

Parkin Precipitates on Mitochondria via Aggregation and Autoubiquitination


Abstract
1. Introduction
2. Results
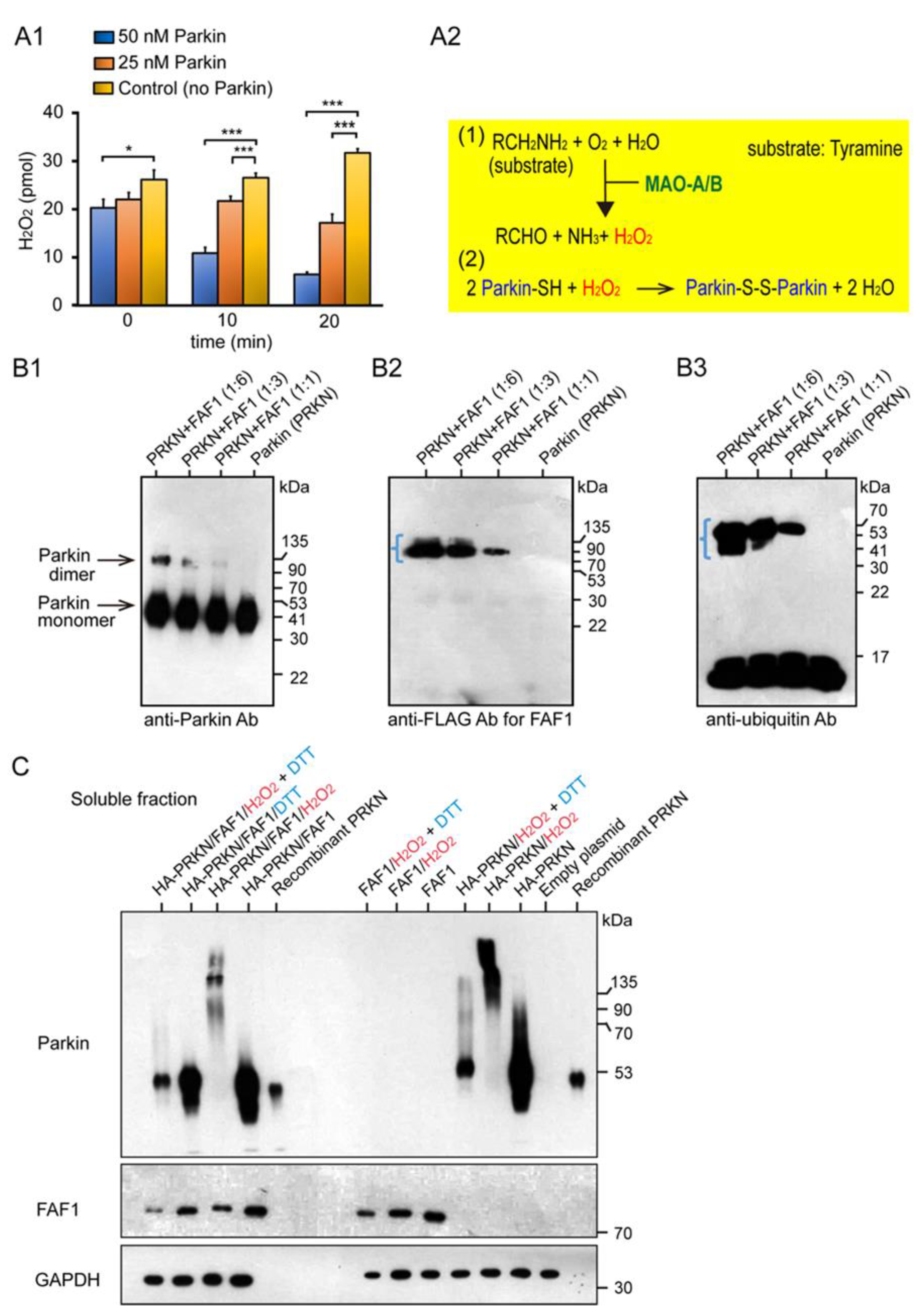
2.1. Characterization of the Basic Properties of the Redox Molecule, PARKIN and Its Substrate FAF1
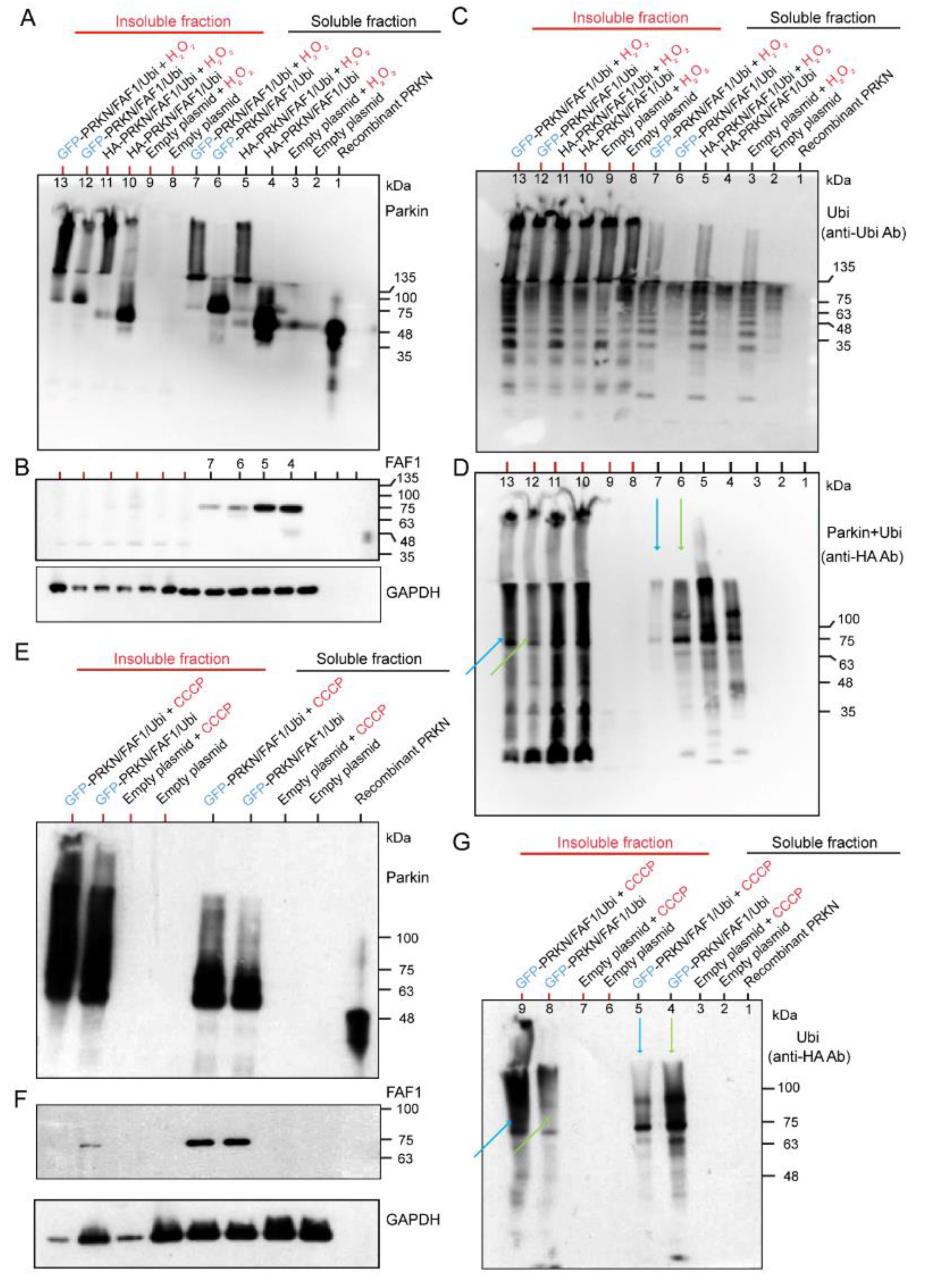
2.2. Overexpression of Three Genes—HA- or GFP-Tagged Parkin/FLAG-FAF1/HA-Ubi—In HEK Cell System and Exposure to H2O2 or CCCP
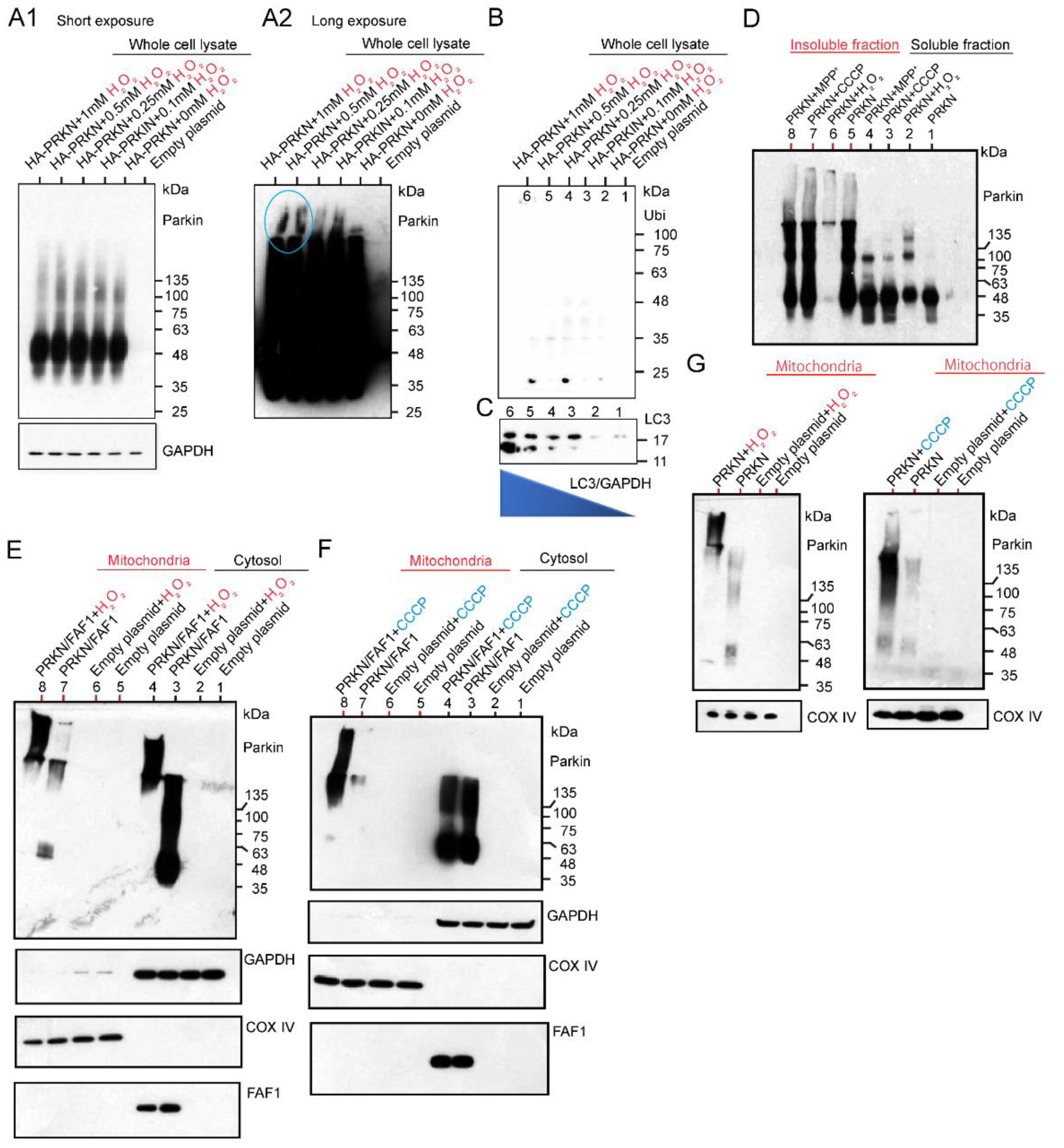
2.2.1. Effects of H2O2 and CCCP Treatment on Different Fractions of HEK Cell System Overexpressing HA-Tagged or Untagged Parkin/FAF1 or Untagged Parkin Alone
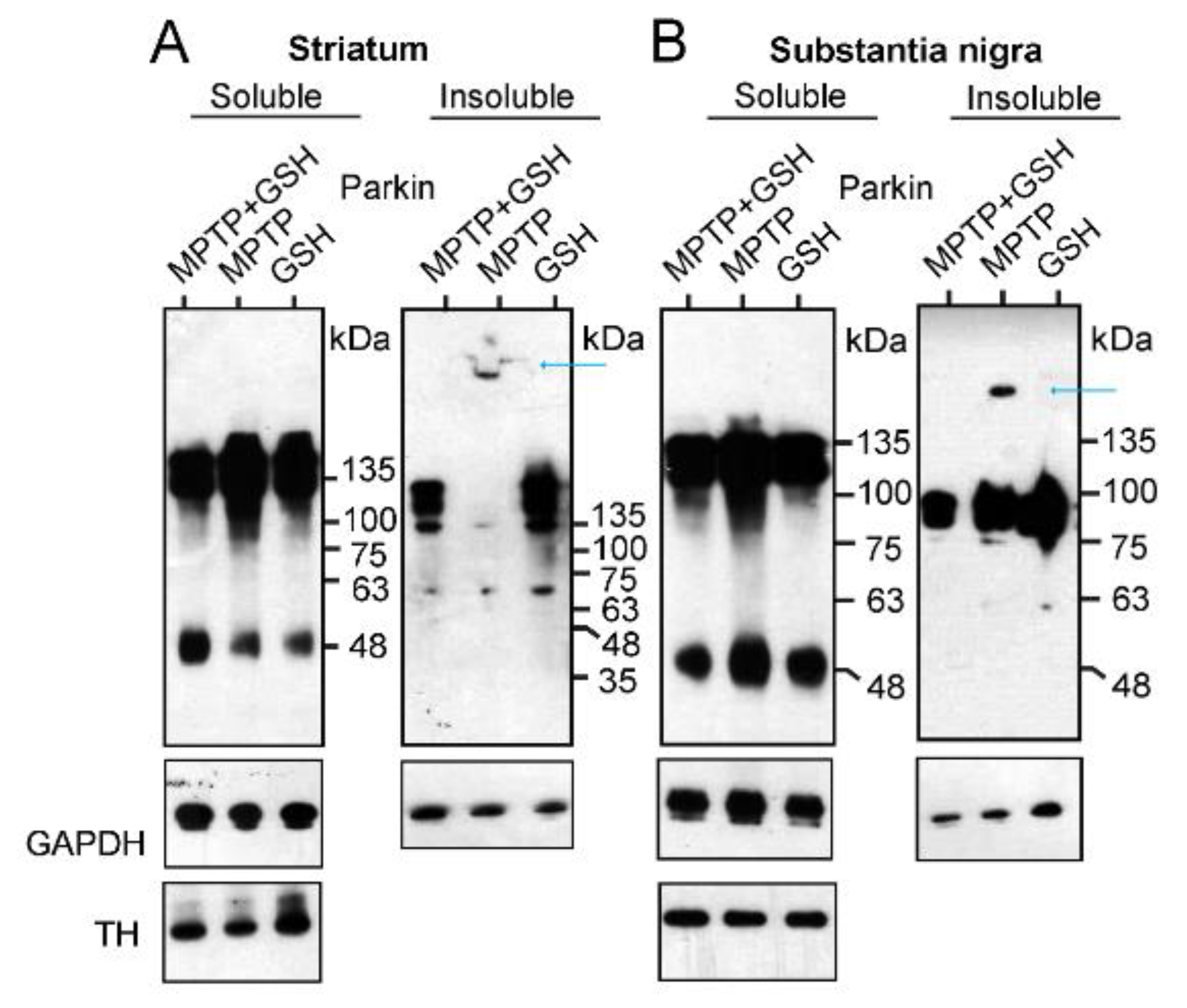
2.2.2. Investigation of Aggregation and Insolubility of Parkin Protein Using MPTP-Treated PD Mouse Model
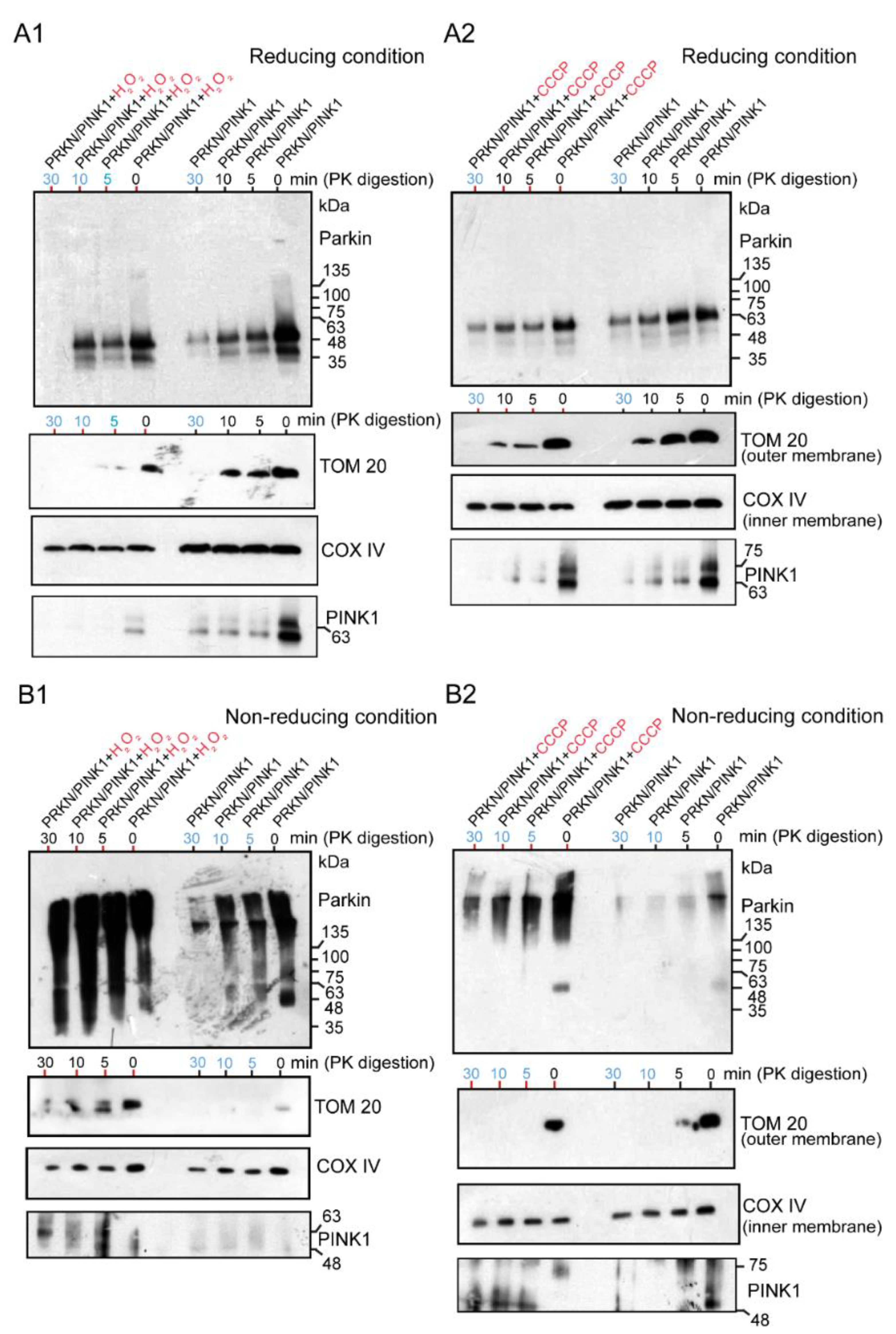
2.3. WB Analysis of Mitochondrial Subfractions after H2O2 or CCCP Exposure to Cultured Cells Overexpressing Untagged Parkin + FLAG-PINK1, Which Was Performed under Reducing or Non-Reducing Conditions
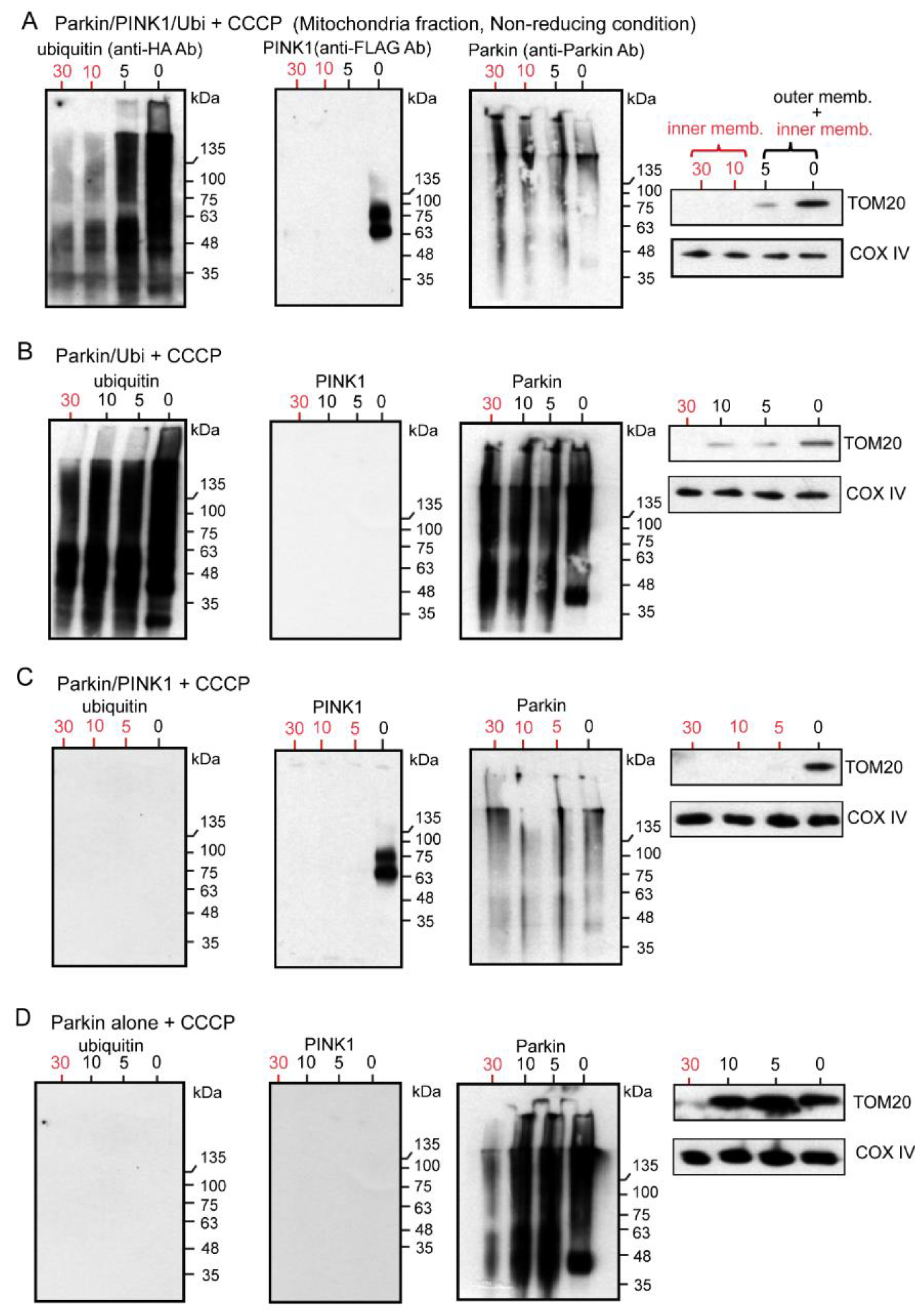
2.4. WB Analysis of Mitochondrial Subfractions after CCCP Exposure to Cultured Cells Overexpressing Various Combinations of Un-Tagged Parkin, FLAG-PINK1, and HA-Ubi, Which Was Performed under Non-Reducing Condition
2.5. Fluorescent Immunocytochemistry (ICC) to Analyze Accumulation of Parkin in Mitochondria of Cultured Cells Overexpressing Several Combinations of GFP-Parkin, HA-Ubi, and myc-PINK1 after Exposure to CCCP
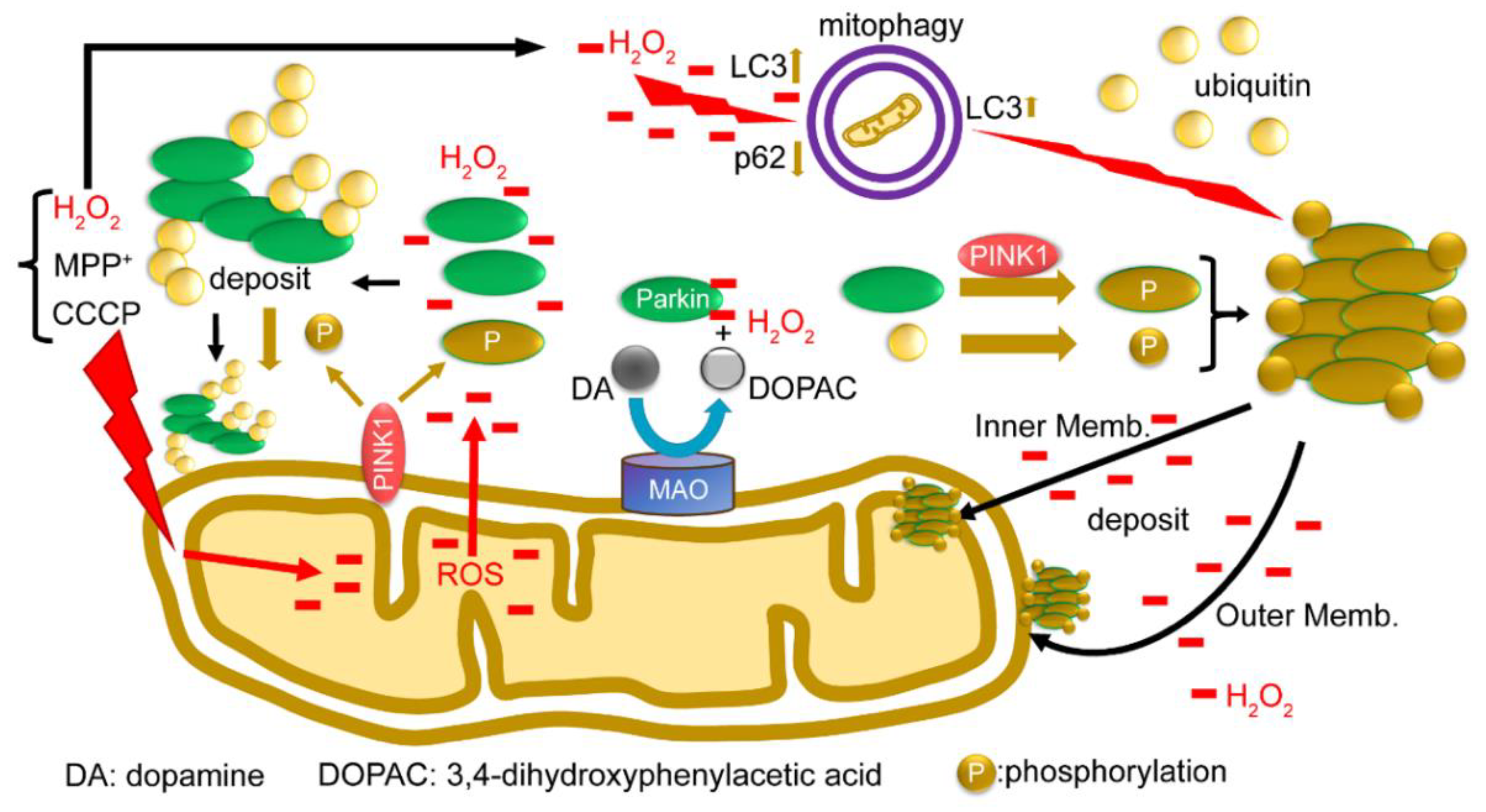
2.6. Summary of Results: Functional Model of the Redox Molecule Parkin around Mitochondria
3. Discussion
4. Materials and Methods
4.1. In Vitro Fas Associated Factor 1 (FAF1) Polyubiquitination Assay by Parkin
4.2. The Monoamine Oxidase (MAO) Assay
4.3. Cell Culture, Treatment and Antibodies
4.4. Cells Treatment and Preparing Cell Lysate
4.5. Fluorescent Immunocytochemistry (ICC)
4.6. Mitochondria and Cytosolic Fraction Isolation and Proteinase K (PK) Digestion
4.7. In Vivo MPTP Injection
4.8. SDS-PAGE and Immunoblotting
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and acti-vates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein ag-gregation. Mol. Neurodegener. 2011, 6, 34–48. [Google Scholar] [CrossRef]
- Kitada, T.; Hikita, R.; Hirose, H. Parkin, Parkinson disease gene product, directly reduces hydrogen peroxide (mitochondrial oxidant), and forms dimerization reversibly. Int. J. Latest Res. Sci. Technol. 2016, 5, 1–3. [Google Scholar]
- Tokarew, J.M.; El-Kodsi, D.N.; Lengacher, N.A.; Fehr, T.K.; Nguyen, A.P.; Shutinoski, B.; O’nuallain, B.; Jin, M.; Khan, J.M.; Ng, A.C.H.; et al. Age-associated insolubility of parkin in human midbrain is linked to redox balance and sequestration of reactive dopamine metabolites. Acta Neuropathol. 2021, 141, 725–754. [Google Scholar] [CrossRef]
- El Kodsi, D.N.; Tokarew, J.M.; Sengupta, R.; Lengacher, N.A.; Ng, A.C.; Boston, H.; Jiang, Q.; Palmberg, C.; Pileggi, C.; Shutinoski, B.; et al. Parkinson disease-linked Parkin mediates redox reactions that lower oxidative stress in mammalian brain. bioRxiv 2020, preprint. [Google Scholar] [CrossRef]
- Sul, J.W.; Park, M.Y.; Shin, J.; Kim, Y.R.; Yoo, S.E.; Kong, Y.Y.; Kwon, K.S.; Lee, Y.H.; Kim, E. Accumulation of the parkin substrate, FAF1, plays a key role in the dopaminergic neurodegeneration. Hum. Mol. Genet. 2013, 22, 1558–1573. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinsons Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Edmondson, D. Hydrogen Peroxide Produced by Mitochondrial Monoamine Oxidase Catalysis: Biological Implications. Curr. Pharm. Des. 2014, 20, 155–160. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Matsuda, N.; Kitami, T.; Suzuki, T.; Mizuno, Y.; Hattori, N.; Tanaka, K. Diverse Effects of Pathogenic Mutations of Parkin That Catalyze Multiple Monoubiquitylation in Vitro. J. Biol. Chem. 2006, 281, 3204–3209. [Google Scholar] [CrossRef]
- Burchell, L.; Chaugule, V.K.; Walden, H. Small, N-terminal tags activate Parkin E3 ubiquitin ligase activity by disrupting its autoinhibited confor-mation. PLoS ONE 2012, 7, e34748. [Google Scholar] [CrossRef]
- Yamamura, Y.; Kohriyama, T.; Kawakami, H.; Kaseda, Y.; Kuzuhara, S.; Nakamura, S. Autosomal recessive early-onset parkinsonism with diurnal fluctuation (AR-EPDF)--clinical character-istics. Rinsho Shinkeigaku 1996, 36, 944–950. [Google Scholar]
- Yamamura, Y.; Hattori, N.; Matsumine, H.; Kuzuhara, S.; Mizuno, Y. Autosomal recessive early-onset parkinsonism with diurnal fluctuation: Clinicopathologic characteris-tics and molecular genetic identification. Brain Dev. 2000, 22 (Suppl. S1), 87–91. [Google Scholar] [CrossRef]
- Ishikawa, A.; Takahashi, H. Clinical and neuropathological aspects of autosomal recessive juvenile parkinsonism. J. Neurol. 1998, 245, P4–P9. [Google Scholar] [CrossRef] [PubMed]
- Takanashi, M.; Mochizuki, H.; Yokomizo, K.; Hattori, N.; Mori, H.; Yamamura, Y.; Mizuno, Y. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP). Park. Relat. Disord. 2001, 7, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(−/−) mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Kang, M.J.; Kim, K.H.; Han, P.L.; Kim, H.S.; Ha, J.Y.; Son, J.H. Nitric oxide induction of Parkin translocation in PTEN-induced putative kinase 1 (PINK1) deficiency: Func-tional role of neuronal nitric oxide synthase during mitophagy. J. Biol. Chem. 2015, 290, 10325–10335. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449. [Google Scholar] [CrossRef]
- Allen, G.F.; Toth, R.; James, J.; Ganley, I.G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef]
- Wang, C.; Ko, H.S.; Thomas, B.; Tsang, F.; Chew, K.C.; Tay, S.P.; Ho, M.W.; Lim, T.M.; Soong, T.W.; Pletnikova, O.; et al. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protec-tive function. Hum. Mol. Genet. 2005, 14, 3885–3897. [Google Scholar] [CrossRef]
- Shiiba, I.; Takeda, K.; Nagashima, S.; Ito, N.; Tokuyama, T.; Yamashita, S.; Kanki, T.; Komatsu, T.; Urano, Y.; Fujikawa, Y.; et al. MITOL promotes cell survival by degrading Parkin during mitophagy. EMBO Rep. 2021, 22, e49097. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal imag-es. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid DNA | Source | Catalogue |
|---|---|---|
| pRK5-HA-Parkin | Addgene, MA, USA | 17613 |
| pCDNA3-Flag-FAF1 | Genscript, NJ, USA | Custome designe |
| pRK5-HA-Ubi | Addgene, MA, USA | 17608 |
| pEGFP-Parkin | Addgene, MA, USA | 45875 |
| pCMV3-non-tagged Parkin | Sino Biological, Beijing, PR China | HG12092-UT |
| pAdtrack-CMV3-FLAG-PINK1 FL | Haque et al. | PNAS, 5 February 2008; 105 (5): 1716–1721. ?? |
| Chemical | Source | Catalogue |
|---|---|---|
| proteinase K | Sigma-Aldrich-USA | |
| MOA kit | Abcam-USA | Ab241031 |
| Mitochondria isolation Kit for mammalian Cells | ThermoFisher Scientific-USA | 89874 |
| SuperSignal West Pico Plus Chemiluminescent Substrate | ThermoFisher Scientific-USA | 34580 |
| Pierce BCA protein Assay Kit | ThermoFisher Scientific-USA | 23225 |
| UltraCruz Autoradiography film | Santa Cruz-USA | SC-201697 |
| Recombinant Human His6-Ubiquitin E1 Enzyme (UBE1), CF | Boston Biochem-USA | E-304-050 |
| Recombinant Human Parkin pS65, CF | Boston Biochem-USA | E3-166-025 |
| Recombinant Human Ubiquitin Biotin Protein, CF | Boston Biochem-USA | UB-570-100 |
| MgATP Solution | Boston Biochem_USA | B-20 |
| Recombinant Human Parkin, CF | Boston BiochemUSA | E3-160-025 |
| Recombinant Human UbcH7/UBE2L3, CF | Boston Biochem-USA | E2-640-100 |
| Recombinant Human Ubiquitin | Boston BiochemUSA | U-100H |
| 10X E3 Ligase buffer | Boston BiochemUSA | B-71 |
| Recombinant Human Phospho-Ubiquitin (S65) Protein, CF | Boston BiochemUSA | U-102 |
| Carbonyl cyanide 3-chlorophenylhydrazone “CCCP” | Abcam-USA | ab14122 |
| Antibody | Host | Source | Catalogue |
|---|---|---|---|
| Parkin | Mouse | Santa Cruz-USA | SC-32282 |
| Flag | Rabbit | Cell Signaling Technologies-USA | 2368S |
| GAPDH | Rabbit | Cell Signaling Technologies-USA | 2118S |
| HA | Mouse | Cell Signaling Technologies-USA | 2367S |
| Ubiquitin | Mouse | Cell Signaling Technologies-USA | 3936S |
| Ubiquitin | Mouse | Boston Biochem-USA | A-104 |
| LC3 | Rabbit | Cell Signaling Technologies-USA | 12741S |
| COX IV | Mouse | Abcam-USA | ab33985 |
| TOM 20 | Rabbit | Santa Cruz-USA | Sc-11415 |
| TH | Mouse | Immunostar-USA | 22941 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardah, M.T.; Radwan, N.; Khan, E.; Kitada, T.; Haque, M.E. Parkin Precipitates on Mitochondria via Aggregation and Autoubiquitination. Int. J. Mol. Sci. 2023, 24, 9027. https://doi.org/10.3390/ijms24109027
Ardah MT, Radwan N, Khan E, Kitada T, Haque ME. Parkin Precipitates on Mitochondria via Aggregation and Autoubiquitination. International Journal of Molecular Sciences. 2023; 24(10):9027. https://doi.org/10.3390/ijms24109027
Chicago/Turabian StyleArdah, Mustafa T., Nada Radwan, Engila Khan, Tohru Kitada, and M Emdadul Haque. 2023. "Parkin Precipitates on Mitochondria via Aggregation and Autoubiquitination" International Journal of Molecular Sciences 24, no. 10: 9027. https://doi.org/10.3390/ijms24109027
APA StyleArdah, M. T., Radwan, N., Khan, E., Kitada, T., & Haque, M. E. (2023). Parkin Precipitates on Mitochondria via Aggregation and Autoubiquitination. International Journal of Molecular Sciences, 24(10), 9027. https://doi.org/10.3390/ijms24109027