.; Goll, P.; Morellini, F.; Mandelkow, E.; et al. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol. Commun. 2015, 3, 25. [Google Scholar] [CrossRef]
- Abel, T.; Nguyen, P.V. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog. Brain Res. 2008, 169, 97–115. [Google Scholar] [CrossRef]
- Chiang, M.C.; Chen, H.M.; Lai, H.L.; Chen, H.W.; Chou, S.Y.; Chen, C.M.; Tsai, F.J.; Chern, Y. The A2A adenosine receptor rescues the urea cycle deficiency of Huntington’s disease by enhancing the activity of the ubiquitin-proteasome system. Hum. Mol. Genet. 2009, 18, 2929–2942. [Google Scholar] [CrossRef]
- Zhang, F.; Paterson, A.J.; Huang, P.; Wang, K.; Kudlow, J.E. Metabolic control of proteasome function. Physiology 2007, 22, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Anglada-Huguet, M.; Vidal-Sancho, L.; Giralt, A.; Garcia-Diaz Barriga, G.; Xifro, X.; Alberch, J. Prostaglandin E2 EP2 activation reduces memory decline in R6/1 mouse model of Huntington’s disease by the induction of BDNF-dependent synaptic plasticity. Neurobiol. Dis. 2016, 95, 22–34. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Delhaye, S.; Bardoni, B. Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders. Mol. Psychiatry 2021, 26, 4570–4582. [Google Scholar] [CrossRef]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van Der Jeugd, A.; et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry 2016, 21, 97–107. [Google Scholar] [CrossRef]
- Mayford, M.; Bach, M.E.; Huang, Y.-Y.; Wang, L.; Hawkins, R.D.; Kandel, E.R. Control of memory formation through regulated expression of a CaMKII transgene. Science 1996, 274, 1678–1683. [Google Scholar] [CrossRef]
- Bachmanov, A.A.; Reed, D.R.; Beauchamp, G.K.; Tordoff, M.G. Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav. Genet. 2002, 32, 435–443. [Google Scholar] [CrossRef]
- Hocher, B. Adenosine A1 receptor antagonists in clinical research and development. Kidney Int. 2010, 78, 438–445. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nonakay, H.; Ichimura, M.; Takeda, M.; Kanda, T.; Shimada, J.; Suzuki, F.; Kase, H. KW-3902, a selective high affinity antagonist for adenosine A1 receptors. Br. J. Pharmacol. 1996, 117, 1645–1652. [Google Scholar] [CrossRef]
- Glaser, E.M.; Van der Loos, H. Analysis of thick brain sections by obverse-reverse computer microscopy: Application of a new, high clarity Golgi-Nissl stain. J. Neurosci. Methods 1981, 4, 117–125. [Google Scholar] [CrossRef] [PubMed]
- D’Hooge, R.; Lüllmann-Rauch, R.; Beckers, T.; Balschun, D.; Schwake, M.; Reiss, K.; von Figura, K.; Saftig, P. Neurocognitive and psychotiform behavioral alterations and enhanced hippocampal long-term potentiation in transgenic mice displaying neuropathological features of human α-mannosidosis. J. Neurosci. 2005, 25, 6539–6549. [Google Scholar] [CrossRef] [PubMed]
- Anglada-Huguet, M.; Xifro, X.; Giralt, A.; Zamora-Moratalla, A.; Martin, E.D.; Alberch, J. Prostaglandin E2 EP1 receptor antagonist improves motor deficits and rescues memory decline in R6/1 mouse model of Huntington’s disease. Mol. Neurobiol. 2014, 49, 784–795. [Google Scholar] [CrossRef]
- Smith, S.M.; Nichols, T.E. Threshold-free cluster enhancement: Addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage 2009, 44, 83–98. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anglada-Huguet, M.; Endepols, H.; Sydow, A.; Hilgers, R.; Neumaier, B.; Drzezga, A.; Kaniyappan, S.; Mandelkow, E.; Mandelkow, E.-M. Reversal of Tau-Dependent Cognitive Decay by Blocking Adenosine A1 Receptors: Comparison of Transgenic Mouse Models with Different Levels of Tauopathy. Int. J. Mol. Sci. 2023, 24, 9260. https://doi.org/10.3390/ijms24119260
Anglada-Huguet M, Endepols H, Sydow A, Hilgers R, Neumaier B, Drzezga A, Kaniyappan S, Mandelkow E, Mandelkow E-M. Reversal of Tau-Dependent Cognitive Decay by Blocking Adenosine A1 Receptors: Comparison of Transgenic Mouse Models with Different Levels of Tauopathy. International Journal of Molecular Sciences. 2023; 24(11):9260. https://doi.org/10.3390/ijms24119260
Chicago/Turabian StyleAnglada-Huguet, Marta, Heike Endepols, Astrid Sydow, Ronja Hilgers, Bernd Neumaier, Alexander Drzezga, Senthilvelrajan Kaniyappan, Eckhard Mandelkow, and Eva-Maria Mandelkow. 2023. "Reversal of Tau-Dependent Cognitive Decay by Blocking Adenosine A1 Receptors: Comparison of Transgenic Mouse Models with Different Levels of Tauopathy" International Journal of Molecular Sciences 24, no. 11: 9260. https://doi.org/10.3390/ijms24119260
APA StyleAnglada-Huguet, M., Endepols, H., Sydow, A., Hilgers, R., Neumaier, B., Drzezga, A., Kaniyappan, S., Mandelkow, E., & Mandelkow, E.-M. (2023). Reversal of Tau-Dependent Cognitive Decay by Blocking Adenosine A1 Receptors: Comparison of Transgenic Mouse Models with Different Levels of Tauopathy. International Journal of Molecular Sciences, 24(11), 9260. https://doi.org/10.3390/ijms24119260