

X-Linked Myotubular Myopathy in a Female Patient with a Pathogenic Variant in the MTM1 Gene
 , , ,
, , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
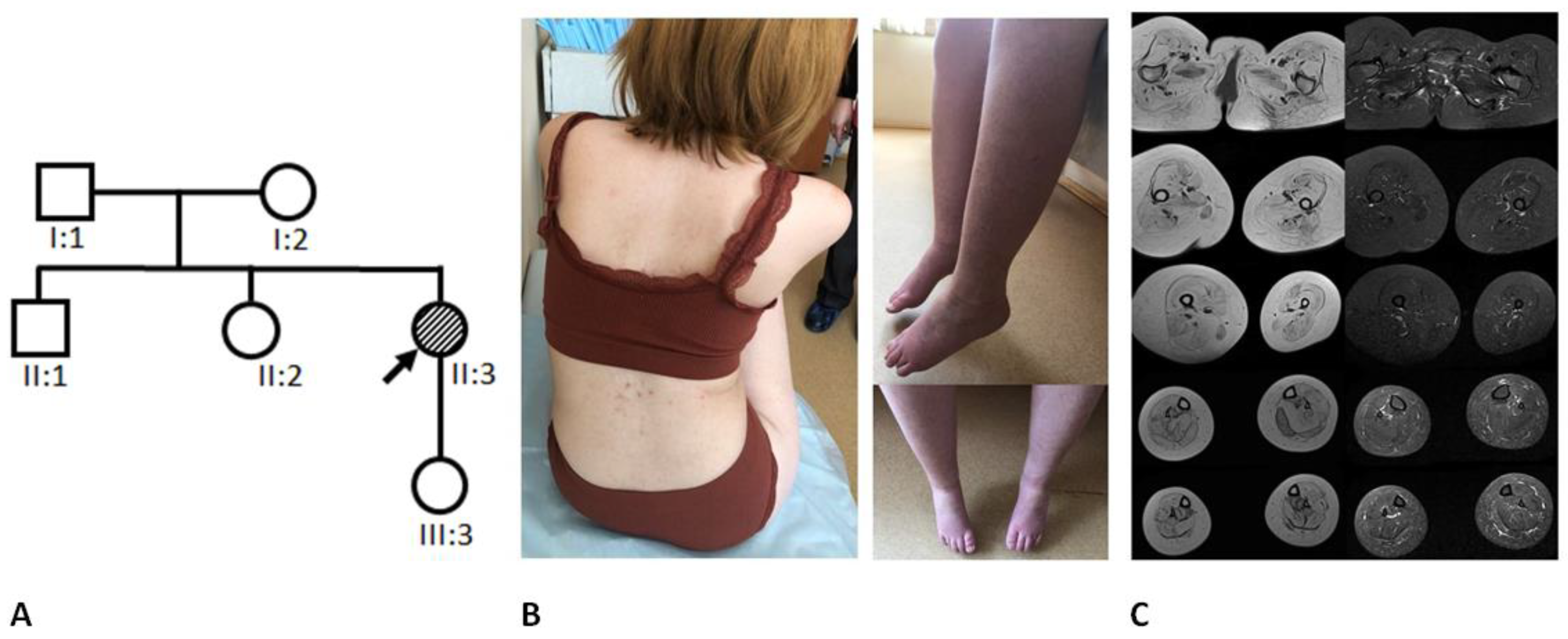
2. General Characteristics of the Family and Examination Methods
3. Results
3.1. Clinical Data
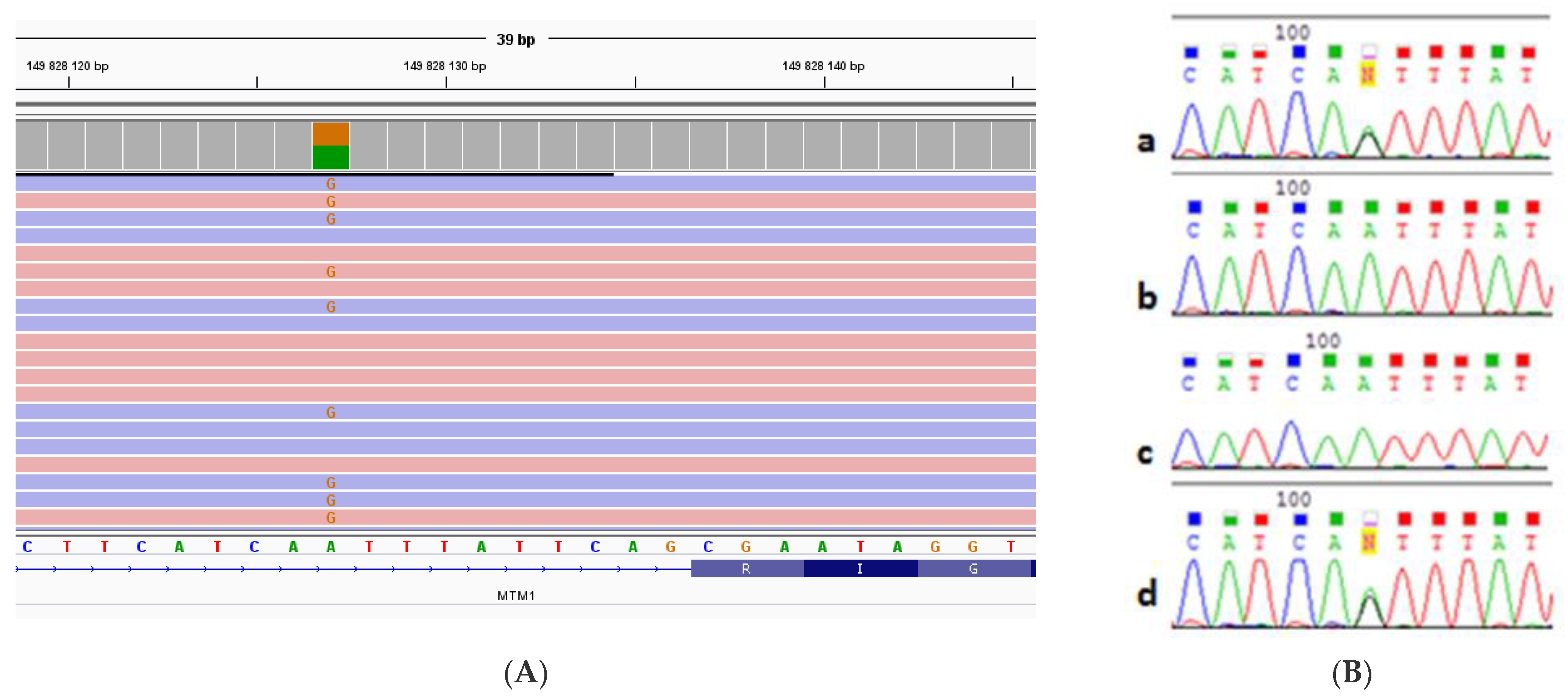
3.2. Molecular Genetic Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Annoussamy, M.; Lilien, C.; Gidaro, T.; Gargaun, E.; Chê, V.; Schara, U.; Gangfuß, A.; D’Amico, A.; Dowling, J.J.; Darras, B.T.; et al. X-linked myotubular myopathy: A prospective international natural history study. Neurology 2019, 92, e1852–e1867. [Google Scholar] [CrossRef]
- Blondeau, F.; Laporte, J.; Bodin, S.; Superti-Furga, G.; Payrastre, B.; Mandel, J.-L. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum. Mol. Genet. 2000, 9, 2223–2229. [Google Scholar] [CrossRef]
- Lawlor, M.W.; Dowling, J.J. X-linked myotubular myopathy. Neuromuscul. Disord. 2021, 31, 1004–1012. [Google Scholar] [CrossRef]
- Gómez-Oca, R.; Cowling, B.S.; Laporte, J. Common Pathogenic Mechanisms in Centronuclear and Myotubular Myopathies and Latest Treatment Advances. Int. J. Mol. Sci. 2021, 22, 11377. [Google Scholar] [CrossRef]
- The Human Gene Mutation Database, v.20.19.4. Available online: https://portal.biobase-international.com (accessed on 10 February 2022).
- Badalyan, L.O.; Guzeva, V.I.; Petrukhina, A.S.; Boris, N.W.; Daruna, J.H.; Owens, J.A. Myotubular Myopathy in Children. Diagnosis and Treatment. MedUnivercom 2021. Available online: https://meduniver.com/Medical/Neurology/miotubuliarnaia_miopatia_u_detei.html (accessed on 15 March 2023).
- Claeys, K.G. Congenital myopathies: An update. Dev. Med. Child Neurol. 2019, 62, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, V.; Scheidecker, S.; Miguet, M.; Laquerrière, A.; Romero, N.B.; Stojkovic, T.; Neto, O.A.; Mercier, S.; Voermans, N.; Tanner, L.; et al. Affected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: Delineating diagnostic clues. Acta Neuropathol. 2017, 134, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Reumers, S.F.; Braun, F.; Spillane, J.E.; Böhm, J.; Pennings, M.; Schouten, M.; van der Kooi, A.J.; Foley, A.R.; Bönnemann, C.G.; Kamsteeg, E.-J.; et al. Spectrum of Clinical Features in X-Linked Myotubular Myopathy Carriers: An International Questionnaire Study. Neurology 2021, 97, e501–e512. [Google Scholar] [CrossRef]
- Ryzhkova, O.P.; Kardymon, O.L.; Prohorchuk, E.B.; Konovalov, F.A.; Maslennikov, A.B.; Stepanov, V.A.; Afanasyev, A.A.; Zaklyazminskaya, E.V.; Rebrikov, D.V.; Savostianov, K.V.; et al. Guidelines for the interpretation of massive parallel sequencing variants (update 2018, v2). Med. Genet. 2019, 18, 3–23. [Google Scholar] [CrossRef]
- De Gouyon, B.M.; Zhao, W.; Laporte, J.; Mandel, J.-L.; Metzenberg, A.; Herman, G.E. Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Hum. Mol. Genet. 1997, 6, 1499–1504. [Google Scholar] [CrossRef]
- Morales, R.J.; Perrin, A.; Solé, G.; Lacourt, D.; Pegeot, H.; Walther-Louvier, U.; Cintas, P.; Cances, C.; Espil, C.; Theze, C.; et al. An Integrated Clinical-Biological Approach to Identify Interindividual Variability and Atypical Phenotype-Genotype Correlations in Myopathies: Experience on A Cohort of 156 Families. Genes 2021, 12, 1199. [Google Scholar] [CrossRef]
- Gangfuss, A.; Schmitt, D.; Roos, A.; Braun, F.; Annoussamy, M.; Servais, L.; Schara-Schmidt, U. Diagnosing X-linked Myotubular Myopathy—A German 20-year Follow Up Experience. J. Neuromuscul. Dis. 2021, 8, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.M.; Tiwari, V.K.; Abdullaev, Z.; Vostrov, A.A.; Flanagan, P.T.; Quitschke, W.W.; Loukinov, D.I.; Ohlsson, R.; Lobanenkov, V.V. Familial cases of point mutations in the XIST promoter reveal a correlation between CTCF binding and pre-emptive choices of X chromosome inactivation. Hum. Mol. Genet. 2005, 14, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Plenge, R.M.; Hendrich, B.D.; Schwartz, C.; Arena, J.F.; Naumova, A.; Sapienza, C.; Winter, R.M.; Willard, H.F. A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nat. Genet. 1997, 17, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Schara, U.; Kress, W.; Tücke, J.; Mortier, W. X-linked myotubular myopathy in a female infant caused by a new MTM1 gene mutation. Neurology 2003, 60, 1363–1365. [Google Scholar] [CrossRef]
- Sutton, I.J.; Winer, J.B.; Norman, A.N.; Liechti-Gallati, S.; MacDonald, F. Limb girdle and facial weakness in female carriers of X-linked myotubular myopathy mutations. Neurology 2001, 57, 900–902. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I.; Minami, N.; Kobayashi, O.; Ikezawa, M.; Goto, Y.-I.; Arahata, K.; Nonaka, I. MTM1 gene mutations in Japanese patients with the severe infantile form of myotubular myopathy. Neuromuscul. Disord. 1998, 8, 453–458. [Google Scholar] [CrossRef]
- Tanner, S.M.; Laporte, J.; Guiraud-Chaumeil, C.; Liechti-Gallati, S. Confirmation of prenatal diagnosis results of X-linked recessive myotubular myopathy by mutational screening, and description of three new mutations in the MTM1 gene. Hum. Mutat. 1998, 11, 62–68. [Google Scholar] [CrossRef]
- Buj-Bello, A.; Biancalana, V.; Moutou, C.; Laporte, J.; Mandel, J.L. Identification of novel mutations in the MTM1 gene causing severe and mild forms of X-linked myotubular myopathy. Hum. Mutat. 1999, 14, 320–325. [Google Scholar] [CrossRef]
- RuExAc. Available online: http://ngs-data.ru/vcfdb (accessed on 30 January 2023).
- RuSeq. Available online: http://ruseq.ru/# (accessed on 30 January 2023).
- Souza, L.S.; Almeida, C.F.; Yamamoto, G.L.; Pavanello, R.D.C.M.; Gurgel-Giannetti, J.; da Costa, S.S.; Anequini, I.P.; Carmo, S.A.D.; Wang, J.Y.T.; Scliar, M.D.O.; et al. Manifesting carriers of X-linked myotubular myopathy: Genetic modifiers modulating the phenotype. Neurol. Genet. 2020, 6, e513. [Google Scholar] [CrossRef] [PubMed]
- Panova, A.V.; Bogomazova, A.N.; Lagarkova, M.A.; Kiselev, S.L. Methylation of the human AR locus does not correlate with the presence of an Inactivated X chromosome in cells with Induced pluripotency. Russ. J. Genet. 2020, 56, 321–327. [Google Scholar] [CrossRef]
- Bertelsen, B.; Tümer, Z.; Ravn, K. Three new loci for determining x chromosome inactivation patterns. J. Mol. Diagn. 2011, 13, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Gale, R.; Wheadon, H.; Boulos, P.; Linch, D. Tissue specificity of X-chromosome inactivation patterns. Blood 1994, 83, 2899–2905. [Google Scholar] [CrossRef] [PubMed]
- Puck, J.M.; Willard, H.F. X inactivation in females with X-linked disease. N. Engl. J. Med. 1998, 338, 325–328. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chausova, P.; Murtazina, A.; Stepanova, A.; Borovicov, A.; Kovalskaia, V.; Ryadninskaya, N.; Chukhrova, A.; Ryzhkova, O.; Poliakov, A. X-Linked Myotubular Myopathy in a Female Patient with a Pathogenic Variant in the MTM1 Gene. Int. J. Mol. Sci. 2023, 24, 8409. https://doi.org/10.3390/ijms24098409
Chausova P, Murtazina A, Stepanova A, Borovicov A, Kovalskaia V, Ryadninskaya N, Chukhrova A, Ryzhkova O, Poliakov A. X-Linked Myotubular Myopathy in a Female Patient with a Pathogenic Variant in the MTM1 Gene. International Journal of Molecular Sciences. 2023; 24(9):8409. https://doi.org/10.3390/ijms24098409
Chicago/Turabian StyleChausova, Polina, Aysylu Murtazina, Anna Stepanova, Artem Borovicov, Valeriia Kovalskaia, Nina Ryadninskaya, Alena Chukhrova, Oxana Ryzhkova, and Aleksander Poliakov. 2023. "X-Linked Myotubular Myopathy in a Female Patient with a Pathogenic Variant in the MTM1 Gene" International Journal of Molecular Sciences 24, no. 9: 8409. https://doi.org/10.3390/ijms24098409
APA StyleChausova, P., Murtazina, A., Stepanova, A., Borovicov, A., Kovalskaia, V., Ryadninskaya, N., Chukhrova, A., Ryzhkova, O., & Poliakov, A. (2023). X-Linked Myotubular Myopathy in a Female Patient with a Pathogenic Variant in the MTM1 Gene. International Journal of Molecular Sciences, 24(9), 8409. https://doi.org/10.3390/ijms24098409