
Modelling Microglial Innate Immune Memory In Vitro: Understanding the Role of Aerobic Glycolysis in Innate Immune Memory

Abstract
1. Introduction
2. Results
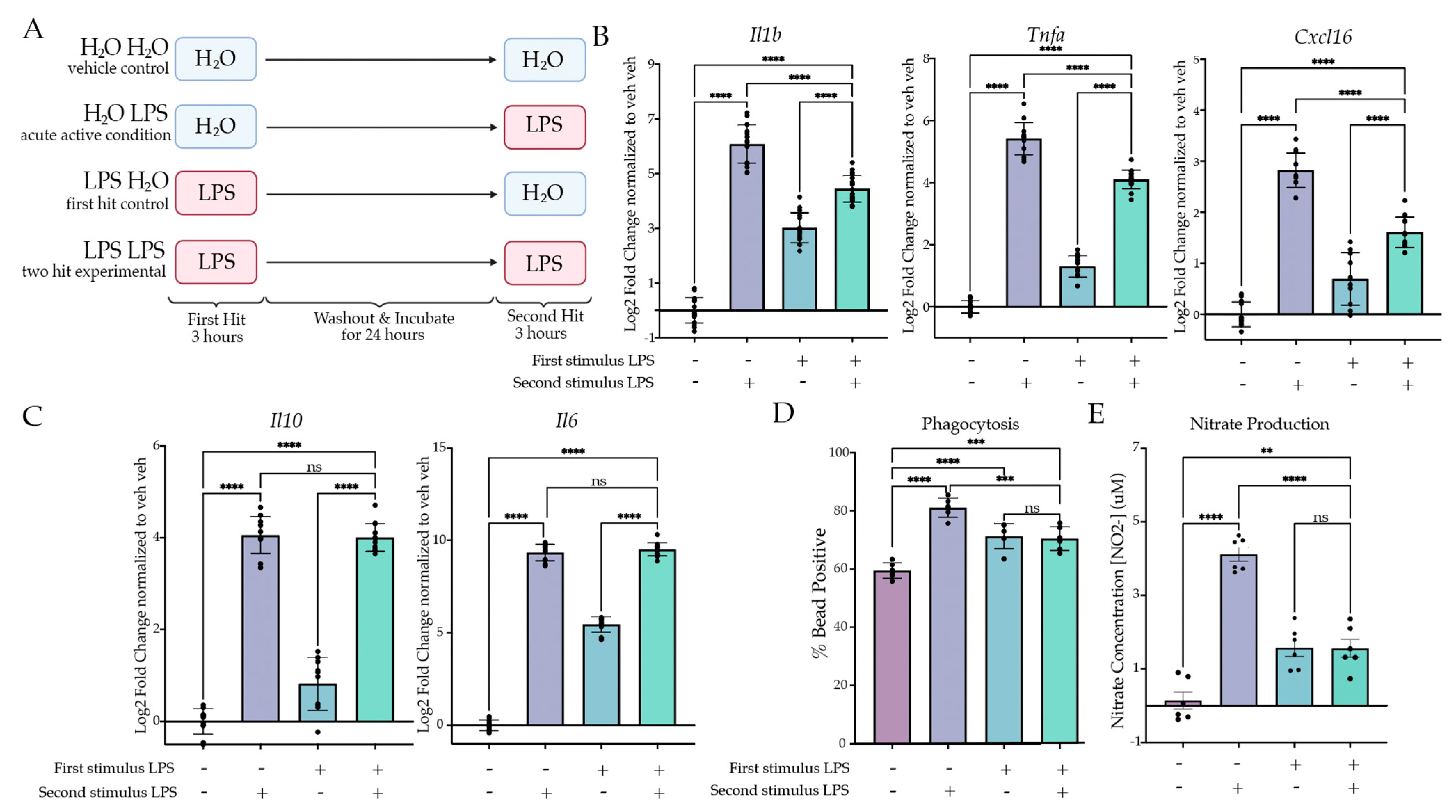
2.1. Two Hits of LPS Produce Tolerance In Vitro
2.1.1. Two Hits of LPS Result in Attenuation of Inflammatory Gene Expression
2.1.2. Two Hits of LPS Result in Attenuation of Microglial Phagocytosis and NO Release
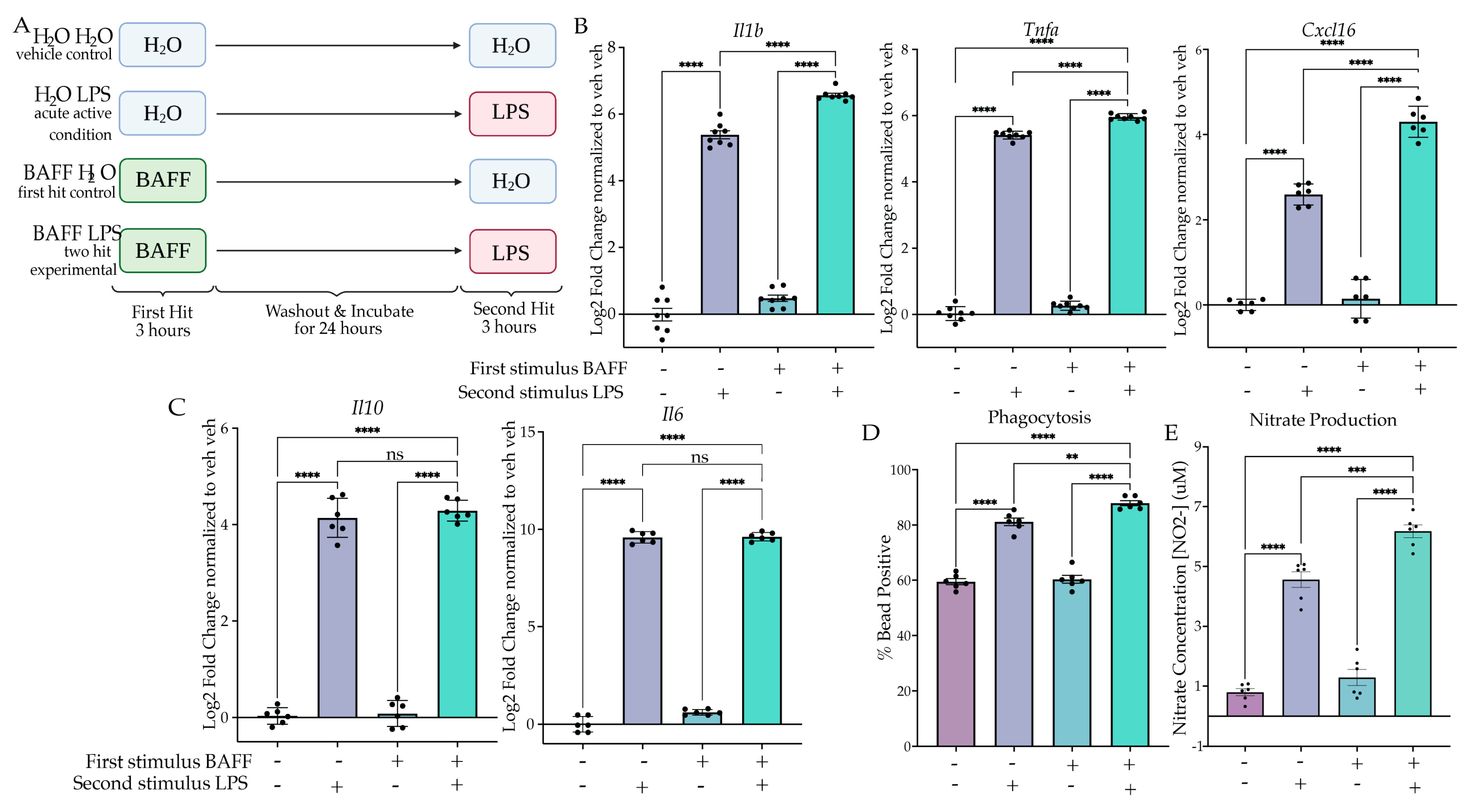
2.2. BAFF Pre-Treatment Produces Training In Vitro
2.2.1. BAFF Pre-Treatment Results in Excessive Induction of Inflammatory Gene Expression upon LPS Stimulus
2.2.2. BAFF Pre-Treatment Results in Excessive Induction of Phagocytosis and NO Release
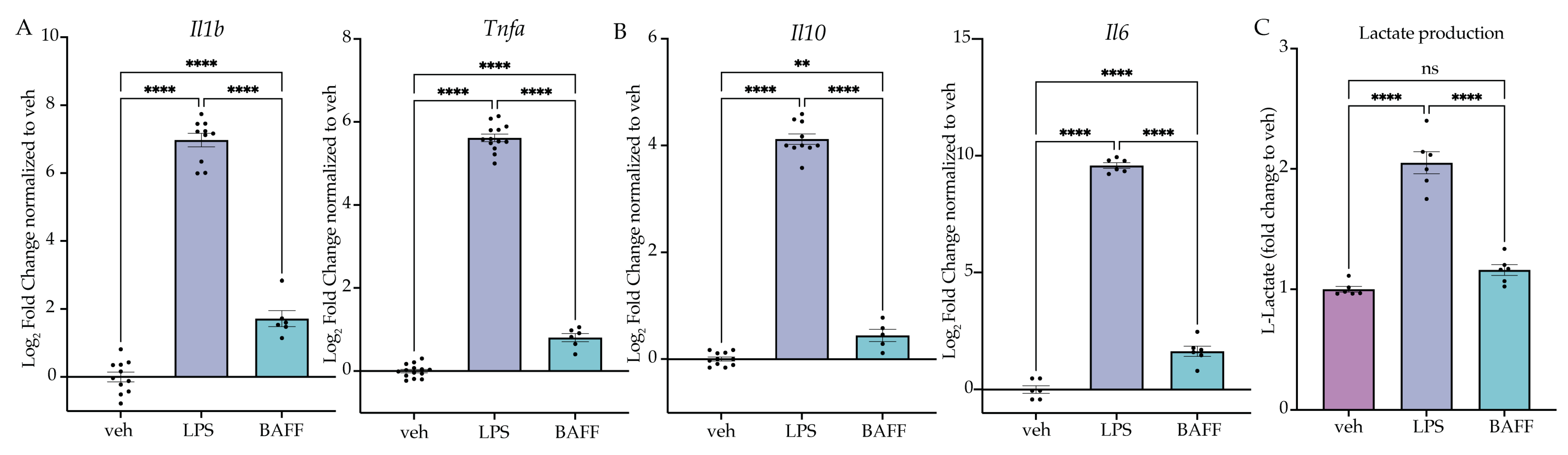
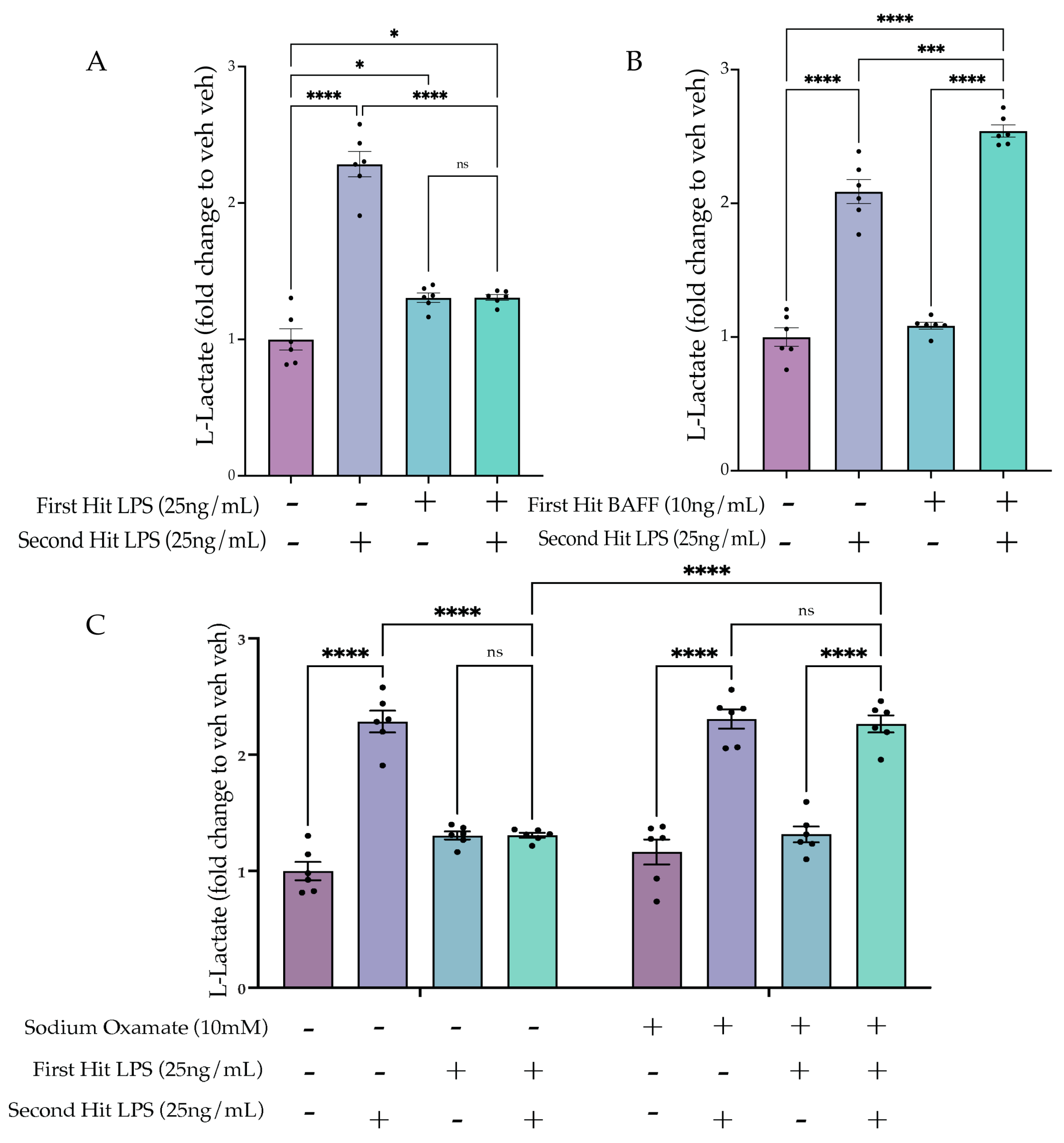
2.3. LPS but Not BAFF Results in Rapid Changes in Cellular Metabolism and Induction of Aerobic Glycolysis
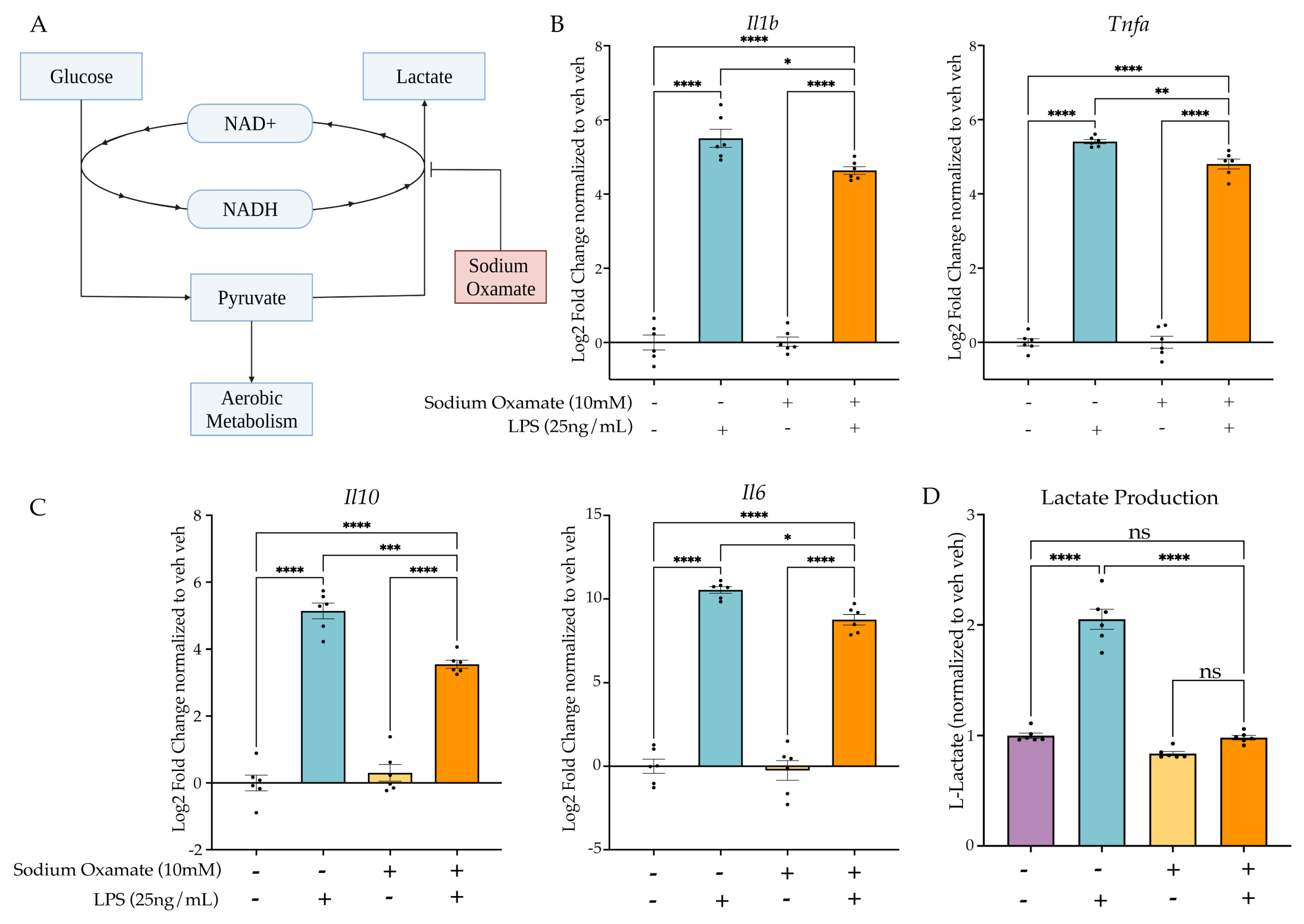
2.4. Inhibition of LPS-Induced Aerobic Glycolysis Impairs Establishment of Microglial Memory
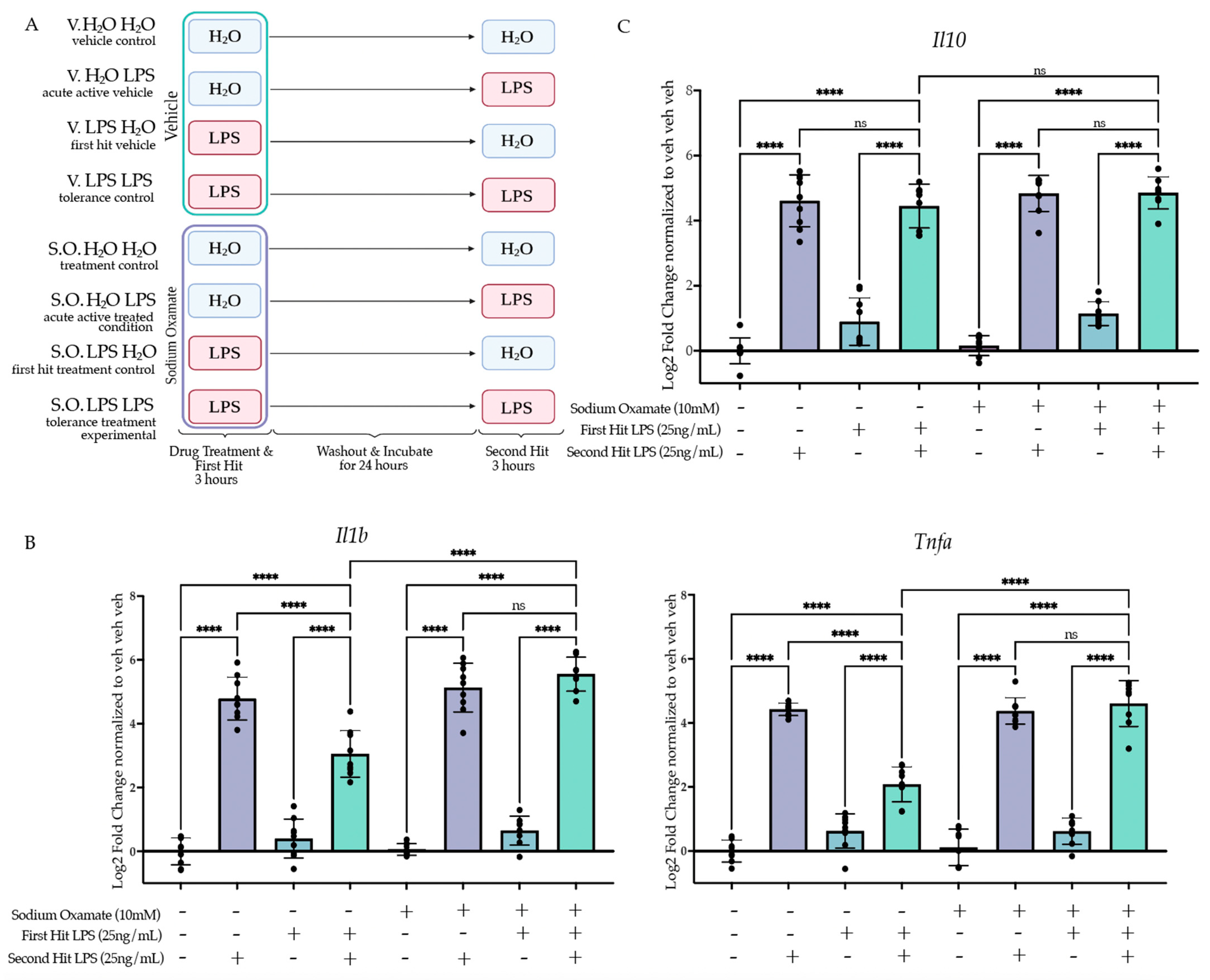
2.4.1. Sodium Oxamate Attenuates LPS-Induced Gene Expression and Blocks Lactate Production
2.4.2. Treatment of Sodium Oxamate during the First Stimulus Blocks Establishment of the Tolerance Memory State Gene Expression but Not the Training Memory State
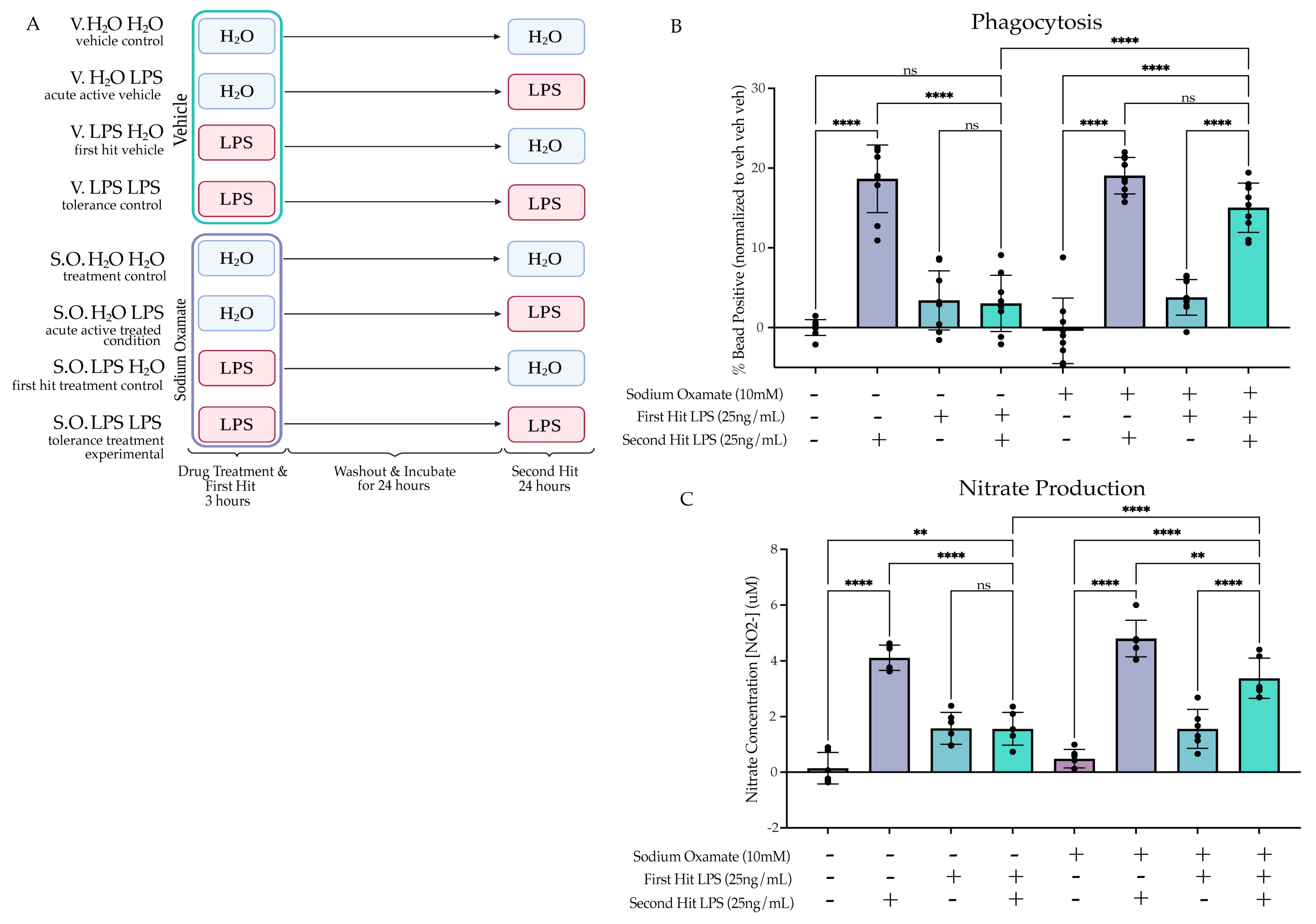
2.4.3. Sodium Oxamate Rescues Attenuation of Phagocytosis and NO Production
2.5. Tolerized Microglial Metabolic State Is Rescued by Sodium Oxamate Treatment during the First Hit
2.5.1. Two Hit of LPS Block Aerobic Glycolysis, whereas BAFF Pre-Treatment Enhances Aerobic Glycolysis
2.5.2. Sodium Oxamate Treatment Prevents the Tolerance-Induced Metabolic State
3. Discussion
4. Materials and Methods
4.1. BV2 Microglial Culture + Treatments
4.2. RNA-Extraction and RT-qPCR Analysis
4.3. pHrodo E. coli Phagocytosis Assay Quantified by Flow Cytometry
4.4. Griess Reagent Assay
4.5. Lactate Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Atladottir, H.O.; Thorsen, P.; Ostergaard, L.; Schendel, D.E.; Lemcke, S.; Abdallah, M.; Parner, E.T. Maternal Infection Requiring Hospitalization during Pregnancy and Autism Spectrum Disorders. J. Autism Dev. Disord. 2010, 40, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Mednick, S.A.; Machon, R.A.; Huttunen, M.O.; Bonett, D. Adult Schizophrenia Following Prenatal Exposure to an Influenza Epidemic. Arch. Gen. Psychiatry 1988, 45, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.J.; Reynolds, R.M.; Hardy, D.B. Developmental Origins of Health and Disease: Current Knowledge and Potential Mechanisms. Nutr. Rev. 2017, 75, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.J.; Powell, T.L.; Barrett, E.S.; Hardy, D.B. Developmental Origins of Metabolic Diseases. Physiol. Rev. 2021, 101, 739–795. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Schwarz, J.M. Early-Life Programming of Later-Life Brain and Behavior: A Critical Role for the Immune System. Front. Behav. Neurosci. 2009, 3, 14. [Google Scholar] [CrossRef]
- Guerrin, C.G.J.; Doorduin, J.; Sommer, I.E.; de Vries, E.F.J. The Dual Hit Hypothesis of Schizophrenia: Evidence from Animal Models. Neurosci. Biobehav. Rev. 2021, 131, 1150–1168. [Google Scholar] [CrossRef]
- Lee, Y.H.; Cherkerzian, S.; Seidman, L.J.; Papandonatos, G.D.; Savitz, D.A.; Tsuang, M.T.; Goldstein, J.M.; Buka, S.L. Maternal Bacterial Infection during Pregnancy and Offspring Risk of Psychotic Disorders: Variation by Severity of Infection and Offspring Sex. Am. J. Psychiatry 2020, 177, 66–75. [Google Scholar] [CrossRef]
- Zerbo, O.; Qian, Y.; Yoshida, C.; Grether, J.K.; Van de Water, J.; Croen, L.A. Maternal Infection During Pregnancy and Autism Spectrum Disorders. J. Autism Dev. Disord. 2013. [Google Scholar] [CrossRef]
- Nee, L.E.; Lippa, C.F. Alzheimer’s Disease in 22 Twin Pairs--13-Year Follow-up: Hormonal, Infectious and Traumatic Factors. Dement. Geriatr. Cogn. Disord. 1999, 10, 148–151. [Google Scholar] [CrossRef]
- Perry, V.H.; Newman, T.A.; Cunningham, C. The Impact of Systemic Infection on the Progression of Neurodegenerative Disease. Nat. Rev. Neurosci. 2003, 4, 103–112. [Google Scholar] [CrossRef]
- Plassman, B.L.; Havlik, R.J.; Steffens, D.C.; Helms, M.J.; Newman, T.N.; Drosdick, D.; Phillips, C.; Gau, B.A.; Welsh-Bohmer, K.A.; Burke, J.R.; et al. Documented Head Injury in Early Adulthood and Risk of Alzheimer’s Disease and Other Dementias. Neurology 2000, 55, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head Injury as a Risk Factor for Alzheimer’s Disease: The Evidence 10 Years on; a Partial Replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate Immune Memory in the Brain Shapes Neurological Disease Hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Zhang, X.; Kracht, L.; Lerario, A.M.; Dubbelaar, M.L.; Brouwer, N.; Wesseling, E.M.; Boddeke, E.W.G.M.; Eggen, B.J.L.; Kooistra, S.M. Epigenetic Regulation of Innate Immune Memory in Microglia. J. Neuroinflamm. 2022, 19, 111. [Google Scholar] [CrossRef]
- Hayes, L.N.; An, K.; Carloni, E.; Li, F.; Vincent, E.; Trippaers, C.; Paranjpe, M.; Dölen, G.; Goff, L.A.; Ramos, A.; et al. Prenatal Immune Stress Blunts Microglia Reactivity, Impairing Neurocircuitry. Nature 2022, 610, 327–334. [Google Scholar] [CrossRef]
- Neher, J.J.; Cunningham, C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019, 40, 358–374. [Google Scholar] [CrossRef]
- Schaafsma, W.; Basterra, L.B.; Jacobs, S.; Brouwer, N.; Meerlo, P.; Schaafsma, A.; Boddeke, E.W.G.M.; Eggen, B.J.L. Maternal Inflammation Induces Immune Activation of Fetal Microglia and Leads to Disrupted Microglia Immune Responses, Behavior, and Learning Performance in Adulthood. Neurobiol. Dis. 2017, 106, 291–300. [Google Scholar] [CrossRef]
- Williamson, L.L.; Sholar, P.W.; Mistry, R.S.; Smith, S.H.; Bilbo, S.D. Microglia and Memory: Modulation by Early-Life Infection. J. Neurosci. 2011, 31, 15511–15521. [Google Scholar] [CrossRef]
- Bilbo, S.D.; Rudy, J.W.; Watkins, L.R.; Maier, S.F. A Behavioural Characterization of Neonatal Infection-Facilitated Memory Impairment in Adult Rats. Behav. Brain Res. 2006, 169, 39–47. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.V.; Domiguéz-Andrés, J.; Netea, M.G. The Role of Cell Metabolism in Innate Immune Memory. J. Innate Immun. 2022, 14, 42–50. [Google Scholar] [CrossRef]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-Specific Control of Inflammation by TLR-Induced Chromatin Modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368.e14. [Google Scholar] [CrossRef] [PubMed]
- Seeley, J.J.; Baker, R.G.; Mohamed, G.; Bruns, T.; Hayden, M.S.; Deshmukh, S.D.; Freedberg, D.E.; Ghosh, S. Induction of Innate Immune Memory via MicroRNA Targeting of Chromatin Remodelling Factors. Nature 2018, 559, 114–119. [Google Scholar] [CrossRef]
- Ciernia, A.V.; Link, V.M.; Careaga, M.; LaSalle, J.M.; Ashwood, P. Genetic Variants Drive Altered Epigenetic Regulation of Endotoxin Response in BTBR Macrophages. Brain Behav. Immun. 2020, 89, 20–31. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsem, K.; Van Der Ent, M.A.; et al. Epigenetic Programming of Monocyte-to-Macrophage Differentiation and Trained Innate Immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef]
- Bauer, M.; Weis, S.; Netea, M.G.; Wetzker, R. Remembering Pathogen Dose: Long-Term Adaptation in Innate Immunity. Trends Immunol. 2018, 39, 438–445. [Google Scholar] [CrossRef]
- Maher, P. Investigations into the Role of Metabolism in the Inflammatory Response of BV2 Microglial Cells. Antioxidants 2021, 10, 109. [Google Scholar] [CrossRef]
- Xavier, A.M.; Distéfano-Gagné, F.; Belhamiti, N.; Belhocine, S.; Bitarafan, S.; Falle, A.; Fiola, S.; Rivest, S.; Gosselin, D. Systematic Delineation of Signaling and Epigenomic Mechanisms Underlying Microglia Inflammatory Activity in Acute and Chronic Brain Pathologies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Huang, M.; Malovic, E.; Ealy, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Microglial Immune Regulation by Epigenetic Reprogramming through Histone H3K27 Acetylation in Neuroinflammation. Front. Immunol. 2023, 14, 1052925. [Google Scholar] [CrossRef] [PubMed]
- Bernier, L.-P.; York, E.M.; MacVicar, B.A. Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends Neurosci. 2020, 43, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Bernier, L.-P.; York, E.M.; Kamyabi, A.; Choi, H.B.; Weilinger, N.L.; MacVicar, B.A. Microglial Metabolic Flexibility Supports Immune Surveillance of the Brain Parenchyma. Nat. Commun. 2020, 11, 1559. [Google Scholar] [CrossRef] [PubMed]
- Joshi, L.; Plastira, I.; Bernhart, E.; Reicher, H.; Koyani, C.N.; Madl, T.; Madreiter-Sokolowski, C.; Koshenov, Z.; Graier, W.F.; Hallström, S.; et al. Lysophosphatidic Acid Induces Aerobic Glycolysis, Lipogenesis, and Increased Amino Acid Uptake in BV-2 Microglia. Int. J. Mol. Sci. 2021, 22, 1968. [Google Scholar] [CrossRef] [PubMed]
- Lauro, C.; Limatola, C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 2020, 11, 493. [Google Scholar] [CrossRef]
- York, E.M.; Zhang, J.; Choi, H.B.; MacVicar, B.A. Neuroinflammatory Inhibition of Synaptic Long-term Potentiation Requires Immunometabolic Reprogramming of Microglia. Glia 2021, 69, 567–578. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is an Inflammatory Signal That Induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The Suitability of BV2 Cells as Alternative Model System for Primary Microglia Cultures or for Animal Experiments Examining Brain Inflammation. ALTEX 2009, 26, 83–94. [Google Scholar] [CrossRef]
- Pollock, T.B.; Cholico, G.N.; Isho, N.F.; Day, R.J.; Suresh, T.; Stewart, E.S.; McCarthy, M.M.; Rohn, T.T. Transcriptome Analyses in BV2 Microglial Cells Following Treatment with Amino-Terminal Fragments of Apolipoprotein E. Front. Aging Neurosci. 2020, 12, 256. [Google Scholar] [CrossRef]
- Meleady, L.; Towris, M.; Bacarac, V.; Kim, J.; Rowland, M.; Ciernia, A.V. Histone Deacetylase 3 Regulates Microglial Function through Histone Deacetylation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Grubman, A.; Choo, X.Y.; Chew, G.; Ouyang, J.F.; Sun, G.; Croft, N.P.; Rossello, F.J.; Simmons, R.; Buckberry, S.; Landin, D.V.; et al. Transcriptional Signature in Microglia Associated with Aβ Plaque Phagocytosis. Nat. Commun. 2021, 12, 3015. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Z.; Song, J.; Wang, H.; Li, J.-X.; Xiao, Q.; Yu, Z.; Liu, J.-X.; Liu, Z. B Cell-Activating Factor Promotes B Cell Survival in Ectopic Lymphoid Tissues in Nasal Polyps. Front. Immunol. 2020, 11, 625630. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhang, R.; Xu, Z.; Ke, Y.; Sun, R.; Yang, H.; Zhang, X.; Zhen, X.; Zheng, L.-T. Early Glycolytic Reprogramming Controls Microglial Inflammatory Activation. J. Neuroinflamm. 2021, 18, 129. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhang, H.; Qin, J.; Xu, Z.; Gui, L.; Liu, B.; Liu, C.; Xu, C.; Liu, W.; Zhang, S.; et al. Rapamycin Inhibits BAFF-Stimulated Cell Proliferation and Survival by Suppressing MTOR-Mediated PP2A-Erk1/2 Signaling Pathway in Normal and Neoplastic B-Lymphoid Cells. Cell Mol. Life Sci. 2015, 72, 4867–4884. [Google Scholar] [CrossRef]
- Wang, J.; Yang, C.; Hou, X.; Xu, J.; Yun, Y.; Qin, L.; Yang, P. Rapamycin Modulates the Proinflammatory Memory-Like Response of Microglia Induced by BAFF. Front. Immunol. 2021, 12, 639049. [Google Scholar] [CrossRef]
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal Immune Activation and Abnormal Brain Development across CNS Disorders. Nat. Rev. Neurol. 2014, 10, 643–660. [Google Scholar] [CrossRef]
- Schwartzer, J.J.; Careaga, M.; Coburn, M.A.; Rose, D.R.; Hughes, H.K.; Ashwood, P. Behavioral Impact of Maternal Allergic-Asthma in Two Genetically Distinct Mouse Strains. Brain Behav. Immun. 2017, 36, 99–107. [Google Scholar] [CrossRef]
- Schwartzer, J.J.; Careaga, M.; Chang, C.; Onore, C.E.; Ashwood, P. Allergic Fetal Priming Leads to Developmental, Behavioral and Neurobiological Changes in Mice. Transl. Psychiatry 2015, 5, e543. [Google Scholar] [CrossRef]
- Schwartzer, J.J.; Careaga, M.; Onore, C.E.; Rushakoff, J.A.; Berman, R.F.; Ashwood, P.; Schwartzer, J.J.; Careaga, M.; Berman, R.F.; Ashwood, P. Maternal Immune Activation and Strain Specific Interactions in the Development of Autism-like Behaviors in Mice. Transl. Psychiatry 2013, 3, 220–226. [Google Scholar] [CrossRef]
- Tamayo, J.M.; Rose, D.; Church, J.S.; Schwartzer, J.J.; Ashwood, P. Maternal Allergic Asthma Induces Prenatal Neuroinflammation. Brain Sci. 2022, 12, 1041. [Google Scholar] [CrossRef] [PubMed]
- Church, J.S.; Tamayo, J.M.; Ashwood, P.; Schwartzer, J.J. Repeated Allergic Asthma in Early versus Late Pregnancy Differentially Impacts Offspring Brain and Behavior Development. Brain Behav. Immun. 2021, 93, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Vogel Ciernia, A.; Careaga, M.; LaSalle, J.M.; Ashwood, P. Microglia from Offspring of Dams with Allergic Asthma Exhibit Epigenomic Alterations in Genes Dysregulated in Autism. Glia 2018, 66, 505–521. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Kopec, A.M.; Smith, C.J.; Ayre, N.R.; Sweat, S.C.; Bilbo, S.D. Microglial Dopamine Receptor Elimination Defines Sex-Specific Nucleus Accumbens Development and Social Behavior in Adolescent Rats. Nat. Commun. 2018, 9, 3769. [Google Scholar] [CrossRef] [PubMed]
- Schalbetter, S.M.; von Arx, A.S.; Cruz-Ochoa, N.; Dawson, K.; Ivanov, A.; Mueller, F.S.; Lin, H.-Y.; Amport, R.; Mildenberger, W.; Mattei, D.; et al. Adolescence Is a Sensitive Period for Prefrontal Microglia to Act on Cognitive Development. Sci. Adv. 2022, 8, eabi6672. [Google Scholar] [CrossRef] [PubMed]
- Block, C.L.; Eroglu, O.; Mague, S.D.; Smith, C.J.; Ceasrine, A.M.; Sriworarat, C.; Blount, C.; Beben, K.A.; Malacon, K.E.; Ndubuizu, N.; et al. Prenatal Environmental Stressors Impair Postnatal Microglia Function and Adult Behavior in Males. Cell Rep. 2022, 40, 111161. [Google Scholar] [CrossRef]
- Bolton, J.L.; Short, A.K.; Othy, S.; Kooiker, C.L.; Shao, M.; Gunn, B.G.; Beck, J.; Bai, X.; Law, S.M.; Savage, J.C.; et al. Early Stress-Induced Impaired Microglial Pruning of Excitatory Synapses on Immature CRH-Expressing Neurons Provokes Aberrant Adult Stress Responses. Cell Rep. 2022, 38, 110600. [Google Scholar] [CrossRef]
- Dayananda, K.K.; Ahmed, S.; Wang, D.; Polis, B.; Islam, R.; Kaffman, A. Early Life Stress Impairs Synaptic Pruning in the Developing Hippocampus. Brain Behav. Immun. 2023, 107, 16–31. [Google Scholar] [CrossRef]
- Smith, C.J.; Lintz, T.; Clark, M.J.; Malacon, K.E.; Abiad, A.; Constantino, N.J.; Kim, V.J.; Jo, Y.C.; Alonso-Caraballo, Y.; Bilbo, S.D.; et al. Prenatal Opioid Exposure Inhibits Microglial Sculpting of the Dopamine System Selectively in Adolescent Male Offspring. Neuropsychopharmacology 2022, 47, 1755–1763. [Google Scholar] [CrossRef]
- Hui, C.W.; Vecchiarelli, H.A.; Gervais, É.; Luo, X.; Michaud, F.; Scheefhals, L.; Bisht, K.; Sharma, K.; Topolnik, L.; Tremblay, M.-È. Sex Differences of Microglia and Synapses in the Hippocampal Dentate Gyrus of Adult Mouse Offspring Exposed to Maternal Immune Activation. Front. Cell Neurosci. 2020, 14, 558181. [Google Scholar] [CrossRef] [PubMed]
- Fernández de Cossío, L.; Guzmán, A.; van der Veldt, S.; Luheshi, G.N. Prenatal Infection Leads to ASD-like Behavior and Altered Synaptic Pruning in the Mouse Offspring. Brain Behav. Immun. 2017, 63, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Chen, C.; Liu, A.; Shan, Q.; Zhu, X.; Jia, C.; Peng, X.; Zhang, M.; Farzinpour, Z.; Zhou, W.; et al. Early-Life Inflammation Promotes Depressive Symptoms in Adolescence via Microglial Engulfment of Dendritic Spines. Neuron 2021, 109, 2573–2589.e9. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Glaros, T.; Zhu, M.; Wang, P.; Wu, Z.; Tyson, J.J.; Li, L.; Xing, J. Network Topologies and Dynamics Leading to Endotoxin Tolerance and Priming in Innate Immune Cells. PLoS Comput. Biol. 2012, 8, e1002526. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-C.; Scicluna, B.P.; Arts, R.J.W.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.L.; Cremer, O.L.; et al. Broad Defects in the Energy Metabolism of Leukocytes Underlie Immunoparalysis in Sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef]
- Lajqi, T.; Stojiljkovic, M.; Williams, D.L.; Hudalla, H.; Bauer, M.; Witte, O.W.; Wetzker, R.; Bauer, R.; Schmeer, C. Memory-Like Responses of Brain Microglia Are Controlled by Developmental State and Pathogen Dose. Front. Immunol. 2020, 11, 546415. [Google Scholar] [CrossRef]
- Jeon, S.-M. Regulation and Function of AMPK in Physiology and Diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. MTOR- and HIF-1α-Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Das, A.; Kim, S.H.; Arifuzzaman, S.; Yoon, T.; Chai, J.C.; Lee, Y.S.; Park, K.S.; Jung, K.H.; Chai, Y.G. Transcriptome Sequencing Reveals That LPS-Triggered Transcriptional Responses in Established Microglia BV2 Cell Lines Are Poorly Representative of Primary Microglia. J. Neuroinflamm. 2016, 13, 182. [Google Scholar] [CrossRef]
- Aktories, P.; Petry, P.; Kierdorf, K. Microglia in a Dish-Which Techniques Are on the Menu for Functional Studies? Front. Cell Neurosci. 2022, 16, 908315. [Google Scholar] [CrossRef] [PubMed]
- García-Cáceres, C.; Balland, E.; Prevot, V.; Luquet, S.; Woods, S.C.; Koch, M.; Horvath, T.L.; Yi, C.-X.; Chowen, J.A.; Verkhratsky, A.; et al. Role of Astrocytes, Microglia, and Tanycytes in Brain Control of Systemic Metabolism. Nat. Neurosci. 2019, 22, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate Metabolism in the Brain Focusing on Astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Mason, S. Lactate Shuttles in Neuroenergetics-Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef]
- Sullivan, O.; Ciernia, A.V. Work Hard, Play Hard: How Sexually Differentiated Microglia Work to Shape Social Play and Reproductive Behavior. Front. Behav. Neurosci. 2022, 16, 989011. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Sequence (5′→3′) | Reverse Sequence (5′→3′) |
|---|---|---|
| Hprt | CAGTACAGCCCCAAAATGGTTA | AGTCTGGCCTGTATCCAACA |
| Il1b | TGGCAACTGTTCCTGAACTCA | GGGTCCGTCAACTTCAAAGAAC |
| Il6 | CGATGATGCACTTGCAGAAA | ACTCCAGAAGACCAGAGGAA |
| Cxcl16 | ATCAGGTTCCAGTTGCAGTC | TTCCCATGACCAGTTCCAC |
| Il10 | ACAAAGGACCAGCTGGACAA | TAAGGCTTGGCAACCCAAGTA |
| Tnfa | GGGTGATCGGTCCCCAAA | TGAGGGTCTGGGCCATAGAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Towriss, M.; MacVicar, B.; Ciernia, A.V. Modelling Microglial Innate Immune Memory In Vitro: Understanding the Role of Aerobic Glycolysis in Innate Immune Memory. Int. J. Mol. Sci. 2023, 24, 8967. https://doi.org/10.3390/ijms24108967
Towriss M, MacVicar B, Ciernia AV. Modelling Microglial Innate Immune Memory In Vitro: Understanding the Role of Aerobic Glycolysis in Innate Immune Memory. International Journal of Molecular Sciences. 2023; 24(10):8967. https://doi.org/10.3390/ijms24108967
Chicago/Turabian StyleTowriss, Morgan, Brian MacVicar, and Annie Vogel Ciernia. 2023. "Modelling Microglial Innate Immune Memory In Vitro: Understanding the Role of Aerobic Glycolysis in Innate Immune Memory" International Journal of Molecular Sciences 24, no. 10: 8967. https://doi.org/10.3390/ijms24108967
APA StyleTowriss, M., MacVicar, B., & Ciernia, A. V. (2023). Modelling Microglial Innate Immune Memory In Vitro: Understanding the Role of Aerobic Glycolysis in Innate Immune Memory. International Journal of Molecular Sciences, 24(10), 8967. https://doi.org/10.3390/ijms24108967